

Abstract
Murine splenic dendritic cells (DCs) can be divided into two subsets based on CD8α expression, but the specific role of each subset in stimulation of T cells is largely unknown. An important function of DCs is the ability to take up exogenous antigens and cross-present them in the context of major histocompatibility complex (MHC) class I molecules to CD8+ T cells. We previously demonstrated that, when cell-associated ovalbumin (OVA) is injected into mice, only the CD8+ DC subset cross-presents OVA in the context of MHC class I. In contrast to this selectivity with cell-associated antigen, we show here that both DC subsets isolated from mice injected with OVA/anti-OVA immune complexes (OVA-IC) cross-present OVA to CD8+ T cells. The use of immunoglobulin G Fc receptor (FcγR) common γ-chain–deficient mice revealed that the cross-presentation by CD8− DCs depended on the expression of γ-chain–containing activating FcγRs, whereas cross-presentation by CD8+ DCs was not reduced in γ-chain–deficient mice. These results suggest that although CD8+ DCs constitutively cross-present exogenous antigens in the context of MHC class I molecules, CD8− DCs only do so after activation, such as via ligation of FcγRs. Cross-presentation of immune complexes may play an important role in autoimmune diseases and the therapeutic effect of antitumor antibodies.
Keywords: antigen presentation, cytotoxic T lymphocyte, cross-priming, dendritic cell, Fc receptors
Introduction
Dendritic cells (DCs)* have the capacity to take up, process, and present exogenous antigens in association with MHC class I molecules (1). This pathway is termed cross-presentation and the resulting CD8+ T cell priming is referred to as cross-priming. It was first noted when mice primed with cells that expressed foreign minor histocompatibility antigens but not host MHC molecules, generated minor antigen specific, host MHC-restricted CD8+ T cells (2, 3). This indicated that host APCs had taken up the exogenous cell-associated minor histocompatibility antigens and presented them in the context of their own MHC class I molecules. As cross-presentation allows animals to mount CD8+ T cell responses to antigens not expressed by the APCs, this pathway is essential for many immune responses to viral, bacterial, and tumor antigens (4–8).
Murine splenic DCs can be divided into two subsets on the basis of CD8α expression. CD8− DCs have high CD11b expression and can be further subdivided into a CD4+ and a CD4− subset, whereas CD8+ DCs express less CD11b (9–12). These two subsets of CD8− and CD8+ DCs have also been referred to as myeloid and lymphoid DCs respectively. The role of these different DC subsets in the induction of immune responses is not completely understood. Initially it was suggested that CD8− DCs were stimulatory APCs and CD8+ DCs were tolerizing APCs (13, 14). Later reports indicated a role for CD8+ and CD8− DC subsets in the selective stimulation of Th1 and Th2 cells respectively (15–17). However, recently both DC subsets have been shown to produce the Th1 stimulatory cytokine IL-12 and it has been suggested that the DC subtypes express a different repertoire of pattern recognition receptors which lead to different immune responses when engaged (18–22). When we evaluated MHC class I–restricted cross-presentation of antigen introduced in a cell-associated form in vivo, we found that antigen was presented only by the CD8+ DC subset (23). Because both types of DCs were able to stimulate CD8+ T cells when coated with peptide, this result suggested that the CD8+ DC subset had a specialized cross-presentation function that the CD8− DC subset lacked. More evidence for this hypothesis was provided by a recent study in which cross-presentation of soluble antigen was evaluated (24).
In vitro studies have shown that antigens complexed with IgG are much more efficiently cross-presented by DCs than soluble antigen. This cross-presentation in the context of MHC class I molecules coincides with activation of DCs and with presentation of immune complex antigen in the context of MHC class II molecules (25–27). Subsequent in vitro studies showed that DCs, but not macrophages, possess a specialized cross-presentation transport system in which immune complexes are transferred from the endosome to the cytosol where they become available for the classical MHC class I antigen processing pathway (28). For both the activation of DCs by immune complexes, as well as the cross-presentation of immune complexes, the common γ-chain of the Fcγ receptors (FcγR) was essential (26).
In the mouse, three FcγRs exist that can interact with IgG (for reviews, see references 29 and 30). FcγRI is a receptor with high affinity for monomeric IgG2a, whereas FcγRII and FcγRIII have been described to interact preferentially with IgG1, IgG2a, and IgG2b immune complexes. Both FcγRI and FcγRIII are multimeric complexes including the common γ-chain that contains an activating immune receptor tyrosine activation motif (ITAM). Binding to FcγRI and FcγRIII leads to phagocytosis by macrophages, antibody dependent cell-mediated cytotoxity by NK cells and mast cell degranulation (31). In contrast, murine FcγRII consists of one α chain that contains an inhibitory immune receptor tyrosine inhibition motif (ITIM). Four isoforms of murine FcγRII exist and ligation of these receptors generally results in an inhibitory signal. Interaction of the b1 isoform of FcγRII on B cells and mast cells results in inhibitory effects on B cell receptor and FcεRI-mediated activation respectively. Myeloid cells express the b2 isoform of FcγRII which mediates endocytosis of immune complexes and subsequent antigen presentation (32), but which also has a down-regulatory action on the activation status of macrophages (33).
Although in vitro studies have shown expression of all three FcγRs by bone marrow–derived DCs and DC cell lines, relatively little is known of FcγR expression on DC subsets and their role in the uptake and presentation of immune complexes in vivo. In addition to FcγRs, complement receptors may play an important role in cross-presentation of immune complexes in vivo, as DCs express the complement receptors C3 (CD18/CD11b) and C4 (CD18/CD11c) and possibly also the C1q receptor (34). We set out to determine the cross-presentation of immune complexes by splenic CD8+ and CD8− DC subsets and to define the role of FcγRs in this process.
Materials and Methods
Mice.
C57BL/6 (B6) mice, FcγRII-deficient mice, Fcγ-chain–deficient mice lacking FcγRI and FcγRIII, and FcγR deficient mice (FcγRII × Fcγ-chain–deficient mice) were purchased from Taconic Farms. OT-I/Thy1.1 and OT-I/RAG1-deficient mice were bred in our specific pathogen free facility and have a transgenic Vα2Vβ5 TCR specific for the OVA257–264 epitope in the context of H2-Kb. OT-II mice were a gift from Dr. A. Rudensky (University of Washington, Seattle, WA) and have a transgenic Vα2Vβ5 TCR specific for the OVA323–339 epitope in the context of I-Ab (35).
Antibodies.
CD11c-, CD8α-, FcγRII/III-specific, biotinylated anti–mouse IgG2a antibodies and mouse IgG2a and IgG2b were purchased from BD Biosciences. Purified anti-FcγRII antibody (K9.361) was a gift from Dr. U. Hämmerling (Memorial Sloan-Kettering Cancer Center, New York, NY; references 36–38). For FcγRI staining, cells were first incubated with biotinylated mouse IgG2a followed by a blocking step with nonlabeled anti-FcγRII/III antibody containing supernatant with 10 μg/ml mouse IgG, 10 μg/ml rat IgG, and 10 μg/ml hamster IgG (ICN Biomedicals). After the blocking step, cells were incubated with streptavidin-allophycocyanin (APC), CD11c-FITC, and CD8α-PE. For FcγRII staining, cells were first incubated with anti-FcγRII antibody or isotype control followed by a blocking step. After the blocking step, cells were incubated with biotinylated anti–mouse IgG2a antibody followed by streptavidin-APC, CD11c-FITC, and CD8α-PE. For FcγRII/III staining cells were first stained with biotinylated anti-FcγRII/III antibody or isotype control. Subsequently, cells were incubated with streptavidin-APC, CD11c-FITC, and CD8α-PE. Flow cytometry was conducted on a FACSCalibur™ and analyzed using CELLQuest™ software (Becton Dickinson).
DC Purification.
Spleens from 10–20 mice were cut into small pieces and digested by stirring at 37°C for 45 min in RPMI-1640 containing 1 WU/ml Liberase RI (Roche) and 50 μg/ml DNase I (Roche). EDTA was added to a final concentration of 10 mM, and the cell suspension was incubated for an additional 10 min at room temperature. RPMI-1640 with 10% FCS/10 mM EDTA/20 mM Hepes (RP10/HE) was added and the cells were pelleted. Red blood cells were lysed with ACK lysis buffer. Cells were washed once with RP10/HE and undigested material was removed by filtration through a 100 μm cell strainer. CD11c+ DCs were purified using anti-CD11c microbeads and the autoMACS system (Miltenyi Biotec) according to manufacturer's instructions. To purify DC subsets, FcγRII and III on CD11c+ DCs were blocked with 2.4G2 for 10 min and subsequently stained with CD11c-FITC and CD8-PE for 20 min at 4°C in RP10/HE under continuous rotation. Cells were washed twice with RP10/HE and resuspended in RPMI with 20% FCS/10 mM EDTA/20 mM Hepes. Cell sorting was performed in HBSS with 25 mM Hepes using a FACS Vantage™ (Becton Dickinson). Autofluorescent cells were gated out using the FL3 channel and CD8+ and CD8− CD11chigh DCs were sorted. After sorting the cells were washed twice in RP10 to remove EDTA. The purity of sorted CD8+ and CD8− DC subsets was minimally 95%.
Generation of OVA Immune Complexes.
OVA (Calbiochem) was dissolved in PBS (3 mg/ml). Rabbit anti-OVA IgG fraction and rabbit anti-HRP IgG fraction (ICN Biomedicals) were purified over Protein A columns and diluted in PBS (4 mg/ml). OVA and anti-OVA antibodies or anti-HRP antibodies were mixed together at a concentration of 0.38 mg/ml OVA and 1.49 mg/ml anti-OVA or anti-HRP in PBS and incubated for 30 min at 37°C. This mixture was further diluted in PBS to 0.25 mg/ml OVA and 1mg/ml anti-OVA or anti-HRP. Mice were primed with 50 μg OVA/200 μg anti-OVA or anti-HRP in 200 μl by tail vein injection. For uptake of fluorescent OVA-immune complexes, mice were primed with 100 μg DQ OVA (Molecular Probes) complexed to 400 μg anti-OVA. DQ OVA is a self-quenched conjugate of ovalbumin that exhibits bright green fluorescence upon proteolytic degradation.
Proliferation Assay.
To detect OVA presentation, different DC preparations were used as stimulators for naive RAG1-deficient OT-I cells or OT-II cells in a [3H]thymidine incorporation assay. In some experiments CD8+ T cells or CD4+ T cells were purified from RAG1-deficient OT-I or OT-II spleen and lymph node cells using anti-CD8 or anti-CD4 microbeads and the autoMACS system. Titered numbers of nonirradiated DCs were incubated with 105 OT-I RAG1−/− or OT-II cells in flat bottom plates in 200 μl RP10. As a positive control, stimulator cells were coated with 1 μM OVA257–264 or with 10 μM OVA323–339 peptide for 1 h and washed three times. After 48 h, the plates were pulsed for 16 h with 1 μCi/well of [3H]thymidine and harvested.
Carboxyfluorescein Diacetate Succinimidyl Ester Labeling of OT-I Cells.
Spleen and lymph node cells from OT-I mice were washed twice in PBS containing 0.1% BSA. To label cells with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes) the cells were resuspended at 5 × 107 cells/ml in PBS, 0.1% BSA with 10 μM CFSE for 10 min at 37°C. Cells were washed twice with cold RP10 followed by two washes in PBS. 4 × 106 CD8+Va2+ CFSE+ OT-I cells in 200 μl of PBS were injected into the tail vein.
Results
In Vivo Processing and Presentation of Immune Complexes by DCs.
Previous in vitro studies have shown that immune complexes are much more efficiently taken up by DCs and cross-presented to T cells than soluble antigens (26, 27). To determine whether this enhancement also occurs in vivo, we injected B6 mice with 50 μg soluble OVA alone, OVA coincubated with a control anti-HRP IgG antibody, or OVA coincubated with anti-OVA IgG antibody (OVA-IC). 14 h after injection CD11c+ DCs were isolated from spleens and used as stimulators for naive MHC class I–restricted OVA-specific TCR transgenic OT-I, and MHC class II restricted OVA-specific TCR transgenic OT-II T cells in an in vitro proliferation assay. As shown by the data in Fig. 1 , DCs from mice injected with soluble OVA alone or together with the irrelevant antibody were not able to stimulate T cells. In contrast, injection of OVA-IC resulted in strong proliferation of OT-I and OT-II T cells, indicating an efficient uptake and presentation of OVA-IC by DCs in vivo.
Figure 1.
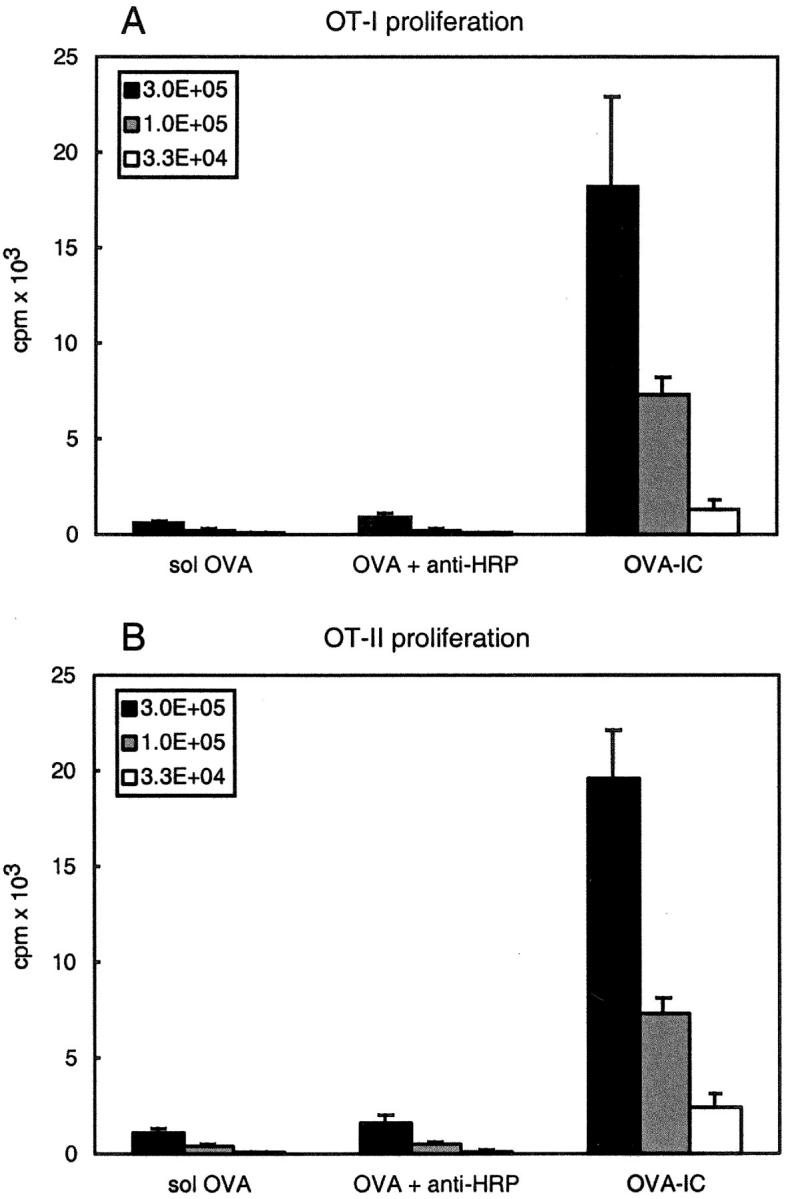
DCs present OVA epitopes in association with both MHC class I and II after in vivo injection of OVA-IC. B6 mice were primed with 50 μg soluble OVA, 50 μg OVA incubated with 200 μg anti-HRP antibodies, or 50 μg OVA with 200 μg anti-OVA antibodies (OVA-IC). CD11c+ DCs were isolated 14 h after injection and analyzed for their ability to stimulate T cells in vitro. The indicated numbers of DCs were coincubated with RAG1-deficient OT-I cells (A) or purified CD4+ OT-II cells (B). Proliferation of cells was determined by [3H]thymidine incorporation. Error bars indicate SEM of triplicate wells.
Cross-presentation of immune complexes by DCs in vitro has been shown to involve an endosome-to-cytosol transport (26, 28). In the cytosol the antigens follow the ‘classical’ MHC class I antigen processing pathway in which the antigens are processed by the proteasome and transported into the ER by the transporter associated with antigen processing (TAP) transporter. In the ER the antigens subsequently bind to nascent MHC class I molecules. Evidence for another MHC class I cross-presentation pathway suggests that peptides can bind to recycled MHC class I molecules that are present in the endosomal–lysosomal compartments and this does not require the presence of a TAP transporter. To investigate which pathway is used by in vivo primed DCs, we injected TAP-deficient mice with OVA-IC. TAP-deficient DCs lacked MHC class I–restricted OVA presentation, whereas MHC class II–restricted presentation of immune complexes remained intact (unpublished data). This indicates that DCs transport OVA-IC antigens from the endosome to the cytosol in vivo, where they follow the ‘classical’ MHC class I processing pathway.
Expression of FcγRs by DC Subsets.
Uptake of immune complexes by DCs can be mediated by FcγRs or by complement receptors. A major role for FcγRs was implicated by in vitro studies concerning cross-presentation by DCs and by in vivo studies on inflammatory responses induced by immune complexes (26, 39, 40). However, the expression of activating and inhibitory FcγRs on the CD8+ and CD8− DC subsets was unknown. Because the relative ratio of these receptors may be of importance for the effect of immune complexes, we set out to determine the expression of the three FcγRs on DC subsets.
As FcγRI is the only receptor capable of binding monomeric mouse IgG2a, we determined functional FcγRI expression using biotinylated mouse IgG2a. We observed strong binding of mouse IgG2a to splenic autofluorescent cells (AFCs) that are mostly macrophages (unpublished data; reference 10). In contrast, much lower levels of binding to both DC subsets was observed (Fig. 2 A). This suggests that the CD8+ and CD8− DC subsets have low expression of FcγRI. RT-PCR analysis of sorted CD8+ and CD8− DC subsets also showed very low message for the FcγRIα chain (unpublished data).
Figure 2.

Both CD8+ and CD8− DCs express FcγRII and RIII. CD11c+ DCs were purified from B6 (A–C), and from FcγRII-deficient (FcγRIIko; C), and mice deficient in all three FcγRs (FcγRko; C) and stained with antibodies specific for CD11c and CD8. DCs were gated on high expression of CD11c and the absence or presence of CD8. (A) FcγRI expression was determined by FACS® analysis of mouse IgG2a binding to CD8+ and CD8− DCs. Bold line depicts mouse IgG2a binding compared with background staining in the absence of mouse IgG2a. (B) FcγRII expression on CD8+ and CD8− DCs was determined by staining with specific anti-FcγRII antibody (bold line) compared with isotype control antibody (fine line). (C) The histograms show FcγRII/III expression of CD8+ and CD8− DCs from B6 mice (bold line), FcγRIIko (shaded histogram), and FcγRko mice (fine line).
Next we analyzed the expression of FcγRII by using the Ly-17.2 antibody K9.361 (Fig. 2 B) (36–38). Both the CD8+ and the CD8− DC subsets showed clear staining with this antibody, although the staining of CD8− DCs was lower and more heterogenous than that of CD8+ DCs. This indicates that both subsets express FcγRII. Using the 2.4G2 antibody we analyzed the expression of FcγRII and FcγRIII on the DC subsets (Fig. 2 C). CD8+ DCs from B6 mice expressed high levels of FcγRII/III, whereas CD8− DCs showed lower, but clear expression of FcγRII/III (Fig. 2 C). To analyze the expression of FcγRIII, we purified CD11c+ DCs from mice deficient in FcγRII. CD8+ and CD8− DC subsets from mice that lacked FcγRII showed staining with 2.4G2 indicating the presence of FcγRIII. DCs from mice deficient in all three FcγRs did not stain with 2.4G2 (Fig. 2 C). Both the CD8+ and CD8− DC subsets from FcγRII-deficient mice exhibited significantly lower staining with 2.4G2 compared with wild-type DC subsets. This indicates that FcγRII is more highly expressed than FcγRIII on both CD8+ and CD8− DC subsets from normal mice.
In conclusion, both DC subsets express low levels FcγRI and significant levels of FcγRII and FcγRIII. The level of expression of both FcγRII and FcγRIII appears to be higher on CD8+ DC than on CD8− DCs, but this may partly be explained by differences in cell size as CD8+ DCs are larger than CD8− DCs. However, the two DC subsets do not appear to differ substantially in the relative ratio of expression of the activating and inhibitory FcγRs as both express higher levels of FcγRII compared with FcγRIII. Therefore, the CD8+ and CD8− DC subsets are not likely to receive substantially different activating and/or inhibitory signals after immune complex–FcγR interaction.
Presentation of Immune Complexes by CD8+ and CD8− DC Subsets.
Both cell-associated and soluble antigens are preferentially cross-presented by CD8+ DCs (23, 24). To determine the cross-presentation of immune complexes by DC subsets, we purified CD8+ and CD8− DC subsets from mice that had been injected with OVA-IC and used them as stimulators for OVA specific OT-I and OT-II T cells. In contrast to cell-associated and soluble antigens, uptake of immune complexes in vivo lead to strong stimulation of OT-I T cells by both CD8− and CD8+ DC subsets (Fig. 3 A).
Figure 3.
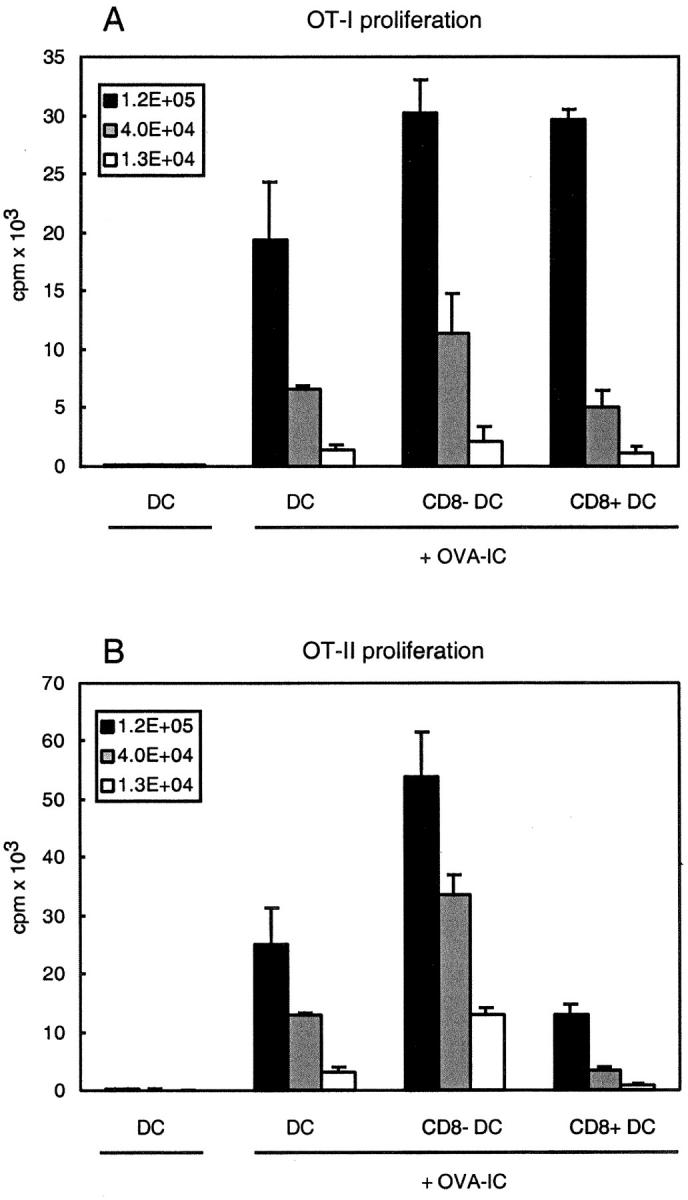
Both CD8+ and CD8- DC subsets cross-present OVA-IC in association with MHC class I molecules, whereas presentation in association with MHC class II molecules is mainly restricted to CD8− DCs. B6 mice were injected with OVA-IC and 14 h after injection CD11c+ DCs were isolated. CD8+ and CD8− DC subsets were FACS® sorted and analyzed for their ability to stimulate T cells in vitro. Indicated numbers of DCs were coincubated with RAG1-deficient OT-I cells (A) or purified CD4+ OT-II cells (B). Proliferation of cells was determined by [3H]thymidine incorporation. Error bars indicate SEM of triplicate wells.
The two DC subsets did differ in their capacity to stimulate OVA-specific MHC class II–restricted OT-II cells: CD8+ DCs exhibited a substantially lower level of stimulation of OVA specific CD4+ T cells compared with CD8− DCs (Fig. 3 B). This preferential presentation of OVA-IC by CD8− DCs to CD4+ T cells was similar to that seen with soluble OVA (24). These results suggest that whereas CD8− DCs transfer OVA-IC to both the MHC class I and class II presentation pathway, CD8+ DCs mainly transport antigens to the cytosol to be presented in MHC class I.
Cross-Presentation in Fcγ-Chain and FcγR-deficient Mice.
FcγRs have been shown to be important in the cross-presentation of immune complexes, as in vitro–generated DCs from mice that were deficient in the FcγRI and FcγRIII-associated γ-chain lost their capacity to cross-present antigen in the context of MHC class I (26). In addition, FcγRs play an important role in the development of inflammatory reactions caused by immune-complexes in vivo (for reviews, see references 29, 30, and 41). To determine whether DCs from mice that are deficient in the FcγRI and FcγRIII-associated γ-chain loose their capacity to cross-present immune complexes in vivo, we primed Fcγ-chain–deficient mice with OVA-IC and 14 h later isolated CD11c+ DCs to test as stimulators for OT-I and OT-II cells. CD8+ DCs from these mice were still able to activate OVA-specific OT-I cells, but, strikingly, CD8− DCs lost their MHC class I restricted cross-presentation capacity (Fig. 4 A). No effects were seen on the MHC class II restricted OVA presentation by the two DC subsets from the γ-chain–deficient mice compared with wild-type mice (Figs. 4 B and 3 B). This suggests that, although immune complexes are still taken up by both DC subsets, the CD8− DCs lost the capacity to cross-present in the context of MHC class I molecules.
Figure 4.
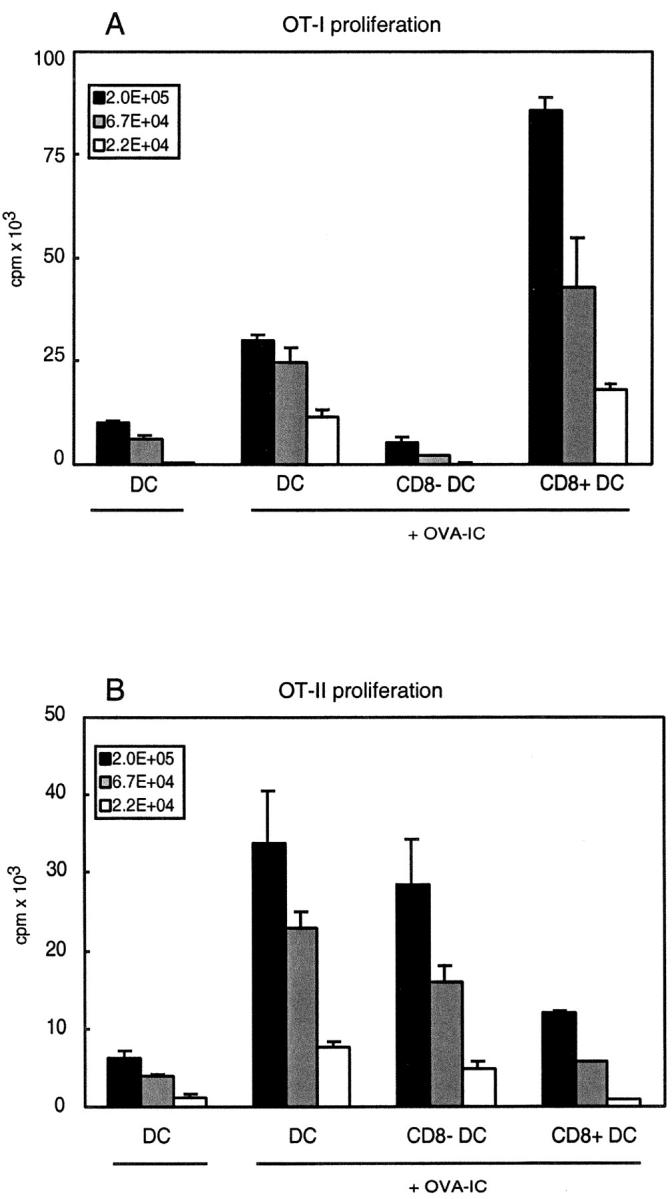
Only CD8+ and not CD8− DCs from Fcγ-chain–deficient mice cross-present OVA-IC to CD8+ T cells. Fcγ-chain–deficient mice were injected with OVA-IC and 14 h after injection CD11c+ DCs were isolated. CD8+ and CD8− DC subsets were FACS® sorted and analyzed for their ability to stimulate T cells in vitro. Indicated numbers of DCs were coincubated with purified CD8+ RAG1-deficient OT-I cells (A) or purified CD4+ OT-II cells (B). Proliferation of cells was determined by [3H]thymidine incorporation. Error bars indicate SEM of triplicate wells.
To determine the role of the inhibitory FcγR in the cross-presentation of immune complexes, we analyzed MHC class I–restricted cross-presentation by CD8− and CD8+ DC subsets from FcγRII-deficient mice (unpublished data). In contrast to the loss of cross-presentation by CD8− DC in Fc γ-chain–deficient mice, the absence of inhibitory FcγRII did not have a major effect on cross-presentation by this DC subset. Cross-presentation by CD8+ DCs appeared to be increased twofold in the absence of FcγRII. This suggests that immune complex interaction with FcγRII does not significantly inhibit cross-presentation in CD8− DCs, but may decrease cross-presentation in CD8+ DCs.
Mice deficient in all three FcγRs have been generated by crossing FcγRII-deficient mice with mice deficient in the common γ-chain. To determine whether the uptake of immune complexes in the absence of activating FcγRs was due to interaction with FcγRII, we isolated DC subsets from mice that were deficient in all three FcγRs that been primed with OVA-IC 14 h previously. Similarly to the γ-chain–deficient mice, CD8+ DCs from FcγR-deficient mice were very good stimulators for OT-I cells, whereas CD8− DCs had a reduced capacity compared with wild-type mice (Fig. 5 A). The deficit of the CD8− DCs, however, seemed less complete compared with the γ-chain–deficient mice. Again no drastic effects were observed for the MHC class II–restricted presentation by the two DC subsets from the FcγR-deficient mice compared with wild-type mice (Fig. 5 B).
Figure 5.
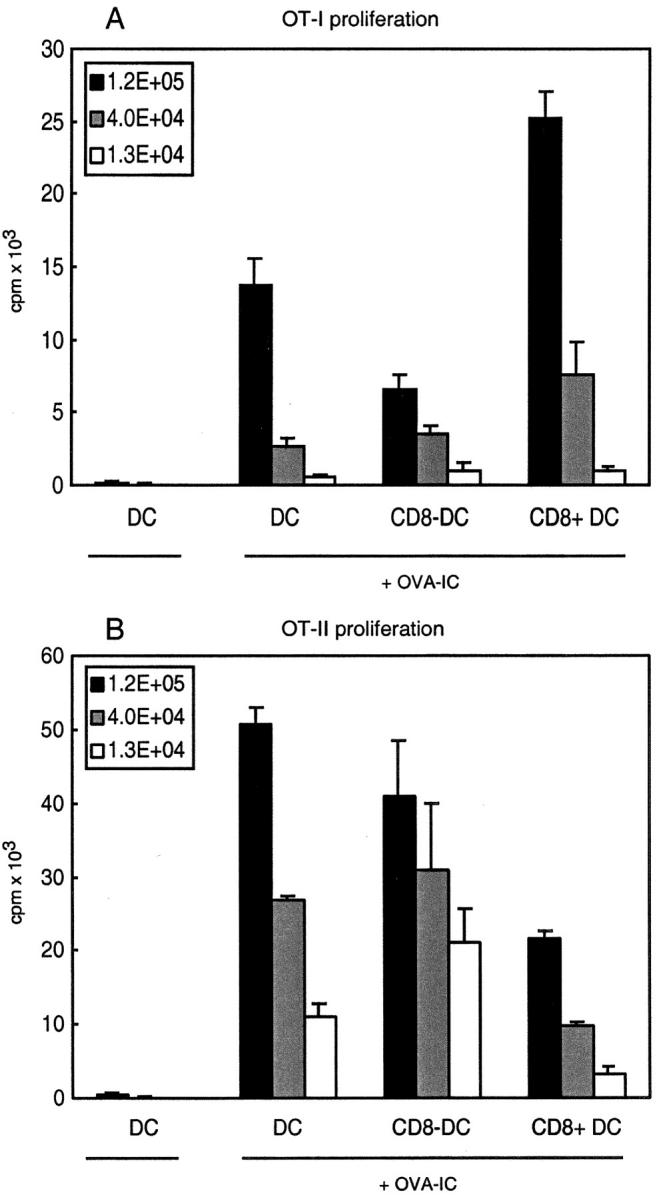
CD8− DCs from FcγR-deficient mice have strongly decreased capacity to cross-present OVA-IC to CD8+ T cells. FcγR-deficient mice were injected with OVA-IC and 14 h after injection CD11c+ DCs were isolated. CD8+ and CD8− DC subsets were FACS® sorted and analyzed for their ability to stimulate T cells in vitro. Indicated numbers of DCs were coincubated with purified CD8+ RAG1-deficient OT-I cells (A) or purified CD4+ OT-II cells (B). Proliferation of cells was determined by [3H]thymidine incorporation. Error bars indicate SEM of triplicate wells.
In conclusion, although both DC subsets express the same set of FcγRs, in the absence of the two activating FcγRs or all three FcγRs, CD8− DCs specifically loose their capacity to cross-present OVA-IC to MHC class I–restricted T cells, whereas the presentation by CD8+ DCs is not affected. In contrast, the pattern of presentation of OVA-IC to MHC class II–restricted T cells is not changed by the absence of FcγRs.
FcγR-deficient DCs Take Up OVA-IC.
To investigate whether the decrease of cross-presentation by CD8− DCs in the absence of activating FcγRs could be due to a selective decrease in uptake of OVA-IC by this subset, we isolated DCs from mice that had been injected with fluorescent OVA-IC (Fig. 6) . Both CD8+ and CD8− DCs isolated from mice that were deficient in the activating γ-chain and in mice that were deficient for all three FcγRs showed similar or even increased uptake of fluorescent OVA-IC compared with wild-type DCs. This indicates that FcγRs are not absolutely required for the uptake of immune complexes by DCs in vivo and that other receptors expressed on DCs, such as C1q, C3 (CD18/CD11b), and C4 (CD18/CD11c) complement receptors, may be involved in this process.
Figure 6.
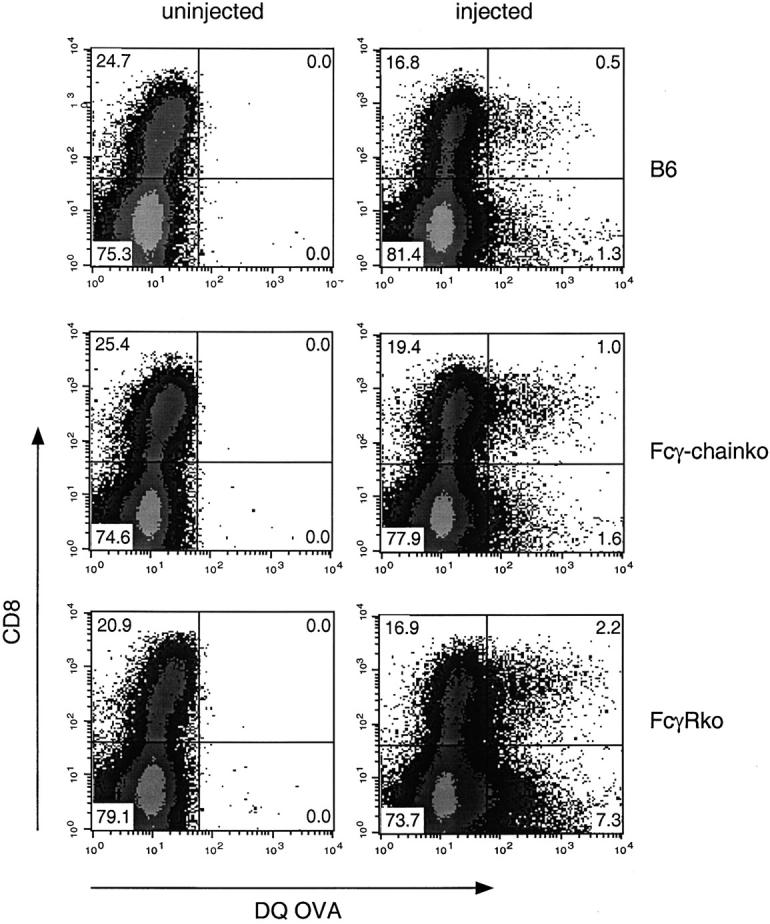
Both DC subsets from B6 mice, Fcγ-chain–deficient mice and FcγR-deficient mice take up OVA-IC in vivo. CD11c+ DCs were isolated from control mice or mice injected with fluorescent DQ OVA-IC 14 h previously. Dot plots depict CD8 expression and uptake of DQ OVA by DCs isolated from B6 mice, Fcγ-chain–deficient mice and FcγR-deficient mice. DCs were gated on high CD11c expression, while autofluorescent cells were excluded.
This result indicates that the loss of cross-presentation in the CD8− DC subset in the absence of activating FcγRs cannot be explained by decreased uptake of immune complexes. Therefore, signaling through the activating FcγRs or changes in intracellular trafficking induced by activating FcγR interactions must be required for MHC class I–restricted presentation by CD8− DCs.
In Vivo Proliferation of CD8+ T Cells by OVA-IC in Wild-Type and FcγR-deficient Mice.
In the absence of all three FcγRs, CD8− DCs exhibited a significantly decreased ability to cross-present OVA to CD8+ T cells, whereas cross-presentation by the CD8+ DCs was unaffected (Fig. 5). To investigate whether the absence of FcγRs had an effect on the proliferative response of OVA-specific CD8+ T cells in vivo, we transferred CFSE-labeled OT-I cells into wild-type mice, into mice that were deficient in FcγRI and FcγRIII, and into mice that were deficient in all three FcγRs. Mice were primed with OVA-IC and 3 d later we determined the proliferation of OT-I cells in the spleen. Whereas nonimmunized mice only contained OT-I cells with CFSE high intensity (Fig. 7 A), all three types of mice that had received OVA-IC contained much higher numbers of OT-I cells with much lower CFSE intensity (Fig. 7 B). Comparison of the CFSE levels indicated that the OT-I cells had divided 4–7 times in the OVA-IC injected hosts, and no difference in the proliferation of OT-I cells in the absence or presence of FcγR was observed. This suggests that the transferred OT-I cells were efficiently activated by CD8+ DCs and that MHC class I–restricted presentation CD8− DCs is redundant in this process.
Figure 7.
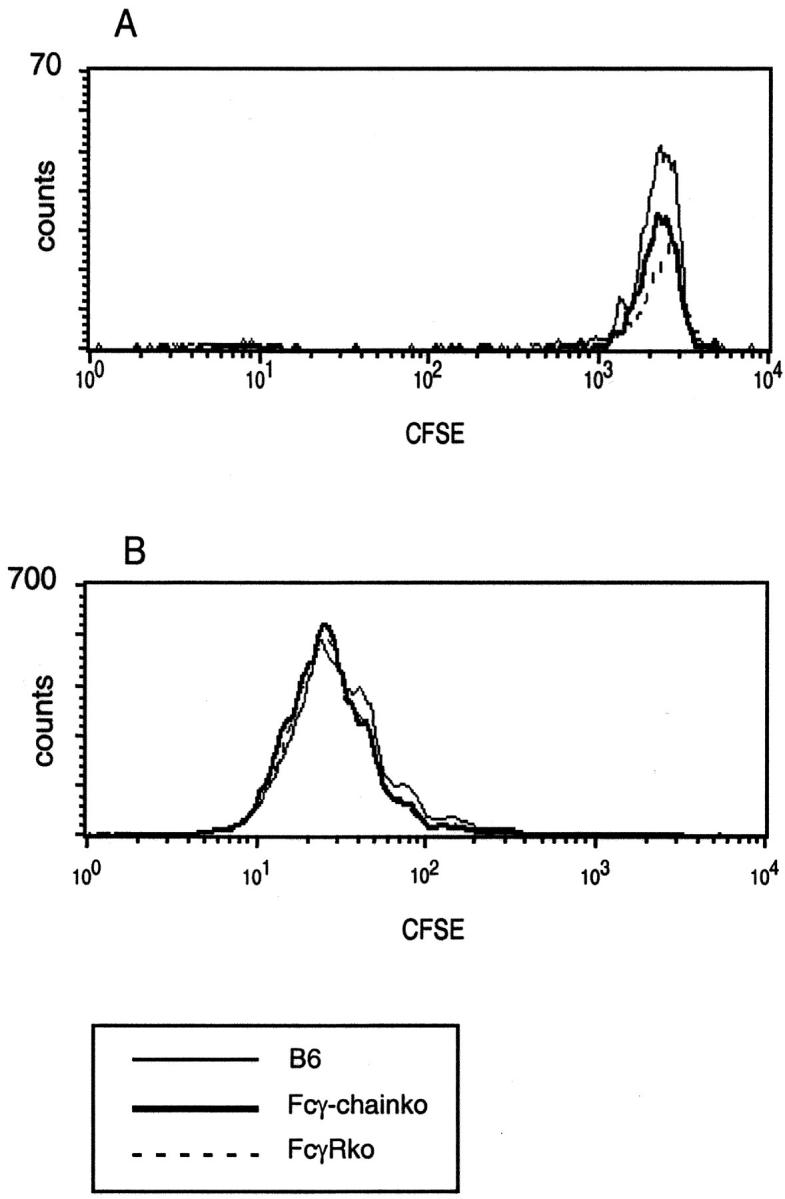
Proliferation of OVA-specific CD8+ T cells in vivo after priming with OVA-IC is comparable in B6, Fcγ-chain–deficient mice and FcγR-deficient mice. CFSE-labeled, Thy1.1+ OT-I cells were transferred into B6, Fcγ-chain–deficient, and FcγR-deficient mice. 3 d later, mice were primed by injection of OVA-IC. Spleen cells were isolated 3 d after priming and stained for Thy1.1 and CD8. (A) CFSE profiles of Thy1.1+CD8+ OT-1 cells from mice that received no priming. (B) Same profiles from mice that were primed with OVA-IC.
Discussion
In mouse spleen, DCs can be divided into CD8+ and CD8− subsets and the latter can be further separated into CD4+ and CD4− subsets (9–12). In addition, there is evidence for an IFNα producing plasmacytoid DCs in mouse spleen (42–44). In mouse lymph nodes, five different DC subsets have been distinguished (45). Although largely unknown, it is likely that these different DC subsets have different functions in the initiation of immune responses.
In this study, we investigated the capacity of the CD8+ and CD8− splenic DC subsets to present immune-complexes in the context of MHC class I and II molecules and the role of FcγRs in this process. Both CD8+ and CD8− DC subsets were found to have similar FcγR expression: clear expression of both FcγRII and FcγRIII, but low expression of FcγRI. Both DC subsets were highly efficient in the cross-presentation of immune complexes in the context of MHC class I molecules in wild-type mice. Unexpectedly, in the absence of activating FcγRI and FcγRIII, CD8− DCs lost the ability to cross-present immune complexes in the context of MHC class I molecules, whereas CD8+ DCs retained this capacity. This selective cross-presentation by the CD8+ DC subset in the absence of activating FcγRs is similar to that seen with the cross-presentation of cell-associated and soluble antigen (23, 24).
What is the role for activating FcγRs in the MHC class I–restricted cross-presentation by CD8- DCs? FcγRs may be involved in cross-presentation of immune complexes at three different levels: they are involved in the uptake of immune complexes, in the intracellular trafficking of immune complexes, and in transmitting activating or inhibitory signals to the cell. At all these levels the CD8− DCs could potentially be affected by the absence of activating FcγRs. Our data regarding the uptake of immune complexes by DCs indicate that FcγRs are not essential for this process in vivo (Fig. 6) and strongly suggest that other receptors, such as complement receptors, are involved in this process. Soluble immune complexes activate complement and bind to complement receptor 1 on erythrocytes (41, 46, 47). This results in removal of immune complexes as erythrocytes travel to the liver and spleen where the immune complexes are removed from the erythrocytes by phagocytes. In addition, binding of complement to antigen has been shown to increase immunogenicity significantly due to binding to complement receptor 2 on B cells (48). Although CD8+ and CD8− DC subsets do not express complement receptor 1 or 2, they strongly express complement receptors 3 (CD18/CD11b) and 4 (CD18/CD11c) and these may be involved in the uptake of immune complexes.
As the uptake of immune complexes is not substantially decreased in the absence of FcγRs, do the FcγRs direct antigen to different processing compartments? In the case of B cell antigen processing for MHC class II, it has been suggested that antigens taken up by activating FcγRs may be targeted to different processing routes than antigens taken up by FcγRII or other receptors (32). It remains to be shown whether activating and inhibitory FcγRs target to different MHC class I processing pathways in DCs. Furthermore this would not explain why this mechanism would be essential in only CD8− DCs and not in CD8+ DCs, as cross-presentation by CD8+ DCs is not affected in the absence of activating FcγRs.
The third known function of γ-chain containing FcγRs is that these receptors can activate cells via their ITAM motif. Activation mediated by γ-chain containing FcγRs could potentially result in changes in the intracellular trafficking and processing of not only FcγR bound antigens, but also of antigens taken up by other mechanisms (49). Bone marrow–derived DCs have been shown to gain the ability to cross-present in the context of MHC class I molecules and to up-regulate costimulatory molecules after interaction of immune complexes with activating FcγRs in vitro (26, 50). As in vivo only a small percentage of the DCs take up immune complexes, we failed to detect consistent and significant upregulation of costimulatory molecules in vivo. However, these in vitro studies clearly suggest that DCs will also be activated by immune-complexes in vivo. DC activation may be achieved by the direct interaction of immune complexes with the activating FcγRs. Alternatively, as FcγRs have been shown to be the main regulators of inflammation by immune complexes (for reviews, see references 29, 30, and 41), DC activation may occur indirectly via inflammatory mediators. We think that this FcRγ-chain–mediated activation of CD8− DCs enables them to cross-present antigens in the context of MHC class I. The hypothesis that activation of CD8− DCs is necessary for their ability to cross-present is further supported by a recent study concerning cross-presentation of soluble antigen. In this study, CD8− DCs could not cross-present soluble OVA unless further activated by LPS (24).
In contrast to CD8− DCs, CD8+ DCs cross-present immune complexes in the context of MHC class I molecules regardless of the presence or absence of activating FcγRs. CD8+ DCs have been shown to be highly efficient in cross-presenting all types of antigens studied so far, which include cell-associated antigens, high doses of soluble antigens, and now immune complexes. This suggests that in this DC subset the MHC class I–restricted ‘cross-presentation pathway’ is constitutive for exogenous antigens and does not depend on additional activating stimuli. The nature of the transport system that shuttles antigens from the endosome to the cytosol, which has been shown to be present in DCs and not in macrophages, remains to be identified (28).
Our data suggest that the activation state of CD8− DCs is essential for the cross-presentation capacity of this DC subset. The activation state of DCs is also crucial for the functional outcome of the T cell–DC interaction. Non-activated DCs tolerize or delete T cells, whereas activation converts the DC to a stimulatory state that results in T cell activation and memory (51). We hypothesize that CD8+ DCs constitutively cross-present exogenous antigens in the context of MHC class I molecules and, in the absence of a DC activation stimulus, this will tolerize CD8+ T cells. As CD8− DCs are more efficient in the presentation of exogenous antigens in the context of MHC class II, these cells may be similarly involved in the tolerization of CD4+ T cells. We think that activation of both types of DCs will enable them to activate T cells. CD8+ DCs may still mainly activate CD8+ T cells, whereas activated CD8− DCs may activate both CD8+ and CD4+ T cells. Activation of DCs can occur via activated CD4+ T cells (52–54), by interaction of pattern recognition receptors on the DC with their ligands (55), or possibly via interaction of immune complexes with activating FcγR (26). The in vivo proliferation of OVA-specific CD8+ T cells was not affected in FcγR-deficient mice compared with wild-type (Fig. 7). However, this short-term assay does not distinguish the eventual induction of T cell tolerance versus activation and memory, as both are preceded by proliferation (56, 57). CD8+ T cell activation after immune complex priming has recently been shown to be CD4+ T cell independent, which suggests that interaction of immune complexes with activating FcγRs is sufficient for DC activation (50, and unpublished data). Our model predicts that immune complex priming in the absence of both CD4+ T cells and FcγRs will result in tolerization, but this remains to be investigated.
In addition to differences in cross-presentation of antigens in the context of MHC class I, the two DC subsets clearly differed in their capacity to present OVA-IC to CD4+ T cells: CD8− DCs were far superior in stimulating OVA-specific CD4+ T cells compared with CD8+ DCs. This is in line with the presentation of soluble OVA to CD4+ T cells by DC subsets, in which the CD8+ DCs also showed reduced CD4+ T cell stimulating capacity compared with the CD8− DC subset (24).
Autoantibodies and immune complexes are detected in many autoimmune diseases. Deposition of these immune complexes initiates inflammatory reactions leading to tissue damage. In addition to these well known directly destructive effects of immune complexes, our data indicate that immune complexes are also highly efficiently cross-presented by DCs in vivo to both CD4+ and CD8+ T cells. This in turn may result in activated autoantigen specific CD8+ T cells that may contribute to tissue destruction. Both autoantibodies and CD8+ T cells are present and possibly involved in the development of diabetes, Hashimoto's thyroiditis, and primary biliary cirrhosis (58–60). Complexes of autoantibodies and the E2 component of pyruvate dehydrogenase were shown to be efficiently cross-presented by human DCs and expanded E2 pyruvate dehydrogenase specific CD8+ T cells from primary biliary cirrhosis patients in vitro (60). In addition to immune complexes, DCs have also been shown to phagocytose antibody coated tumor cells effectively eliciting tumor-specific CD8+ T cells in vitro (61). This suggests that increased DC cross-presentation and CD8+ T cell activation may also play a role in the protective effect of therapeutic antitumor antibodies. Our study demonstrates that in vivo cross-presentation of immune complexes is differentially regulated in the CD8+ and CD8− DC subsets. Further elucidation of the role of CD8+ and CD8− DCs in the cross-priming of MHC class I-restricted T cells will be essential to develop therapeutic strategies for both autoimmune diseases and tumors.
Acknowledgments
We thank the cell analysis facility for help with FACS® sorting and X. Pan for maintaining the mouse colony.
This work was supported by the Howard Hughes Medical Institute, National Institutes of Health Grant AI19335, and by postdoctoral fellowships from the European Molecular Biology Organization (ALTF 115-1998) and from the Dutch Cancer Society to J.M.M. den Haan.
Footnotes
Abbreviations used in this paper: APC, allophycocyanin; DC, dendritic cell; OVA-IC, OVA/anti-OVA immune complexes; TAP, transporter associated with antigen presentation; FcγR, immunoglobulin G Fc receptor.
References
- 1.Mellman, I., and R.M. Steinman. 2001. Dendritic cells: specialized and regulated antigen processing machines. Cell. 106:255–258. [DOI] [PubMed] [Google Scholar]
- 2.Bevan, M.J. 1976. Minor H antigens introduced on H-2 different stimulating cells cross-react at the cytotoxic T cell level during in vivo priming. J. Immunol. 117:2233–2238. [PubMed] [Google Scholar]
- 3.Bevan, M.J. 1976. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 143:1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang, A.Y., P. Golumbek, M. Ahmadzadeh, E. Jaffee, D. Pardoll, and H. Levitsky. 1994. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 264:961–965. [DOI] [PubMed] [Google Scholar]
- 5.Kurts, C., W.R. Heath, F.R. Carbone, J. Allison, J.F. Miller, and H. Kosaka. 1996. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J. Exp. Med. 184:923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sigal, L.J., S. Crotty, R. Andino, and K.L. Rock. 1999. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 398:77–80. [DOI] [PubMed] [Google Scholar]
- 7.Lenz, L.L., E.A. Butz, and M.J. Bevan. 2000. Requirements for bone marrow-derived antigen presenting cells in priming cytotoxic T cell responses to intracellular pathogens. J. Exp. Med. 192:1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.den Haan, J.M., and M.J. Bevan. 2001. Antigen presentation to CD8+ T cells: cross-priming in infectious diseases. Curr. Opin. Immunol. 13:437–441. [DOI] [PubMed] [Google Scholar]
- 9.Vremec, D., M. Zorbas, R. Scollay, D.J. Saunders, C.F. Ardavin, L. Wu, and K. Shortman. 1992. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J. Exp. Med. 176:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vremec, D., J. Pooley, H. Hochrein, L. Wu, and K. Shortman. 2000. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164:2978–2986. [DOI] [PubMed] [Google Scholar]
- 11.Anjuere, F., P. Martin, I. Ferrero, M.L. Fraga, G.M. del Hoyo, N. Wright, and C. Ardavin. 1999. Definition of dendritic cell subpopulations present in the spleen, Peyer's patches, lymph nodes, and skin of the mouse. Blood. 93:590–598. [PubMed] [Google Scholar]
- 12.Pulendran, B., J. Lingappa, M.K. Kennedy, J. Smith, M. Teepe, A. Rudensky, C.R. Maliszewski, and E. Maraskovsky. 1997. Developmental pathways of dendritic cells in vivo: distinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand-treated mice. J. Immunol. 159:2222–2231. [PubMed] [Google Scholar]
- 13.Kronin, V., K. Winkel, G. Suss, A. Kelso, W. Heath, J. Kirberg, H. von Boehmer, and K. Shortman. 1996. A subclass of dendritic cells regulates the response of naive CD8 T cells by limiting their IL-2 production. J. Immunol. 157:3819–3827. [PubMed] [Google Scholar]
- 14.Suss, G., and K. Shortman. 1996. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 183:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pulendran, B., J.L. Smith, G. Caspary, K. Brasel, D. Pettit, E. Maraskovsky, and C.R. Maliszewski. 1999. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc. Natl. Acad. Sci. USA. 96:1036–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith, A.L., and B.F. de St Groth. 1999. Antigen-pulsed CD8alpha+ dendritic cells generate an immune response after subcutaneous injection without homing to the draining lymph node. J. Exp. Med. 189:593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maldonado-Lopez, R., T. De Smedt, P. Michel, J. Godfroid, B. Pajak, C. Heirman, K. Thielemans, O. Leo, J. Urbain, and M. Moser. 1999. CD8alpha+ and CD8alpha− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 189:587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hochrein, H., K. Shortman, D. Vremec, B. Scott, P. Hertzog, and M. O'Keeffe. 2001. Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J. Immunol. 166:5448–5455. [DOI] [PubMed] [Google Scholar]
- 19.Huang, L.Y., C. Reis e Sousa, Y. Itoh, J. Inman, and D.E. Scott. 2001. IL-12 induction by a TH1-inducing adjuvant in vivo: dendritic cell subsets and regulation by IL-10. J. Immunol. 167:1423–1430. [DOI] [PubMed] [Google Scholar]
- 20.Liu, Y.J., H. Kanzler, V. Soumelis, and M. Gilliet. 2001. Dendritic cell lineage, plasticity and cross-regulation. Nat. Immunol. 2:585–589. [DOI] [PubMed] [Google Scholar]
- 21.Kadowaki, N., S. Ho, S. Antonenko, R. de Waal Malefyt, R.A. Kastelein, F. Bazan, and Y.J. Liu. 2001. Subsets of human dendritic cell precursors express different Toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194:863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarrossay, D., G. Napolitani, M. Colonna, F. Sallusto, and A. Lanzavecchia. 2001. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 31:3388–3393. [DOI] [PubMed] [Google Scholar]
- 23.den Haan, J.M., S.M. Lehar, and M.J. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pooley, J.L., W.R. Heath, and K. Shortman. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 166:5327–5330. [DOI] [PubMed] [Google Scholar]
- 25.Sallusto, F., and A. Lanzavecchia. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Regnault, A., D. Lankar, V. Lacabanne, A. Rodriguez, C. Thery, M. Rescigno, T. Saito, S. Verbeek, C. Bonnerot, P. Ricciardi-Castagnoli, and S. Amigorena. 1999. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I–restricted antigen presentation after immune complex internalization. J. Exp. Med. 189:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Machy, P., K. Serre, and L. Leserman. 2000. Class I-restricted presentation of exogenous antigen acquired by Fcgamma receptor-mediated endocytosis is regulated in dendritic cells. Eur. J. Immunol. 30:848–857. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez, A., A. Regnault, M. Kleijmeer, P. Ricciardi-Castagnoli, and S. Amigorena. 1999. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1:362–368. [DOI] [PubMed] [Google Scholar]
- 29.Ravetch, J.V., and S. Bolland. 2001. IgG Fc receptors. Annu. Rev. Immunol. 19:275–290. [DOI] [PubMed] [Google Scholar]
- 30.Gessner, J.E., H. Heiken, A. Tamm, and R.E. Schmidt. 1998. The IgG Fc receptor family. Ann. Hematol. 76:231–248. [DOI] [PubMed] [Google Scholar]
- 31.Takai, T., M. Li, D. Sylvestre, R. Clynes, and J.V. Ravetch. 1994. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 76:519–529. [DOI] [PubMed] [Google Scholar]
- 32.Amigorena, S., D. Lankar, V. Briken, L. Gapin, M. Viguier, and C. Bonnerot. 1998. Type II and III receptors for immunoglobulin G (IgG) control the presentation of different T cell epitopes from single IgG-complexed antigens. J. Exp. Med. 187:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clynes, R., J.S. Maizes, R. Guinamard, M. Ono, T. Takai, and J.V. Ravetch. 1999. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 189:179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim, T.S., M. Park, R.R. Nepomuceno, G. Palmarini, S. Winokur, C.A. Cotman, U. Bengtsson, and A.J. Tenner. 2000. Characterization of the murine homolog of C1qR(P): identical cellular expression pattern, chromosomal location and functional activity of the human and murine C1qR(P). Mol. Immunol. 37:377–389. [DOI] [PubMed] [Google Scholar]
- 35.Barnden, M.J., J. Allison, W.R. Heath, and F.R. Carbone. 1998. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76:34–40. [DOI] [PubMed] [Google Scholar]
- 36.Kimura, S., N. Tada, E. Nakayama, Y. Liu, and U. Hammerling. 1981. A new mouse cell-surface antigen (Ly-m20) controlled by a gene linked to Mls locus and defined by monoclonal antibodies. Immunogenetics. 14:3–14. [DOI] [PubMed] [Google Scholar]
- 37.Holmes, K.L., R.G. Palfree, U. Hammerling, and H.C. Morse III. 1985. Alleles of the Ly-17 alloantigen define polymorphisms of the murine IgG Fc receptor. Proc. Natl. Acad. Sci. USA. 82:7706–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schiller, C., I. Janssen-Graalfs, U. Baumann, K. Schwerter-Strumpf, S. Izui, T. Takai, R.E. Schmidt, and J.E. Gessner. 2000. Mouse FcgammaRII is a negative regulator of FcgammaRIII in IgG immune complex-triggered inflammation but not in autoantibody-induced hemolysis. Eur. J. Immunol. 30:481–490. [DOI] [PubMed] [Google Scholar]
- 39.Sylvestre, D.L., and J.V. Ravetch. 1994. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science. 265:1095–1098. [DOI] [PubMed] [Google Scholar]
- 40.Sylvestre, D., R. Clynes, M. Ma, H. Warren, M.C. Carroll, and J.V. Ravetch. 1996. Immunoglobulin G-mediated inflammatory responses develop normally in complement-deficient mice. J. Exp. Med. 184:2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dijstelbloem, H.M., J.G. van de Winkel, and C.G. Kallenberg. 2001. Inflammation in autoimmunity: receptors for IgG revisited. Trends Immunol. 22:510–516. [DOI] [PubMed] [Google Scholar]
- 42.Nakano, H., M. Yanagita, and M.D. Gunn. 2001. CD11c+ B220+Gr-1+ cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 194:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 44.Bjorck, P. 2001. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood. 98:3520–3526. [DOI] [PubMed] [Google Scholar]
- 45.Henri, S., D. Vremec, A. Kamath, J. Waithman, S. Williams, C. Benoist, K. Burnham, S. Saeland, E. Handman, and K. Shortman. 2001. The dendritic cell populations of mouse lymph nodes. J. Immunol. 167:741–748. [DOI] [PubMed] [Google Scholar]
- 46.Cornacoff, J.B., L.A. Hebert, W.L. Smead, M.E. VanAman, D.J. Birmingham, and F.J. Waxman. 1983. Primate erythrocyte-immune complex-clearing mechanism. J. Clin. Invest. 71:236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nardin, A., M.A. Lindorfer, and R.P. Taylor. 1999. How are immune complexes bound to the primate erythrocyte complement receptor transferred to acceptor phagocytic cells? Mol. Immunol. 36:827–835. [DOI] [PubMed] [Google Scholar]
- 48.Dempsey, P.W., and D.T. Fearon. 1996. Complement: instructing the acquired immune system through the CD21/CD19 complex. Res. Immunol. 147:71–75; discussion 119–120. [DOI] [PubMed] [Google Scholar]
- 49.Shen, L., M. van Egmond, K. Siemasko, H. Gao, T. Wade, M.L. Lang, M. Clark, J.G. van De Winkel, and W.F. Wade. 2001. Presentation of ovalbumin internalized via the immunoglobulin-A Fc receptor is enhanced through Fc receptor gamma-chain signaling. Blood. 97:205–213. [DOI] [PubMed] [Google Scholar]
- 50.Schuurhuis, D.H., A. Ioan-Facsinay, B. Nagelkerken, J.J. van Schip, C. Sedlik, C.J. Melief, J.S. Verbeek, and F. Ossendorp. 2002. Antigen-antibody immune complexes empower dendritic cells to efficiently prime specific CD8+ CTL responses in vivo. J. Immunol. 168:2240–2246. [DOI] [PubMed] [Google Scholar]
- 51.Hawiger, D., K. Inaba, Y. Dorsett, M. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett, S.R., F.R. Carbone, F. Karamalis, R.A. Flavell, J.F. Miller, and W.R. Heath. 1998. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 393:478–480. [DOI] [PubMed] [Google Scholar]
- 53.Schoenberger, S.P., R.E. Toes, E.I. van der Voort, R. Offringa, and C.J. Melief. 1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 393:480–483. [DOI] [PubMed] [Google Scholar]
- 54.Ridge, J.P., F. Di Rosa, and P. Matzinger. 1998. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 393:474–478. [DOI] [PubMed] [Google Scholar]
- 55.Pulendran, B., K. Palucka, and J. Banchereau. 2001. Sensing pathogens and tuning immune responses. Science. 293:253–256. [DOI] [PubMed] [Google Scholar]
- 56.Adler, A.J., C.T. Huang, G.S. Yochum, D.W. Marsh, and D.M. Pardoll. 2000. In vivo CD4+ T cell tolerance induction versus priming is independent of the rate and number of cell divisions. J. Immunol. 164:649–655. [DOI] [PubMed] [Google Scholar]
- 57.Mintern, J.D., G.M. Davey, G.T. Belz, F.R. Carbone, and W.R. Heath. 2002. Cutting edge: precursor frequency affects the helper dependence of cytotoxic T cells. J. Immunol. 168:977–980. [DOI] [PubMed] [Google Scholar]
- 58.Tomer, Y. 1997. Anti-thyroglobulin autoantibodies in autoimmune thyroid diseases: cross-reactive or pathogenic? Clin. Immunol. Immunopathol. 82:3–11. [DOI] [PubMed] [Google Scholar]
- 59.Atkinson, M.A., and G.S. Eisenbarth. 2001. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 358:221–229. [DOI] [PubMed] [Google Scholar]
- 60.Kita, H., Z.X. Lian, J. Van de Water, X.S. He, S. Matsumura, M. Kaplan, V. Luketic, R.L. Coppel, A.A. Ansari, and M.E. Gershwin. 2002. Identification of HLA-A2-restricted CD8+ cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J. Exp. Med. 195:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dhodapkar, K.M., J. Krasovsky, B. Williamson, and M.V. Dhodapkar. 2002. Antitumor monoclonal antibodies enhance cross-presentation of cellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J. Exp. Med. 195:125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]