

Abstract
Current treatments for autoantibody-mediated diseases, such as lupus, can cause nonspecific immune suppression. In this paper, we used a bioinformatic approach to identify major histocompatibility complex class I–binding epitopes in the heavy chain variable region of anti-DNA antibodies from lupus-prone (NZB/NZW F1) mice. Vaccination of such mice with plasmid DNA vectors encoding these epitopes induced CD8+ T cells that killed anti-DNA antibody-producing B cells, reduced serum anti-DNA antibody levels, retarded the development of nephritis, and improved survival. Vaccine-mediated induction of anti-VH cytotoxic T lymphocytes that ablate autoreactive B cells represents a novel approach to treat autoantibody-mediated diseases.
Keywords: animal models, peptides, systemic lupus erythematosus, T cell epitopes, T cells
Introduction
B cells are essential for the development of systemic autoimmune disease such as lupus. Genetic deletion of B cells protects mice from developing lupus (1). Drugs such as cyclophosphamide that are currently used to treat lupus deplete B cells (2); the treatment is effective but causes nonspecific immune suppression. As organ damage in lupus is caused, at least in part, by autoantibodies such as anti-dsDNA Ab (3, 4), depletion of dsDNA-specific B cells might inhibit lupus without causing nonspecific immune suppression.
The (NZB × NZW) F1 (BWF1)* mouse is a model of autoantibody-mediated disease that shares many features with human lupus (5). Although these mice carry a diverse autoreactive B cell repertoire, there is a limited clonality for anti-DNA B cells (4, 6–8). In fact, some heavy chain variable region (VH) genes are markedly increased in Ig from BWF1 mice, but are rarely used in Ig of normal mice (7, 8). Additionally, VH regions of Ig molecule contain sequences that can bind MHC class II molecules (9); such sequences are frequently shared among autoantibodies (10, 11). For example, all anti-chromatin autoantibody clones tested express a shared replacement mutation in their framework region, which creates an MHC class II–binding motif in all clones (10). Similarly, MHC class II–binding epitopes frequently occur in anti-DNA and other lupus-related autoantibodies, but are uncommon in the normal Ig repertoire (11). Thus, it may be possible to target autoantibody-producing B cells while sparing normal immune defenses.
Ig molecules contain epitopes that can function as targets for T cell–mediated immune responses (9–13). B cells can process and present such epitopes from the VH of their own Ig (14–16) and activate CD4+ and CD8+ T cells (9–16). Here, we hypothesize that CD8+ CTLs that are specific for an Ig VH epitope will recognize and lyse B cells that can process and display the relevant VH epitope on their surface MHC class I molecules. To test this idea, we identified MHC class I–binding epitopes in the VH of BWF1-derived anti-DNA mAbs. Vaccination of BWF1 mice with plasmid DNA vectors that encode such epitopes activated CTL response against anti-DNA Ab-producing B cells, inhibited anti-DNA Ab production and lupus nephritis, and prolonged survival.
Materials and Methods
Mice.
BWF1 and (BALB/c × NZW) F1 (CWF1) mice were obtained from the The Jackson Laboratory or bred locally. Female mice were used in all experiments.
Peptides.
8- to 15-mer peptides (see Fig. 1 C) were synthesized using F-moc chemistry, purified to a single peak, and tested for mol wt by mass spectrometry (Sigma-Aldrich).
Figure 1.

Identification and selection of VH peptides to induce CTL response in BWF1 mice. (A) Prediction of MHC class I–binding epitopes in the VH of an anti-DNA mAb, A6.1. The cleavage probability, MHC–peptide binding, and overall scores were assigned by computer prediction software (reference 26). The overall score combines the calculated cleavage probability and the binding score (reference 26). (B) Confirmation of predicted epitopes in a cellular binding assay. Kd expression was analyzed in T2.Ld (Control) or T2.Kd cells that were stained with an anti-Kd mAb after overnight incubation without (No peptide) or with peptide VH2 (Peptide KL). Results are from one of three similar experiments. (C) Selection of peptides that have both MHC class I– and class II–binding motifs in the VH of an anti-DNA mAb, A6.1. The MHC class I–binding epitopes are underlined (thick lines); thin lines over the VH sequence denote MHC class II-binding epitopes. Mapping of MHC class II–binding epitopes is described elsewhere (reference 9).
B Cell Hybridomas.
B cell hybridomas, including dsDNA-specific (A6.1 and BWds3) and non-DNA binding (375–57 and 375–100), were established from BWF1 mice by fusion of their spleen cells with a nonsecreting murine myeloma cell line (4, 17).
MHC Binding Assay.
Transporter associated with antigen processing (TAP)-deficient T2.Kd, T2.Ld, or T2.Dd cells were pulsed with peptides at 26°C overnight, washed, and stained with conjugated Abs against relevant MHC class I molecules (18, 19). MHC class I expression was analyzed by flow cytometry. The fluorescence index was calculated by the mean fluorescent intensity (MFI) of MHC class I on T2 cells, using the following formula (13): fluorescence index = MFI (T2 cells pulsed with peptide)/MFI (T2 cells not pulsed with the peptide) − 1.
Construction of Plasmid DNA Vectors.
First, we substituted the human CMV promoter in a mammalian expression vector, pCI (Promega) with the murine CMV promoter that was excised from a plasmid pDK6 (20). The resultant plasmid, termed pCD, was used to construct two minigenes, namely pCD.VH1, that encodes a single peptide (VH1), and pCD.III that encodes three peptides, VH1 plus VH2 plus VH3 (Fig. 1 C). The individual peptide units in the multi-epitope minigene, pCD.III, were separated by glycine (Gly)-Gly linkers, as proteasomal cleavage of cellular proteins generally occurs after the second Gly (21).
To construct pCD.III, four oligonucleotides (oligo), each ∼60 nucleotides with a 20-mer overlapping complementary sequence (underlined), were synthesized at the University of Cincinnati DNA Core as follows: Oligo 1: 5′-CAGAGAA TTCGC-CGCCGCCATGGGCTACTTTATGAACTGGGTGAAGC-AGAGCCA -3′; Oligo 2: 5′-CCCTTGAACTTCTGGTTG-TAGAAACCACCAAGGCTCTTTCCATGGCTCTGCTTCA-CCCAGTTC -3′; Oligo 3: 5′-TACAACCAGAAGTTCAAGG-GCAAGGCCACATTGGGTGGTTCTGAGGACTCTGCACT-CTA -3′; and Oligo 4: 5′-ACGCGGATCCTTAATCTCT-TGCACAATAATAGAGTGCAGAGTCCTCAGA -3′. The final minigenes were assembled by extending the overlapping oligos using PCR reaction under the following conditions: 94°C for 15 s, 55°C for 30 s, 72°C for 50 s, for a total of 35 cycles, then 72°C extension for 5 min. The PCR product was purified using a QIAEXII gel extraction Kit (QIAGEN), and digested with EcoRI and BamHI, gel purified, and cloned in frame downstream to a Kozak sequence (22) and an initiation codon into the multicloning site of the pCD. The single epitope minigene, pCD.VH1, was constructed using a similar approach. For large-scale preparations, plasmid DNA was purified from transformed Escherichia coli, JM109, and concentrated using endo-free columns (QIAGEN). The concentrates were analyzed at A260 and A280 and by agarose gel electrophoresis and ethidium bromide staining, and suspended in endo-free PBS (Sigma-Aldrich) for in vivo injections.
Isolation of T and B Cells.
Spleen cells were enriched for T and B cells using a VarioMACS system and microbead-coated Abs against mouse CD90 (Thy1.2) and CD45R (B220), respectively (Miltenyi Biotech). The purity of isolated populations ranged from 92–98%.
CTL Assay.
CTL activity was tested by the lactate dehydrogenase (LDH) release assay using a CytoTox 96® nonradioactive cytotoxicity assay kit (Promega). Briefly, target cells (1–3 × 104 B cells) were cocultured with effector cells (T cells from vaccinated or control mice) at various ratios for 6–8 h in serum-free AIMV medium (GIBCO BRL). Spontaneous LDH release from the effector and target cells was controlled by separate incubation of the respective cell populations. The percent cytotoxicity was calculated using the formula: % cytotoxicity = ([E−Se−St]) / ([Mt−St]) × 100, where E stands for the experimental LDH release in effector plus target cell cocultures; Se, the spontaneous release by effector cells alone; St, the spontaneous release by target cells alone; and Mt, the maximal release by target cells.
ELISA for the Detection of Anti-DNA and Antipeptide Abs, Total IgG, and Cytokines.
For detection of anti-DNA Ab (4, 9), nitrocellulose-filtered, sonicated calf thymus DNA (Sigma-Aldrich) was coated onto the high-binding EIA/RIA plates (Costar). After blocking, samples were added, followed by alkaline phosphatase-conjugated anti–mouse IgG. Substrate para-nitrophenyl phosphate (Sigma-Aldrich) was then added; plates were read at 405 nm using a Labsystems Multiskan ELISA reader. Anti-DNA Ab levels are expressed as U/ml, which was calculated using a positive standard serum. Antipeptide Abs were detected using a similar protocol, as described previously (23). Sandwich ELISA using Ab pairs and recombinant cytokines (BD Biosciences) or mouse IgG (Southern Biotechnology Associates, Inc.) determined the quantities of IFN-γ, TGF-β, IL-4, IL-13, and IgG (9, 23).
Assessment of Lupus.
Proteinuria was estimated using Albustix (Ames). Severe proteinuria was defined as ≥300 mg/dl (3+) on two consecutive examinations (9). Kidneys were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned to stain with periodic acid Schiff (PAS) and hematoxylin and eosin (H&E). Stained sections were scored for histological changes in a blinded fashion.
Statistical Analysis.
Significance of difference was examined using Student's t test for anti-DNA Ab and cytokines. A log-rank test was performed to compare the proportion of surviving mice and mice with severe proteinuria.
Results
Applying Bioinformatics to Identify the VH-derived CTL Epitopes.
The VH sequence of an anti-dsDNA mAb, A6.1, that was derived from a nephritic BWF1 mouse (4) was screened for the presence of epitopes: (a) that would strongly activate CTLs; and (b) that can be efficiently processed to bind MHC class I molecules in target B cells (24–26). To select the epitopes that can be efficiently processed, we determined the proteasomal cleavage probability using an MHC class I antigenic peptide processing prediction program (MAPPP; reference 26). The calculated cleavage probability and MHC binding scores were combined to determine an overall score (26). In the VH of an anti-DNA mAb, A6.1, four epitopes were predicted to strongly bind BWF1 MHC class I molecule, Kd, and one epitope was predicted to bind Ld (Fig. 1 A).
These predictions were confirmed in a cellular binding assay where we incubated peptides shown in Fig. 1 A with T2.Kd, T2.Ld, or T2.Dd cell lines, which are T2 cells transfected with MHC class I molecules, H2-Kd, Ld, or Dd, respectively (Fig. 1 B). MHC class I expression was analyzed by flow cytometry, and the fluorescence index was calculated, as described in Materials and Methods. As T2 cells are deficient in TAP molecules, exposure to exogenous peptides will increase MHC class I expression on T2 cells that are transfected with the relevant MHC class I molecule (18, 19). For example, the second epitope, KFKGKATL, induced a high Kd expression when incubated with T2.Kd cells (fluorescence index = 1.04), but caused no increase in Kd expression upon incubation with T2.Ld (fluorescence index = 0.02). Similar experiments were performed to confirm the MHC class I binding of other VH epitopes.
Selection of VH Peptides to Induce CTL Response in BWF1 Mice.
As CD8+ CTL priming is generally more efficient in the presence of CD4+ T cell help (27), we selected peptides that could activate both CD4+ and CD8+ T cells. Three such peptides, denoted as VH1, VH2, and VH3 were identified in the VH of A6.1 (Fig. 1 C). The peptide VH1 binds a MHC class II molecule, I-Ed (9), and contains a 9-mer Kd-binding epitope that has high cleavage probability and overall scores (Fig. 1 A). Peptide VH2 binds MHC class II molecules, I-Ed and I-Ez (9), and contains an 8-mer Kd-binding epitope that has moderate cleavage probability and overall scores. Peptide VH3 binds MHC class II, I-Az (9), and is predicted to bind MHC class I of the H-2z haplotype based on its ability to activate CD8+ T cells in NZW parents of BWF1 mice. As MHC class I prediction and binding assays are not available for the H-2z haplotype, we immunized NZW mice that carry H-2z haplotype with the A6.1 heavy chain and determined recall in vitro proliferation of their splenic CD8+ T cells to overlapping peptides from the VH of A6.1 (unpublished data). An 8-mer epitope (underlined sequence in the VH3 peptide, Fig. 1 C) activated strong CD8+ T cell proliferation in NZW mice.
Upon immunization with the synthetic VH1 peptide in CFA, normal CWF1 mice exhibited strong CD8+ T cell proliferative responses; lupus-prone BWF1 mice, on the other hand, had a weak or no response (unpublished data). As antigens administered as genes via plasmid DNA or viral vectors generally activate strong CD8+ T cell responses (28, 29), this mode of peptide delivery might overcome the impairment in CD8+ CTL response in lupus mice. Therefore, we assembled the cDNA encoding a single peptide, VH1, or multiple peptides, VH1 plus VH2 plus VH3, by PCR and cloned them into a modified plasmid DNA vector (pCD) to construct two minigenes, pCD.VH1 and pCD.III, respectively (see Materials and Methods). The presence of gene inserts in the pCD.VH1 and pCD.III was verified by sequencing from the downstream of mCMV promoter using a mCMV primer (5′-CGACCAGCGTCGGTACCGTCGCAG-3′).
Plasmid DNA Vectors that Encode VH Peptides Induce Peptide-specific CTL and Ab Responses.
We first determined if the pCD.VH1 or pCD.III induce peptide-specific responses in vivo. Inclusion of MHC class II–binding epitopes in the minigene constructs allowed us to test peptide-specific Ab responses. BWF1 mice were inoculated with 50 μg each of three intradermal injections of peptide expression vectors (pCD.VH1 or pCD.III) at weekly intervals. Control mice received the same amount of a null vector, pCD, or the same volume of PBS. Sera collected 10 d after the last injection were tested for anti-VH1, anti-VH2, and anti-VH3 Abs (Fig. 2 A). Inoculation of mice with pCD.VH1 increased anti-VH1 but not anti-VH2 or anti-VH3 Abs, and inoculation with pCD.III induced Abs that reacted with all three peptides, VH1, VH2, or VH3.
Figure 2.

VH peptide expression vectors induce peptide-specific Ab and CTL responses in BWF1 mice. 3-mo-old BWF1 mice were inoculated with three intradermal injections of PBS (⋄), a null (pCD, ▪) or peptide expression vectors (pCD.VH1, •; pCD.III, ▴) at weekly intervals (n = 9 mice per group). 10 d after the last injection, mice were bled for detection of serum anti-peptide Abs (A) and killed to harvest spleen cells for CTL assays (B). (A) Serum IgG anti-VH1, anti-VH2, and anti-VH3 Abs are expressed as the mean ± SD OD. (B) Splenic T cells pooled from five mice in each group served as effector cells in CTL assays that used CWF1 B cells as target cells. Minigene-induced anti-VH T cells exhibited CTL activity against peptide-pulsed CWF1 B cells (middle panel), but had little or no CTL activity against unpulsed CWF1 B cells (left panel). Depletion of CD8+ T cells by the addition of an anti-CD8a mAb to the cultures abrogated CTL activity (right panel). Results are from one of three similar experiments.
To determine the induction of peptide-specific CTLs, spleen cells from the vaccinated and control mice were cultured with 10 μg/ml each of the relevant or irrelevant peptides and 100 U/ml hIL-2 for 5 d. Purified T cells from these cultures were tested for CTL activity against B cells from MHC-matched (H-2du), normal CWF1 mice (Fig. 2 B). T cells from the pCD.III-vaccinated mice did not exhibit CTL activity against CWF1 B cells (Fig. 2 B, left panel), unless they were pulsed with the VH peptides (Fig. 2 B, middle panel). The CTL activity was abolished when CD8+ T cells were depleted by an anti-CD8 mAb (0.5 μg/106 cells of clone 53–6.7; BD Biosciences; Fig. 2 B, right panel). T cells from the pCD.VH1-vaccinated mice exhibited CTL activity against CWF1 B cells when pulsed with the VH1 (Fig. 2 B, middle panel), but not when pulsed with an irrelevant (VH3) peptide (unpublished data). Similar results were obtained using target B cells from young male BWF1 mice (unpublished data). Thus, VH peptide-specific CD8+ CTLs recognize and lyse B cells that display the relevant VH epitope.
VH Peptide Expression Vectors Activate CD8+ T Cells that Kill Anti-DNA B Cell Hybridomas.
To test if the minigene-induced anti-VH CTLs recognize and lyse B cells that may express naturally processed VH epitopes, BWF1 mice were inoculated with three intradermal injections of PBS, pCD, or pCD.III at weekly intervals. 10 d after the last injection, splenic T cells from these mice were tested for CTL activity against two BWF1-derived B cell hybridomas: A6.1, a dsDNA-specific hybridoma (4) that expresses Ig that contains the VH1/VH2/VH3 sequences, and 375–57, a nonautoreactive hybridoma (17) that expresses Ig that does not contain these sequences (Fig. 3 A). Results show that T cells from pCD.III-vaccinated mice exhibited CTL activity against A6.1, but not against 375–57. Depletion of CD8+ T cells by the addition of anti-CD8a mAb to the cultures abrogated the CTL activity (unpublished data). Similar results were obtained using another set of dsDNA-specific and nonautoreactive B cell hybridomas. Thus, anti-VH CTLs can kill B cells that express Ig that contains the VH epitopes, but do not kill B cells that do not express the relevant VH epitopes.
Figure 3.

VH minigene vaccination of BWF1 mice induces anti-VH CD8+ CTLs that kill BWF1-derived primary or hybridoma B cells. 3-mo-old female BWF1 mice were inoculated with PBS (⋄), pCD (▪), or pCD.III (▴), as described in the legend of Fig. 2. Splenic T cells pooled from five mice in each group served as effector cells in CTL assays that used BWF1-derived B cell hybridomas, 375–57 and A6.1 (A) or purified splenic B cells from BWF1 mice (B) as target cells. Anti-VH T cells exhibited CTL activity against a dsDNA-specific B cell hybridoma (A6.1) or B cells from aged syngeneic mice (6–9-mo-old BWF1, n = 4) that had high levels of IgG anti-DNA Abs (right panels), but had little or no CTL activity against a nonautoreactive hybridoma (375–57) or B cells from young (2-mo-old) BWF1 mice that did not have detectable anti-DNA Ab-secreting B cells (left panels). Results are from one of four similar experiments.
VH Minigene-induced CTLs Kill B Cells from Diseased BWF1 Mice.
To determine if minigene-induced, VH peptide-specific CD8+ T cells kill autoreactive B cells from lupus mice, T cells from pCD.III-vaccinated or control mice were tested for CTL activity against B cells from untreated BWF1 mice (Fig. 3 B). Results show that T cells from pCD.III-vaccinated BWF1 mice induced CTL activity against B cells from 6–9-mo-old BWF1 mice that had high levels of anti-DNA Abs, but not against B cells from 1–2-mo-old BWF1 that did not have detectable IgG anti-DNA Ab-secreting B cells.
T Cells from VH Minigene-vaccinated Mice Inhibit IgG Anti-DNA Ab Production In Vitro.
To determine if minigene-induced anti-VH CD8+ T cells inhibit anti-DNA Ab production, T cells from pCD.III-vaccinated mice were cocultured with B cells from untreated, 9-mo-old, nephritic BWF1 mice; 5th d culture supernatants were tested for anti-DNA Abs. As shown in Fig. 4 A, IgG anti-DNA Ab production was significantly decreased in the presence of T cells from pCD.III-vaccinated mice compared with pCD- or PBS-injected controls. This decrease in anti-DNA Ab production was abrogated by the depletion of CD8+ (Fig. 4 A), but not of CD4+ (unpublished data) T cells, suggesting a role of anti-VH CD8+ T cells in the inhibition of anti-DNA Ab production.
Figure 4.


Minigene-induced T cells inhibit IgG anti-DNA Ab production in vitro and in vivo. 3-mo-old BWF1 mice were vaccinated with three weekly intradermal injections (three short arrows in panel B) of pCD.III (▴), pCD (▪), or PBS (⋄) (n = 15 per group). (A) Six mice in each group were killed 10 d after the last injection, and their splenic T cells were cocultured with B cells from 9-mo-old untreated BWF1 mice for 5 d in the absence or presence of three peptides. IgG anti-DNA Abs in culture supernatants are shown as mean ± SD U/ml. *P < 0.05, pCD.III versus control (pCD or PBS) groups (Student's t test). (B) The remaining nine mice were challenged once subcutaneously (a long arrow) with 25 μg each of peptide VH1, VH2, and VH3 together in a 1:1 emulsion with CFA 2 wk after the last injection. Their monthly sera were tested for anti-dsDNA Abs (left panel) or total IgG (right panel). Results are expressed as the mean ± SD. *P = 0.01 to 0.003, pCD.III versus both control groups; **P = 0.04, pCD.III versus pCD group (Student's t test). Results are from one of two similar experiments. (C) 10-wk-old BWF1 mice were inoculated thrice on alternate days with pCD.III or pCD (n = 8 per group). A week after the last injection, spleens were harvested and single cell suspensions were cultured with IL-2 and 10 μg/ml each of the three MHC class I–binding epitopes shown in Fig. 1C (cells from pCD.III-injected mice) or with IL-2 alone (cells from pCD-injected mice). After 3 to 7 d in culture, cells were washed, purified as live cells using Ficoll-Hypaque, and then enriched for T cells using MACS. The purified T cells (5 × 106 or 5 × 103) were injected intravenously twice at 3-d intervals into 22-wk-old BWF1 mice (n = 6 mice per group). 5 d after the second transfer, recipient animals were killed and their spleen cells were tested for IgG anti-DNA Ab forming cells in an ELISPOT assay. Results are shown as the mean ± SD IgG anti-DNA Ab forming cells per 106 spleen cells. *P = 0.02, recipients of T cells from pCD.III-vaccinated mice (▴) versus recipients of T cells from pCD-injected mice (▪).
Vaccination with VH Minigenes Reduces IgG Anti-DNA Ab Production In Vivo.
To determine if the VH minigene vaccination inhibits anti-DNA Ab production in vivo, we vaccinated BWF1 mice with pCD.III, pCD, or PBS three times at weekly intervals, and monitored serum IgG anti-dsDNA Ab and total IgG levels (Fig. 4 B). 10 d after the last inoculation, serum IgG anti-DNA Ab levels increased in mice injected with the null vector (pCD) compared with pCD.III-vaccinated mice (P = 0.04), which is consistent with previous reports that showed a transient increase in anti-DNA Ab production after plasmid or bacterial DNA immunizations (30, 31). Subsequent challenge with peptides VH1, VH2, and VH3 at 4-mo of age accelerated anti-DNA Ab production in the control pCD- or PBS-injected mice, but not in the pCD.III-vaccinated mice, which continued to have significantly lower levels of serum anti-DNA Abs at 5 and 6 mo of age (P ≤ 0.01). Total serum IgG levels, however, were not significantly decreased in the pCD.III-vaccinated mice (Fig. 4 B, right panel).
To further examine if the decrease in anti-DNA Ab production in pCD.III-vaccinated mice is mediated by peptide-specific CTLs, we performed adoptive transfer experiments wherein T cells from vaccinated mice were transferred into naive BWF1 mice and anti-DNA Ab-producing B cells were enumerated in spleens of recipients (Fig. 4 C). In brief, BWF1 mice were inoculated thrice with pCD.III or pCD. A week after the third inoculation spleen cells from these mice were expanded in vitro with the relevant MHC class I–binding peptides and IL-2. After one or two cycles of stimulation, purified live T cells were injected into 22-wk-old BWF1 mice that had moderate to high levels of serum IgG anti-DNA Abs. 5 d after the transfer, recipient animals were killed and their spleen cells were tested for IgG anti-DNA Ab producing cells in an ELISPOT assay, as described previously (11). Results show that animals transferred with 5 × 106 T cells from pCD.III-vaccinated mice had significantly decreased numbers of IgG anti-DNA Ab forming cells compared with animals that received smaller numbers (5 × 103) of T cells from the same mice (P = 0.04) or as compared with animals that received the same numbers of T cells from control pCD-injected mice (P = 0.02; Fig. 4 C).
VH Minigene Vaccination Suppresses Lupus Nephritis and Improves Survival.
To determine the effect of vaccination with VH minigenes on the development of lupus, BWF1 mice were vaccinated as described above and monitored for proteinuria and survival (Fig. 5, A and B) . The development of severe proteinuria was delayed and survival was prolonged in the pCD.III-vaccinated mice as compared with control mice (P < 0.01 to < 0.001, log rank test).
Figure 5.

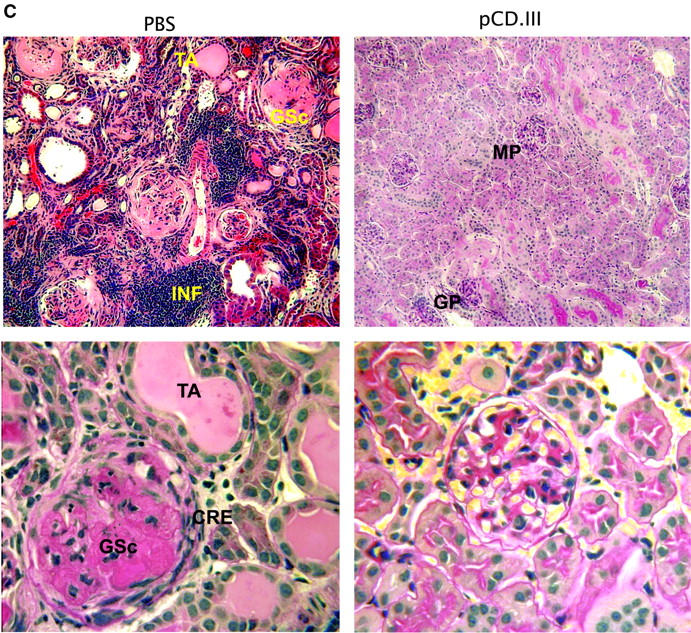
Effect of VH minigene vaccination on lupus nephritis and survival. 3-mo-old BWF1 mice were vaccinated with pCD.III (▴), pCD (▪), or PBS (⋄), as described in Fig. 4 (n = 14 mice per group pooled from two experiments). The percent cumulative incidence of severe (≥300 mg/dl) proteinuria and survival are shown in A and B, respectively. P < 0.01 to < 0.001 by log rank test in pCD.III-vaccinated versus pCD- or PBS-injected mice; P = NS in pCD versus PBS groups. (C) Representative H+E– or PAS-stained renal sections from BWF1 mice inoculated with PBS (left panels) or pCD.III (right panels). Kidneys were harvested at the age of 40 wk, or before in mice that had terminal disease. Findings were similar in the two control groups (PBS and pCD) of mice, which had severe glomerular sclerosis (GSc), interstitial infiltrates (INF), fibrous and cellular crescents (CRE), and tubular atrophy (TA). In contrast, the pCD.III-vaccinated mice had only mild glomerular (GP) or mesangial proliferation (MP). Original magnification: ×200 in the top panels and ×400 in bottom panels.
Kidney sections from the vaccinated and control BWF1 mice were stained with PAS and H&E (Fig. 5 C), and examined for the active (glomerular proliferation, necrosis, crescent formation, and interstitial inflammation) and chronic lesions (glomerulosclerosis, interstitial fibrosis, and tubular atrophy) of lupus nephritis. Findings were similar in the two control groups of mice (PBS- and pCD-injected), all of which had severe glomerular proliferation and/or sclerosis in ≥60% glomeruli per kidney, extensive interstitial infiltrates, fibrous and cellular crescents, and tubular atrophy. The pCD.III-vaccinated mice, on the other hand, had early changes of lupus nephritis, including mesangial or diffuse glomerular proliferation in ≤30% glomeruli per kidney and interstitial infiltrates in ≤3 small foci per kidney. Remarkably, the pCD.III-vaccinated mice had minimal or no chronic changes (glomerulosclerosis, fibrosis, and tubular atrophy). Thus, vaccination with pCD.III markedly retarded the development and progression of lupus nephritis.
Peptide-specific Cytokine Responses in Spleen Cells from the Minigene-vaccinated Mice: Enhanced TGF-β and IFN-γ, but Reduced IL-4 and IL-13, Production.
Cytokines produced during an immune response can influence the generation of CTLs (32–34). To determine if the VH minigene vaccination alters peptide-specific cytokine production in BWF1 mice, we cultured spleen cells from the vaccinated and control mice with or without VH peptides and measured cytokines in supernatants (Fig. 6) . TGF-β and IFN-γ levels were significantly increased, whereas IL-4 and IL-13 were decreased in the pCD.III-vaccinated compared with pCD- or PBS-injected mice. Such cytokine profile may contribute to the induction of potent CD8+ CTL responses by minigene vaccination (32–34).
Figure 6.

Cytokine production by spleen cells is altered in minigene-vaccinated mice. 10 d after the third injection of pCD.III, pCD, or PBS, spleen cells from BWF1 mice were cultured without (Medium) or with three VH peptides (VH1, VH2, and VH3, Peptide) for 48 h, and supernatants were tested for IFN-γ, active TGF-β, IL-4, and IL-13. Results are expressed as the mean ± SD pg/ml in five mice per group from one of three similar experiments. *P < 0.05; **P < 0.01 in pCD-III-vaccinated versus pCD- or PBS-injected groups (Student's t test).
Discussion
In this paper, we show evidence for a novel mechanism of limiting autoantibody production: anti-DNA VH peptide-reactive CD8+ CTLs ablate anti-DNA autoantibody-producing B cells. Vaccination of BWF1 mice with a minigene encoding three VH-derived epitopes inhibits serum autoantibody levels and markedly improves lupus nephritis and survival, suggesting that these anti-VH CTLs may have targeted a broad repertoire of autoreactive B cells.
A major question is how a few CTL epitopes from a single VH inhibit a systemic disease that is apparently caused by autoantibodies of diverse origin. A review of published data on Ig VH gene usage suggests that while there are no autoantibody-specific VH genes, certain VH genes that are rare in Igs of normal mice are commonly used in anti-DNA Ab (7, 8). Moreover, we and others have shown that autoantibody VH framework regions contain T cell epitopes (10, 11) that are commonly present in anti-DNA and other lupus-related autoantibodies, but are uncommon in the normal Ig repertoire (11). Thus, activation of T cells reactive with such shared epitopes might target a broad array of related autoreactive B cells while sparing the unrelated normal B cell repertoire. It is not clear how such specific T cell epitopes are created in the autoantibody VH regions. In a recent paper, a framework (FR1) replacement mutation that was shared among all anti-chromatin autoantibody clones did not enhance chromatin binding, but created a T cell epitope (10). These mutations may not be frequent in nonautoreactive Ab. Indeed, lupus B cells exhibit subtle alterations in the pattern of somatic mutations when compared with normal B cells (35). Such somatic mutations in the autoantibody VH regions may create unique T cell epitopes that would commonly target autoantibody-specific B cells. Consistent with this explanation, the VH peptide-reactive CTLs kill dsDNA-specific B cells that express the VH epitope but do not kill nonautoreactive B cells that do not express the relevant epitopes (Fig. 3 A).
In a GenBank database analysis, 17% of Igs that express three anti-DNA VH–derived T cell epitope sequences are nonself-reactive Ig (11, and unpublished data). The lysis of B cells that express the nonself-reactive Ig containing such T cell epitope sequences would be a tolerable clinical side effect. In our experiments, however, anti-VH CTLs did not kill B cells from nonautoimmune CWF1 (Fig. 2 B, left panel), young, preautoimmune female BWF1 (Fig. 3 B, left panel), or young male BWF1 mice (unpublished data) that did not have detectable autoantibodies in their circulation. B cells from these mice, however, became the target of anti-VH CTLs, when they were pulsed with the appropriate VH–derived peptide (Fig. 2 B, middle panel, and unpublished data). Thus, nonautoreactive B cells can also become the target of anti-VH CTLs, if they express an Ig that contains the epitope and can process and display the peptide–MHC class I complexes, or if the peptide is loaded on to their MHC class I exogenously. The latter scenario can be imagined in mice or humans with SLE who have large amounts of circulating autoantibodies that might undergo physiologic proteolysis, releasing VH peptides into the plasma, which could then bind MHC class I on normal B cells. This would result in VH-specific killing of both autoreactive and normal B cells. This scenario, however, is not supported by our data, as only ∼20% of splenic B cells from BWF1 mice with advanced disease were killed by anti-VH CTLs (Fig. 3 B, right panel). In vivo, the anti-VH CTLs must have specifically targeted anti-DNA Ab-expressing B cells, as serum anti-DNA Ab levels, but not the total serum IgG, were significantly decreased in minigene-vaccinated BWF1 mice (Fig. 4 B).
Inclusion of MHC class II–binding epitopes in minigene constructs allowed us to track the expression of minigenes in vivo by determining serum levels of peptide-specific Abs (Fig. 2 A). The MHC class II–binding epitopes may also activate peptide-specific CD4+ Th cells, which are known to promote CD8+ CTL priming (27), thus improving the efficacy of the vaccination. Additionally, very high levels of expression of MHC class II–binding VH epitopes may induce a high-zone tolerance in CD4+ Th cells (36), which may also improve the efficacy of vaccination. This may be possible, as intravenous administration of large doses of soluble MHC class II–binding VH peptides is known to induce tolerance in CD4+ Th cells and inhibit disease in BWF1 mice (36). In contrast, repeated immunizations by subcutaneous injections of MHC class II–binding VH peptides in adjuvant activates CD4+ Th cells that accelerate disease in BWF1 mice (9). Thus, inclusion of MHC class II–binding epitopes in minigene constructs may also potentially worsen the efficacy of vaccination, especially in older BWF1 mice that exhibit stronger Th (antipeptide Ab) and weaker CTL responses than in younger, prenephritic animals (unpublished data). Indeed, although treatment of 24-wk-old BWF1 mice with pCD.III decreased anti-DNA Ab levels and proteinuria (P = 0.05–0.06 versus pCD- or PBS-treated control mice; n = 12 mice per group; unpublished data), it was far less effective than when the vaccination was initiated at a prediseased stage (Figs. 4 and 5). To address the possibility that decreased efficacy of minigene treatment is due to inclusion of overlapping MHC class II–binding Th epitopes in the construct, we generated another plasmid DNA vector that encodes ‘pure’ MHC class I–binding epitopes (underlined 8–9-mer sequences in Fig. 1 C). Treatment of 24-wk-old BWF1 mice that already had clinical nephritis (proteinuria ≥100 mg/dl) with this vector significantly decreased serum IgG anti-DNA Ab levels (P < 0.05, Student's t test) and proteinuria (P < 0.01, log rank test) compared with the control PBS and pCD injected mice (n = 15 mice per group). By 36 wk of age, only 20% mice in the minigene-treated group compared with 67 and 74% in the PBS and pCD groups, respectively, had developed persistent, severe proteinuria (≥300 mg/dl). Thus, in mice with full-blown lupus, treatment with a minigene encoding CTL epitopes alone is likely to be more effective than a minigene that encodes both CTL and Th epitopes.
The beneficial effect of minigene treatment in 24-wk-old BWF1 mice was associated with a significant decrease in anti-DNA Ab-producing B cells. The mean ± SE IgG anti-DNA Ab-producing B cells were 308 ± 48, 353 ± 70, and 158 ± 28 per 106 spleen cells in 36-wk-old PBS, pCD, and minigene-treated animals, respectively (P < 0.05 in treated versus control mice, n = 6 mice per group). Total CD19+ B and CD4+ T cell numbers and relative percentages were also slightly lower (P = 0.05–0.06), whereas CD8+ T cells were significantly higher (P < 0.01) in minigene-treated than in control mice. Minigene treatment did not result in nonspecific inhibition of all Ig production, as IgG anti–hen egg lysozyme Ab-producing B cell numbers were similar in the treated and control mice immunized with hen egg lysozyme (unpublished data).
The IdGN2 is an idiotype that is highly enriched in glomerular Ig deposits in kidneys from nephritic BWF1 mice (37) and among nephritogenic anti-DNA Ab-producing B cells, including A6.1 mAb, derived from diseased BWF1 mice (4, 38). We have recently found that an anti-IdGN2 Ab that was established by immunizing BWF1 mice with A6.1 selectively binds to 8-mer sequences represented in the VH1 and VH2 peptides (unpublished data). Thus, IdGN2-expressing Ig may represent a subset of nephritogenic autoantibodies; B cells expressing such Ig may be specifically targeted by CTLs reactive against VH1 and VH2 epitopes. Indeed, IdGN2-positive ELISPOTs were dramatically decreased in minigene-treated mice (28 ± 20) as compared with the control mice (PBS, 198 ± 31; pCD, 182 ± 22 per 106 spleen cells, mean ± SE; P < 0.001, n = 6 mice per group). This suggests that VH minigene treatment preferentially targets B cells that secrete nephritogenic Abs.
Overwhelming evidence supports a nephritogenic role of anti-DNA Ab (3, 4). Several recent reports, however, suggest that ‘circulating’ autoantibodies may not be essential for the development of lupus nephritis. For example, MRL-lpr/lpr mice rendered deficient in circulating Ig develop nephritis, although the disease in mutant mice is less marked than in the intact mice (39). Another report showed that treatment of nephritic BWF1 mice with CTLA4Ig and anti-CD40 ligand Ab decreases proteinuria, but does not decrease circulating anti-DNA Ab levels; anti-DNA Ab-producing splenic B cells, however, are reduced in the treated mice (40). Genome-wide studies in lupus-prone mice also suggest a dissociation of some anti-DNA Ab loci from glomerulonephritis or mortality loci (41–43), while other loci such as MHC are linked to all three traits (41). As nephritogenic anti-DNA Abs (4), which may be regulated by MHC, may comprise only a fraction of circulating anti-DNA Abs, it is not surprising that serum anti-DNA Ab levels do not always correlate with the presence of nephritis. Moreover, the deposition of anti-DNA Abs may represent only one step in the development of lupus nephritis; subsequent tissue damage may be caused by other mechanisms. Irrespective of the role of circulating anti-DNA Ab, functional B cells expressing surface Ig are essential for the development of lupus nephritis (39). Thus, our data showing a profound effect of depletion of anti-DNA B cells on the overall disease (Figs. 4 and 5) are consistent with the published literature.
The CD8+ CTL and suppressor responses are known to be defective in patients with SLE (44, 45). A similar impairment was observed in lupus-prone BWF1 mice when they were immunized with synthetic peptides in CFA (unpublished data). The impairment in CD8+ T cell responses in BWF1 mice, however, could be overcome by administration of peptides via a plasmid DNA vector (Fig. 3). This effect of DNA vaccination is due primarily to the increased access of peptides that are synthesized within cells to the MHC class I antigen presentation pathway (28). In addition, cytokines produced during an immune response can influence the generation of CTLs (32–34): in vivo CD8+ CTL activity is enhanced by IFN-γ, and diminished by IL-4. In our experiments, vaccination with the VH peptide minigenes, pCD.VH1 or pCD.III increased the production of IFN-γ and TGF-β and decreased IL-4 and IL-13 production by spleen cells in a peptide-specific manner (Fig. 6). Thus, increased IFN-γ and decreased IL-4 production by minigene vaccination may further facilitate the induction of CTLs and enable mice and humans with lupus to overcome the impairment in CTL responses. Additionally, TGF-β selectively activates CD8+ T cells to proliferate (46) and can condition these cells to become suppressors of Ab production (47). Thus, an increase in TGF-β may augment the priming of CTLs and enhance their capability to inhibit autoreactive B cell Ig production in lupus mice.
Finally, we asked how would a therapeutic approach that depends on restricted MHC types be applicable to the clinical situation? Interestingly, screening of published anti-DNA Ab sequences in BWF1 and MRL-lpr/lpr mice reveals that similar T cell epitope sequences are present in the VH of anti-DNA Abs from genetically unrelated mouse strains with SLE (11). Furthermore, such epitopes can bind MHC class I molecules from many different mouse strains. For example, peptide VH2 (Fig. 1 C) contains epitopes that are predicted to bind MHC class I molecules in NZB (H2-Kd), MRL-lpr/lpr (H2-Kk), and BXSB strains (H2-Kb and H2-Db), and in humans (HLA-A*0201). Vaccination using such promiscuous and shared epitopes may elicit anti-VH CTLs across the MHC barrier. An alternative therapeutic approach is to vaccinate using longer sequences of VH genes, which may obviate the limitation of MHC type-dependence. The longer VH sequences, however, may contain both Th and CTL epitopes. A preferential processing and presentation of Th epitopes might activate autoantibody-promoting Th cells and exacerbate lupus, thus reducing the beneficial effect of CTL epitopes. Indeed, our preliminary results suggest that vaccination using a minigene that encodes a ‘pure’ MHC class II-binding epitope without any overlapping or adjacent MHC class I–binding epitope exacerbates lupus nephritis (unpublished data).
In conclusion, altering the presentation of anti-DNA VH-derived T cell epitopes by administering them as minigenes activates CD8+ CTLs that ablate autoreactive B cells in lupus-prone mice. This approach of inhibiting autoantibody production may pave the way for peptide-specific treatments for patients with SLE (36, 48–55). In preliminary analyses, we have found strong HLA class I–binding epitopes in the published VH gene sequences of human anti-DNA mAbs (unpublished data). In these human anti-DNA VH sequences, we will identify the CTL epitopes that would be shared among lupus-related autoantibodies from different patients, but would be rare in the normal human Ig repertoire. A library of such epitopes would be helpful in generating the designer minigene vaccines for patients with SLE and other autoantibody-mediated diseases.
Acknowledgments
We thank Ted Hansen (Washington University School of Medicine, St. Louis, MO) for providing T2 cell transfectants, Tony Marion (University of Tennessee, Memphis, TN) for providing B cell hybridomas, and Todd Braciak, Hermine Brunner, Fred Finkelman, Bevra Hahn, Evelyn Hess, Christopher Karp, and members of the Autoimmunity and Tolerance Laboratory for helpful discussions and suggestions.
This work was supported by grants from the National Institutes of Health (AR47322) and the Arthritis Foundation.
Footnotes
Abbreviations used in this paper: BWF1, (NZB × NZW) F1; CWF1, (BALB/c × NZW) F1; LDH, lactate dehydrogenase; MFI, mean fluorescent intensity; PAS, periodic acid Schiff.
References
- 1.Chan, O.T., M.P. Madaio, and M.J. Shlomchik. 1999. B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J. Immunol. 163: 3592–3596. [PubMed] [Google Scholar]
- 2.Singh, R.R., A.N. Malaviya, R. Sindhwani, R. Malaviya, and S.D. Khare. 1988. Cyclophosphamide pulse therapy in SLE: Effect on clinical activity and lymphocyte subsets. New Engl. Reg. Allergy Proc. 9:479 (Abstr. [Google Scholar]
- 3.Spatz, L., A. Iliev, V. Saenko, L. Jones, M. Irigoyen, A. Manheimer-Lory, B. Gaynor, C. Putterman, M. Bynoe, C. Kowal, et al. 1997. Studies on the structure, regulation, and pathogenic potential of anti-dsDNA antibodies. Methods. 11:70–78. [DOI] [PubMed] [Google Scholar]
- 4.Ohnishi, K., F.M. Ebling, B. Mitchell, R.R. Singh, B.H. Hahn, and B.P. Tsao. 1994. Comparison of pathogenic and nonpathogenic murine antibodies to DNA: antigen binding and structural characteristics. Int. Immunol. 6:817–830. [DOI] [PubMed] [Google Scholar]
- 5.Theofilopoulos, A.N., R. Kofler, P.A. Singer, and F.J. Dixon. 1989. Molecular genetics of murine lupus models. Adv. Immunol. 46:61–109. [DOI] [PubMed] [Google Scholar]
- 6.Tillman, D.M., N.-T. Jou, R.J. Hill, and T.N. Marion. 1992. Both IgM and IgG anti-DNA antibodies are the products of clonally selective B cell stimulation in (NZB x NZW)F1 mice. J. Exp. Med. 176:761–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gangemi, R.M.R., A.K. Singh, and K.J. Barrett. 1993. Independently derived IgG anti-DNA autoantibodies from two lupus-prone mouse strains express a VH gene that is not present in most murine strains. J. Immunol. 151:4660–4671. [PubMed] [Google Scholar]
- 8.Ash-Lerner, A., M. Ginsberg-Strauss, Y. Pewzner-Jung, D.D. Desai, T.N. Marion, and D. Eilat. 1997. Expression of an anti-DNA-associated VH gene in immunized and autoimmune mice. J. Immunol. 159:1508–1519. [PubMed] [Google Scholar]
- 9.Singh, R.R., V. Kumar, F.M. Ebling, S. Southwood, A. Sette, E.E. Sercarz, and B.H. Hahn. 1995. T cell determinants from autoantibodies to DNA can upregulate autoimmunity in murine systemic lupus erythematosus. J. Exp. Med. 181:2017–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang, X., D.S. Smith, A. Guth, and L.J. Wysocki. 2001. A receptor presentation hypothesis for T cell help that recruits autoreactive B cells. J. Immunol. 166:1562–1571. [DOI] [PubMed] [Google Scholar]
- 11.Singh, R.R., B.H. Hahn, B.P. Tsao, and F.M. Ebling. 1998. Evidence for multiple mechanisms of polyclonal T cell activation in murine lupus. J. Clin. Invest. 102:1841–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartnes, K., and K. Hannestad. 1997. Engagement of the B lymphocyte antigen receptor induces presentation of intrinsic immunoglobulin peptides on major histocompatibility complex class II molecules. Eur. J. Immunol. 27:1124–1130. [DOI] [PubMed] [Google Scholar]
- 13.Trojan, A., J.L. Schultze, M. Witzens, R.H. Vonderheide, M. Ladetto, J.W. Donovan, and J.G. Gribben. 2000. Immunoglobulin framework-derived peptides function as cytotoxic T-cell epitopes commonly expressed in B-cell malignancies. Nat. Med. 6:667–672. [DOI] [PubMed] [Google Scholar]
- 14.Weiss, S., and B. Bogen. 1989. B-lymphoma cells process and present their endogenous immunoglobulin to major histocompatibility complex-restricted T cells. Proc. Natl. Acad. Sci. USA. 86:282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yurin, V.L., A.Y. Rudensky, S.M. Mazel, and J.M. Blechman. 1989. Immunoglobulin-specific T-B cell interaction. II. T cell clones recognize the processed form of B cells' own surface immunoglobulin in the context of the major histocompatibility complex class II molecule. Eur. J. Immunol. 19:1685–1691. [DOI] [PubMed] [Google Scholar]
- 16.Weiss, S., and B. Bogen. 1991. MHC class II-restricted presentation of intracellular antigen. Cell. 64:767–776. [DOI] [PubMed] [Google Scholar]
- 17.Krishnan, M.R., and T.N. Marion. 1998. Comparison of the frequencies of arginines in heavy chain CDR3 of antibodies expressed in the primary B-cell repertoires of autoimmune-prone and normal mice. Scand. J. Immunol. 48:223–232. [DOI] [PubMed] [Google Scholar]
- 18.Alexander-Miller, M.A., K. Burke, U.H. Koszinowski, T.H. Hansen, and J.M. Connolly. 1993. Alloreactive cytotoxic T lymphocytes generated in the presence of viral-derived peptides show exquisite peptide and MHC specificity. J. Immunol. 151:1–10. [PubMed] [Google Scholar]
- 19.Deng, Y., J.W. Yewdell, L.C. Eisenlohr, and J.R. Bennink. 1997. MHC affinity, peptide liberation, T cell repertoire, and immunodominance all contribute to the paucity of MHC class I-restricted peptides recognized by antiviral CTL. J. Immunol. 158:1507–1515. [PubMed] [Google Scholar]
- 20.Addison, C.L., M. Hitt, D. Kunsken, and F.L. Graham. 1997. Comparison of the human versus murine cytomegalovirus immediate early gene promoters for transgene expression by adenoviral vectors. J. Gen. Virol. 78:1653–1661. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Otin, C., C. Simon-Mateo, L. Martinez, and E. Vinuela. 1989. Gly-Gly-X, a novel consensus sequence for the proteolytic processing of viral and cellular proteins. J. Biol. Chem. 264:9107–9110. [PubMed] [Google Scholar]
- 22.Kozak, M. 1987. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15:8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh, R.R., B.H. Hahn, and E.E. Sercarz. 1996. Neonatal peptide exposure can prime T cells, and upon subsequent immunization induce their immune deviation: Implications for antibody vs. T cell–mediated autoimmunity. J. Exp. Med. 183:1613–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Burg, S.H., M.J. Visseren, R.M. Brandt, W.M. Kast, and C.J. Melief. 1996. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide complex stability. J. Immunol. 156:3308–3314. [PubMed] [Google Scholar]
- 25.Parker, K.C., M.A. Bednarek, and J.E. Coligan. 1994. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 152:163–175. [PubMed] [Google Scholar]
- 26.Kuttler, C., A.K. Nussbaum, T.P. Dick, H.G. Rammensee, H. Schild, and K.P. Hadeler. 2000. An algorithm for the prediction of proteasomal cleavages. J. Mol. Biol. 298:417–429. [DOI] [PubMed] [Google Scholar]
- 27.Keene, J.A., and J. Forman. 1982. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J. Exp. Med. 155:768–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanke, T., T.J. Blanchard, J. Schneider, C.M. Hannan, M. Becker, S.C. Gilbert, A.V. Hill, G.L. Smith, and A. McMichael. 1998. Enhancement of MHC class I-restricted peptide-specific T cell induction by a DNA prime/MVA boost vaccination regime. Vaccine. 16:439–445. [DOI] [PubMed] [Google Scholar]
- 29.Ulmer, J.B., T.M. Fu, R.R. Deck, A. Friedman, L. Guan, C. DeWitt, X. Liu, S. Wang, M.A. Liu, J.J. Donnelly, and M.J. Caulfield. 1993. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 259:1745–1749. [DOI] [PubMed] [Google Scholar]
- 30.Gilkeson, G.S., P. Ruiz, A.M. Pippen, A.L. Alexander, J.B. Lefkowith, and D.S. Pisetsky. 1996. Modulation of renal disease in autoimmune NZB/NZW mice by immunization with bacterial DNA. J. Exp. Med. 183:1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mor, G., M. Singla, A.D. Steinberg, S.L. Hoffman, K. Okuda, and D.M. Klinman. 1997. Do DNA vaccines induce autoimmune disease? Hum. Gene Ther. 8:293–300. [DOI] [PubMed] [Google Scholar]
- 32.Erard, F., M.T. Wild, J.A. Garcia-Sanz, and G. Le Gros. 1993. Switch of CD8 T cells to noncytolytic CD8−CD4− cells that make TH2 cytokines and help B cells. Science. 260:1802–1805. [DOI] [PubMed] [Google Scholar]
- 33.Aung, S., Y.W. Tang, and B.S. Graham. 1999. Interleukin-4 diminishes CD8+ respiratory syncytial virus-specific cytotoxic T-lymphocyte activity in vivo. J. Virol. 73:8944–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurunathan, S., L. Stobie, C. Prussin, D.L. Sacks, N. Glaichenhaus, A. Iwasaki, D.J. Fowell, R.M. Locksley, J.T. Chang, C.Y. Wu, and R.A. Seder. 2000. Requirements for the maintenance of Th1 immunity in vivo following DNA vaccination: a potential immunoregulatory role for CD8+ T cells. J. Immunol. 165:915–924. [DOI] [PubMed] [Google Scholar]
- 35.Manheimer-Lory, A.J., G. Zandman-Goddard, A. Davidson, C. Aranow, and B. Diamond. 1997. Lupus-specific antibodies reveal an altered pattern of somatic mutation. J. Clin. Invest. 100:2538–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh, R.R., F.M. Ebling, E.E. Sercarz, and B.H. Hahn. 1995. Immune tolerance to autoantibody-derived peptides delays development of autoimmunity in murine lupus. J. Clin. Invest. 96:2990–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hahn, B.H., and F.M. Ebling. 1987. Idiotype restriction in murine lupus; high frequency of three public idiotypes on serum IgG in nephritic NZB/NZW F1 mice. J. Immunol. 138:2110–2118. [PubMed] [Google Scholar]
- 38.Panosian-Sahakian, N., J.L. Klotz, F. Ebling, M. Kronenberg, and B. Hahn. 1989. Diversity of Ig V gene segments found in anti-DNA autoantibodies from a single (NZB x NZW)F1 mouse. J. Immunol. 142:4500–4506. [PubMed] [Google Scholar]
- 39.Chan, O.T., L.G. Hannum, A.M. Haberman, M.P. Madaio, and M.J. Shlomchik. 1999. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 189:1639–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang, X., W. Huang, M. Mihara, J. Sinha, and A. Davidson. 2002. Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J. Immunol. 168:2046–2053. [DOI] [PubMed] [Google Scholar]
- 41.Kono, D.H., R.W. Burlingame, D.G. Owens, A. Kuramochi, R.S. Balderas, D. Balomenos, and A.N. Theofilopoulos. 1994. Lupus susceptibility loci in New Zealand mice. Proc. Natl. Acad. Sci. USA. 91:10168–10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wakeland, E.K., K. Liu, R.R. Graham, and T.W. Behrens. 2001. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 15:397–408. [DOI] [PubMed] [Google Scholar]
- 43.Waters, S.T., S.M. Fu, F. Gaskin, U.S. Deshmukh, S.S. Sung, C.C. Kannapell, K.S. Tung, S.B. McEwen, and M. McDuffie. 2001. NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clin. Immunol. 100:372–383. [DOI] [PubMed] [Google Scholar]
- 44.Stohl, W., J.E. Elliott, L. Li, E.R. Podack, D.H. Lynch, and C.O. Jacob. 1997. Impaired nonrestricted cytolytic activity in systemic lupus erythematosus: involvement of a pathway independent of Fas, tumor necrosis factor, and extracellular ATP that is associated with little detectable perforin. Arthritis Rheum. 40:1130–1137. [DOI] [PubMed] [Google Scholar]
- 45.Filaci, G., S. Bacilieri, M. Fravega, M. Monetti, P. Contini, M. Ghio, M. Setti, F. Puppo, and F. Indiveri. 2001. Impairment of CD8+ T suppressor cell function in patients with active systemic lupus erythematosus. J. Immunol. 166:6452–6457. [DOI] [PubMed] [Google Scholar]
- 46.Rich, S., M. Seelig, H.M. Lee, and J. Lin. 1995. Transforming growth factor beta 1 costimulated growth and regulatory function of staphylococcal enterotoxin B-responsive CD8+ T cells. J. Immunol. 155:609–618. [PubMed] [Google Scholar]
- 47.Gray, J.D., M. Hirokawa, and D.A. Horwitz. 1994. The role of transforming growth factor beta in the generation of suppression: an interaction between CD8+ T and NK cells. J. Exp. Med. 180:1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaynor, B., C. Putterman, P. Valadon, L. Spatz, M.D. Scharff, and B. Diamond. 1997. Peptide inhibition of glomerular deposition of an anti-DNA antibody. Proc. Natl. Acad. Sci. USA. 94:1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waisman, A., P.J. Ruiz, E. Israeli, E. Eilat, S. Konen-Waisman, H. Zinger, M. Dayan, and E. Mozes. 1997. Modulation of murine systemic lupus erythematosus with peptides based on complementarity determining regions of a pathogenic anti-DNA monoclonal antibody. Proc. Natl. Acad. Sci. USA. 94:4620–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jouanne, C., S. Avrameas, and B. Payelle-Brogard. 1999. A peptide derived from a polyreactive monoclonal anti-DNA natural antibody can modulate lupus development in (NZBxNZW)F1 mice. Immunology. 96:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaliyaperumal, A., M.A. Michaels, and S.K. Datta. 1999. Antigen-specific therapy of murine lupus nephritis using nucleosomal peptides: tolerance spreading impairs pathogenic function of autoimmune T and B cells. J. Immunol. 162:5775–5783. [PubMed] [Google Scholar]
- 52.Eilat, E., H. Zinger, A. Nyska, and E. Mozes. 2000. Prevention of systemic lupus erythematosus-like disease in (NZBxNZW)F1 mice by treating with CDR1- and CDR3-based peptides of a pathogenic autoantibody. J. Clin. Immunol. 20:268–278. [DOI] [PubMed] [Google Scholar]
- 53.Hahn, B.H., R.R. Singh, W.K. Wong, B.P. Tsao, K. Bulpitt, and F.M. Ebling. 2001. Treatment with a consensus peptide based on amino acid sequences in autoantibodies prevents T cell activation by autoantigens and delays disease onset in murine lupus. Arthritis Rheum. 44:432–441. [DOI] [PubMed] [Google Scholar]
- 54.Eilat, E., M. Dayan, H. Zinger, and E. Mozes. 2001. The mechanism by which a peptide based on complementarity-determining region-1 of a pathogenic anti-DNA autoantibody ameliorates experimental systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA. 98:1148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh, R.R. 2000. Potential use of peptides and vaccination to treat systemic lupus erythematosus. Curr. Opin. Rheumatol. 12:399–406. [DOI] [PubMed] [Google Scholar]