

Abstract
Respiratory syncytial virus (RSV) is the leading cause of bronchiolitis and viral pneumonia in infants and young children. Administration of a formalin inactivated vaccine against RSV to children in the 1960s resulted in increased morbidity and mortality in vaccine recipients who subsequently contracted RSV. This incident precluded development of subunit RSV vaccines for infants for over 30 years, because the mechanism of illness was never clarified. An RSV vaccine for infants is still not available.
Here, we demonstrate that enhanced RSV disease is mediated by immune complexes and abrogated in complement component C3 and B cell–deficient mice but not in controls. Further, we show correlation with the enhanced disease observed in children by providing evidence of complement activation in postmortem lung sections from children with enhanced RSV disease.
Keywords: respiratory syncytial virus, enhanced disease, immune complexes, airway hyperresponsiveness, compliment
Introduction
Respiratory syncytial virus (RSV) is the leading cause of viral lower respiratory illness (LRI) in infants and young children worldwide. Its clinical manifestations range from asymptomatic infection to LRI with bronchoconstriction and pneumonia (1). In the 1960s, a formalin inactivated (FI) RSV vaccine (FIRSV) was administered to infants and children in the United States. Exposure of vaccinated children to RSV resulted in increased frequency and severity of RSV LRI and greater incidence of hospitalization compared with children immunized with control preparations. Furthermore, two vaccine recipients died following RSV infection (2). Investigators hypothesized that immune complexes (ICs) had caused enhanced RSV disease (ERD; reference 2), but the mechanism of illness was never clarified. There is still no vaccine licensed against RSV.
A widely accepted hypothesis to explain the pathogenesis of ERD has been that disease was caused by an imbalance in the response to RSV attachment (G) and fusion (F) proteins, because of poor preservation of F during formalin inactivation (3, 4). The predominance of G in FIRSV is thought to have led to a pathologic Th2 polarization of the immune response and pulmonary eosinophilia in immunized individuals infected with RSV. This hypothesis was supported by a relative excess of eosinophils in the lungs of an affected child and high levels of interleukin 4 and pulmonary eosinophilia in BALB/c mice challenged with RSV after immunization with either FIRSV, vaccinia virus encoding G, or purified G protein (2, 4–7). However, ERD in other mouse strains (8), cotton rats (9), and cattle (10) is not associated with pulmonary eosinophilia and rereview of the pulmonary histopathology of affected children revealed a predominance of neutrophils (11). In addition, numerous studies have detected an immune response against F after immunization with FIRSV (12, 13) suggesting that F retains antigenicity after inactivation.
An FI vaccine against measles, another paramyxovirus, was also developed in the 1960s. The FI measles vaccine primed for a severe and atypical form of measles characterized by fever, a petechial rash and pneumonitis after exposure to WT measles virus. It was recently shown that atypical measles was associated with IC deposition in affected tissues, eosinophilia, and a Th2 polarization of the immune response (14). ICs have been shown to promote Th2 skewing (15). Drawing from similarities between the immune response associated with ERD and atypical measles, we sought to test the hypothesis that deposition of IC and complement activation play an important role in ERD.
Materials and Methods
Mice.
4–6-wk-old female BALB/c (The Jackson Laboratory), B6129F2 C3−/−, and control mice and C57BL/10 μMT and control mice (The Jackson Laboratory) housed under laminar flow hoods in an environmentally controlled specific pathogen-free animal facility were used for these experiments.
Preparation of Vaccines and Immunization.
FIRSV was prepared using the RSV A2 strain grown in Hep-2 cells (2) with and without alum. PIV3 and rRSVΔG (16) were grown in Vero cells. Mice received the equivalent of 105 pfu of FIRSV, FIPIV, or FIRSVΔG intramuscularly. The dose of FIRSV was selected to allow pulmonary replication of RSV after challenge. Placebo consisted of supernatant from lysed uninfected Hep-2 cells in DMEM, 10% FCS, and 0.4% formalin incubated for 72 h at 37°C and ultracentrifuged at 30,000 g for 30 min. Intranasal immunization used 105 pfu of live RSV A2. 28 d after immunization, all groups were challenged intranasally with 106 pfu of RSV A2.
Fluorescent Staining.
10–20 μm frozen lung sections were stained for C3 and IgG using a FITC-conjugated Fab goat anti–mouse C3 (Cappell ICN) and rhodamine-conjugated rabbit anti–mouse IgG (Pierce Chemical Co.) and isotype control.
Airway Hyperresponsiveness.
7 d after challenge animals were anesthetized with sodium pentobarbital (90 mg/kg), intubated, ventilated at a rate of 120 breaths/min with a constant tidal volume of air (0.2 ml), and paralyzed with decamethonium bromide (25 μg/kg). After establishing a stable airway pressure, acetylcholine was given intravenously (50 μg/kg) and the dynamic airway pressure measured for 5 min.
Virus Titration in Lung Tissue.
Lungs from BALB/c mice were removed aseptically 4 d after RSV challenge and ground in 3 ml of HBSS (Bio Whittaker). Debris was pelleted by centrifugation at 2,000 rpm/5 min and samples plated on Hep-2 cells. Monolayers were overlayed with Opti-MEM (GIBCO BRL) with 2% FCS, 0.8% methylcellulose, glutamine, and antibiotics, and incubated for 5 d. Plates were stained by the immunoperoxidase method and results expressed in pfu/g of lung tissue.
Antibody Assays.
Sera were tested for antibodies to RSV 28 d after immunization and 7 d after challenge by complement-enhanced 60% plaque reduction neutralization assay using RSV A2 and for IgG antibodies to RSV F and G using protein-specific immunoassays (12).
Statistical Analysis.
Data were analyzed with statistical software (Statview). Comparisons were made using the Mann Whitney U test. All experimentation was approved by and performed according to guidelines of the Johns Hopkins Medical Institutions.
Results
ERD Is Associated with Pulmonary IC Deposition and Increased Airway Hyperresponsiveness.
To determine whether ERD was associated with pulmonary deposition of IC, BALB/c mice were immunized with FIRSV and then challenged with RSV. Mice immunized intramuscularly with placebo or intranasally with live WT RSV served as controls (Fig. 1) . Lungs of mice immunized with FIRSV and challenged with RSV stained with hematoxylin and eosin (H&E) showed a patchy mononuclear cell infiltration of the alveolar walls and a peribronchiolar and perivascular lymphomonocytic infiltration with a moderate number of interspersed neutrophils and eosinophils. Lungs of placebo recipients and mice immunized with live RSV contained fewer mononuclear cells after RSV challenge. To examine the lungs of mice for the presence of IC, frozen lung sections were stained for IgG and complement component C3 (Fig. 1). Colocalization of IgG and C3 was detected in alveolar regions and occasionally in bronchioles of mice with ERD, but not in lungs of control mice.
Figure 1.
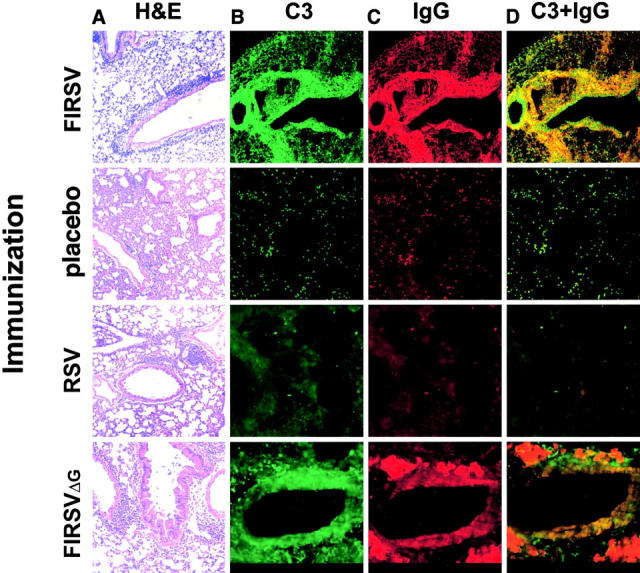
Pulmonary histopathology 7 d after RSV challenge in mice that had received the indicated preimmunization (10×). Hematoxylin and eosin (A); immunofluorescence staining for C3 (FITC; B), IgG (rhodamine; C) and colocalization of C3 and IgG (D).
Elucidation of the pathogenesis of ERD has been complicated because correlates of RSV disease in the mouse model have been limited to measures such as weight loss and the characteristics of the pulmonary cellular infiltrate. Recently, Peebles et al. demonstrated increased airway hyperresponsiveness (AHR) in mice with ERD compared with sham immunized mice and mice immunized with live RSV (17). As bronchoconstriction is a characteristic and serious manifestation of RSV disease and ERD (1, 2), we measured AHR in the lungs of mice as the primary correlate of disease enhancement to compare degrees of illness elicited by typical and ERD (Fig. 2) . After RSV challenge, mice immunized with FIRSV developed ERD with a significant increase in AHR compared with placebo recipients (P < 0.001), mice immunized with a control FI parainfluenza virus type 3 vaccine (FIPIV; P = 0.002), and mice immunized intranasally with live RSV (P < 0.001). The difference between groups was conserved when inactivated vaccines were administered with alum and reproduced in B6129F2 mice (unpublished data).
Figure 2.

AHR 7 d after RSV challenge in previously immunized BALB/c mice. AHR to acetylcholine challenge is defined by the time-integrated rise in peak airway pressure. Results are means ± SEM (error bars) of 7–10 animals per group and are representative of two independent experiments. *P < 0.05 compared with placebo, RSV, and FIPIV. **P < 0.05 compared with placebo, RSV, and FIPIV. * vs. **: P = 0.29.
RSV G Glycoprotein Is Not Required to Prime for ERD.
To determine whether ERD was associated with an abnormal immune response against G in FIRSV, we immunized BALB/c mice with a FI, recombinant RSV that does not express G (rRSVΔG; reference 16). Cellular infiltration in lung biopsies, IC deposition, and AHR after challenge were similar in FIRSVΔG and FIRSV recipients (Figs. 1 and 2) demonstrating that G is not necessary to prime for ERD.
A Role for Antibody and Complement in ERD.
To assess the role of complement in ERD we immunized C3 deficient (C3−/−) and WT mice with FIRSV and challenged them with RSV. Both groups developed similar alveolar, peribronchiolar, and perivascular mononuclear cellular infiltration with many interspersed neutrophils (Fig. 3) . Frozen sections confirmed deposition of C3 and IgG in alveoli and scattered bronchioles of WT mice, but only IgG was present in lungs of C3−/− mice (unpublished data). Although the histopathology in C3−/− and WT mice was similar, pulmonary function studies revealed clear differences between the two groups. FIRSV-immunized, RSV-challenged WT mice had a significant increase in AHR compared with C3−/−mice (P < 0.001), demonstrating that complement is critical for bronchoconstriction in ERD (Fig. 4 A). AHR was not different in naive C3−/− and WT mice (unpublished data).
Figure 3.

Hematoxylin and eosin stained pulmonary sections of C3 ET (+/+) and deficient (−/−) mice preimmunized with FIRSV and challenged with RSV.
Figure 4.

AHR 7 d after RSV challenge previously immunized with FIRSV. (A) B6129F2 WT (C3+/+) and C3 deficient (C3−/−),*: P < 0.001; and (B) C57BL/10 (B+/+) and B10 μMT (B−/−) mice, **: P = 0.009. AHR to acetylcholine challenge is defined by the time-integrated rise in peak airway pressure. Results are means ± SEM (error bars) of 9–12 animals per group and are representative of three independent experiments.
To further explore the role of IC in ERD, B cell–deficient (B−/−), and WT mice were immunized with FIRSV and challenged with RSV. As observed with C3−/− mice, B−/− and WT mice developed similar pulmonary inflammatory infiltrates (unpublished data). However, WT mice challenged with RSV had a significant increase in bronchoconstriction compared with B−/− mice (P = 0.009; Fig. 4 B). This confirmed the role of antibodies in ERD and provided additional evidence that pneumonia and bronchoconstriction are distinct phenomena.
Nonprotective Antibody Response to RSV in Mice Primed with FIRSV.
To determine whether ERD is associated with higher titers of RSV in the lungs of affected mice than in control groups, we compared viral titers after RSV challenge in mice previously immunized with FIRSV, placebo, or live RSV. RSV titers were similar in mice with ERD and placebo recipients infected with RSV, but undetectable in mice protected by prior intranasal infection with live RSV (Table I). These differences could not be explained by differences in serum glycoprotein-specific IgG levels (Table I). Serum anti-G and anti-F IgG levels were high after vaccination and challenge in FIRSV and live RSV groups (P = 0.71 for G and P = 0.44 for F after challenge). However, mice with ERD had no detectable neutralizing antibodies after vaccination or challenge, whereas mice primed with WT RSV had high levels of neutralizing antibodies after vaccination and were protected against challenge. As expected, placebo recipients infected with RSV had lower levels of anti-G and anti-F IgG than either of the previously primed groups.
Table I.
RSV Antibody and Viral Titers in Preimmunized Mice
| After immunization
|
After challenge
|
RSV titer (log pfu/g) |
|||||
|---|---|---|---|---|---|---|---|
| Vaccine | PRNT | EIA F | EIA G | PRNT | EIA F | EIA G | |
| FIRSV | <10 | 658 ± 22 | 139 ± 12 | <10 | 885 ± 18 | 292 ± 16 | 3.96 ± 0.01 |
| Placebo | <10 | <50 | <50 | <10 | 170 ± 24 | 68 ± 4 | 3.98 ± 0.02 |
| RSV IN | 164 ± 3 | 833 ± 12 | 148 ± 8 | 205 ± 7 | 971 ± 10 | 261 ± 8 | <0.6 |
Antibody responses in BALB/c mice determined 28 d after immunization and 7 d after RSV challenge and RSV pulmonary titers 4 d after challenge. PRNT, RSV-specific neutralization as measured by 60% complement-enhanced plaque reduction; EIA-F, immunoassay determination of IgG antibodies against RSV F protein (change in absorbance); EIA G, immunoassay determination of IgG antibodies against RSV G protein (change in absorbance). Results are means ± SEM of 8–10 animals per group.
7 d after RSV challenge, anti-F and anti-G IgM were detectable in naive and FIRSV-immunized mice but not in mice protected by live RSV. Levels of anti-F and -G IgA were similar in all groups. Determination of IgG subclasses in FIRSV and RSV recipients demonstrated that antibody to F was predominantly complement-fixing IgG2a, whereas antibody to the heavily-glycosylated protein G was predominantly IgG2b. However, there were no clear differences in IgG subclass responses between the groups (unpublished data).
Evidence for Activation of the Classic Complement Cascade in Lung Sections of Children with ERD.
To confirm the role of complement in the pathophysiology of ERD we used an antibody against C4d to stain lung sections of the two children who died of ERD (18). The complement cleavage product C4d is a sensitive marker of complement activation mediated by IC using the classical pathway, and can be detected in biopsies obtained years earlier (18). Lung sections of both children had extensive peribronchiolar deposition of C4d (Fig. 5) . No evidence of nonspecific binding of the C4d antibody was observed in sections of two control patients with nonhematologic malignancies (representative example shown in Fig. 5). These controls were used to show the binding specificity of the anti-C4d antibodies, as lung biopsy material from a normal infant with nonfatal RSV infection, paralleling the experimental and control samples from mice shown in Fig. 1, was unavailable.
Figure 5.

Deposition of C4d in pulmonary tissue of both patients (1 and 2) who died of ERD in 1967 and control patient (control). Immunohistochemistry staining for C4d (A) and preimmune rabbit sera (B) (reference 18).
Discussion
This study demonstrates that IC that fix complement are important in the pathogenesis of ERD. Antibody deposition in FIRSV-immunized mice does not result in ERD after RSV challenge in the absence of complement activation and complement cannot elicit ERD in the absence of antibodies. Peribronchiolar C4d deposition in children with ERD provides conclusive evidence of antibody-mediated activation of the classic complement cascade during this process. Moreover, the quality of antibody elicited also appears to be important. IC formation is associated with the presence of large amounts of nonneutralizing antibody at the time of pulmonary RSV replication; large amounts of neutralizing antibody elicited by prior WT infection protect against challenge with RSV and do not lead to ERD, as demonstrated in infants at increased risk of severe RSV disease (19).
These high levels of nonneutralizing antibody suggest that formalin disrupts critical epitopes during the process of inactivation (12, 13). Alternatively, an altered maturation of the B cell response after FIRSV may account for the abundant, nonprotective antibody response leading to IC deposition. Low avidity antibodies have been associated with IC-mediated disease and may explain IC deposition during ERD (20). Data from atypical measles show that B cells of macaques primed with FI measles fail to undergo affinity maturation and display high levels of low avidity, nonprotective antibody when infected with measles (unpublished data). It is likely that ERD and atypical measles have strong parallels. Either process, formalin-mediated disruption of neutralizing epitopes or lack of affinity maturation, could result in the presence of abundant nonprotective RSV-binding antibodies in the face of extensive RSV antigen production and provide the basis for IC formation in the infected airways. This strong nonneutralizing antibody response may be stimulated by the Th2 polarization observed in recipients of FIRSV, an RSV-vaccine that does not elicit Th-modulatory cytotoxic T lymphocyte responses (21, 22). IC themselves may also enhance antibody production and promote a Th2 bias of the immune response (15).
In our study, complement and antibody deficient and WT mice had similar pulmonary inflammatory infiltration, but significantly different airway responses. This suggests that AHR in this mouse model is not associated with the degree of pneumonia, although it may be promoted by cellular degranulation or release of other inflammatory mediators associated with complement activation. A similar dissociation between AHR and pulmonary inflammatory cells has been reported in other murine models of allergic asthma (23). In the past, passive transfer of sera from FIRSV-immunized mice failed to elicit an “enhanced” pneumonia in naive mice upon RSV challenge (bronchoconstriction was not measured). This may be explained by the lack of primed T helper lymphocytes recruiting inflammatory cells to the lungs at the time of challenge (24) and is in agreement with our observation that factors other than complement attract inflammatory cells to the lung during ERD (12, 13).
We do not exclude a role for cellular infiltrate in ERD, a phenomenon that is likely complex and multifactorial. The pneumonia has been shown to be a T cell–dependent manifestation of ERD (4, 24) and the inflammatory infiltrate (2, 11) may play a functional role in other manifestations. The regulatory role of T helper cells in AHR and whether granulocytes can directly cause bronchoconstriction should be further investigated. However, none of these clinical signs appear to require priming by the G glycoprotein.
The complement system may injure tissue through formation of the cell-lytic complex C5b-9, or by action of complement anaphylotoxins (C3a, C4a, and C5a) elicit bronchoconstriction, mucus secretion, or recruit inflammatory cells promoting degranulation and disease. Complement factors can also promote T cell activation (a feature of both ERD and atypical measles [25, 26]) and affect the levels of proinflammatory cytokines. Recent papers have described a role for the C3a anaphylotoxin in the effector phase of allergic bronchoconstriction (27), a modulatory role for C5 in allergic asthma (28), and a critical role for C5a in IC-mediated alveolitis (29). ERD was characterized by bronchoconstriction and pneumonia (2), both signs that may be associated with anaphylotoxin activity (27–30). Preliminary studies in our laboratory demonstrate high AHR in A/J mice with ERD. As A/J mice are deficient in functional C5, these data would support an effector role for C3a in bronchoconstriction.
This study provides a new model for ERD that can be used to screen future candidate vaccines. This model is reproducible in multiple strains of mice, detects ERD by a correlate (bronchoconstriction) that is directly relevant to clinical RSV disease in humans, provides quantitative virologic data and pulmonary histopathology, and differs from another model that measured AHR in that concomitant allergic sensitization is not required (17).
In summary, bronchoconstriction in ERD is mediated by IC and can be elicited by immunization with FIRSV in the absence of the G glycoprotein. Whether this pathologic response is specific to FI vaccines remains to be determined. Therefore, extreme caution should be exercised in the evaluation of nonreplicating RSV vaccines in infants.
Acknowledgments
We thank Bhagvanji Thumar, Liliana Moreno, and Jean Froehlich for expert assistance, Dr. Diane E. Griffin and Dr. Robert M. Chanock for helpful discussions, and Dr. Roma Chandra for access to the human autopsy material and data.
This study was supported in part by The Thomas and Carol McCann Innovative Research Fund for Asthma and Respiratory Disease (FPP).
M. Teng's present address is Department of Biochemistry and Molecular Biology, Penn State University, University Park, PA 16802.
References
- 1.Collins, P.L., R.M. Chanock, and B.R. Murphy. 2001. Fields Virology. D.M. Knipe and P.M. Howley, editors. Lippincott Williams and Wilkins, Philadelphia, PA. 1443–1486.
- 2.Kim, H.W., J.G. Canchola, C.D. Brandt, G. Pyles, R.M. Chanock, K. Jensen, and R.H. Parrott. 1969. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 89:422–434. [DOI] [PubMed] [Google Scholar]
- 3.Graham, B.S. 1995. Pathogenesis of respiratory syncytial virus vaccine-augmented pathology. Am. J. Respir. Crit. Care Med. 152:S63–S66. [DOI] [PubMed] [Google Scholar]
- 4.Connors, M., N.A. Giese, A.B. Kulkarni, C.Y. Firestone, H.C. Morse III, and B.R. Murphy. 1994. Enhanced pulmonary histopathology induced by respiratory syncytial virus (RSV) challenge of FI RSV-immunized BALB/c mice is abrogated by depletion of interleukin-4 (IL-4) and IL-10. J. Virol. 68:5321–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham, B.S., G.S. Henderson, Y.W. Tang, X. Lu, K.M. Neuzil, and D.G. Colley. 1993. Priming immunization determines T helper cytokine mRNA expression patterns in lungs of mice challenged with respiratory syncytial virus. J. Immunol. 151:2032–2040. [PubMed] [Google Scholar]
- 6.Hancock, G.E., D.J. Speelman, K. Heers, E. Bortell, J. Smith, and C. Cosco. 1996. Generation of atypical pulmonary inflammatory responses in BALB/c mice after immunization with the native attachment (G) glycoprotein of respiratory syncytial virus. J. Virol. 70:7783–7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Openshaw, P.J., S.L. Clarke, and F.M. Record. 1992. Pulmonary eosinophilic response to respiratory syncytial virus infection in mice sensitized to the major surface glycoprotein G. Int. Immunol. 4:493–498. [DOI] [PubMed] [Google Scholar]
- 8.Hussell, T., A. Georgiou, T.E. Sparer, S. Matthews, P. Pala, P.J. Openshaw. 1998. Host genetic determinants of vaccine-induced eosinophilia during respiratory syncytial virus infection. J. Immunol. 161:6215–6222. [PubMed] [Google Scholar]
- 9.Prince, G.A., A.B. Jenson, V.G. Hemming, B.R. Murphy, E.E. Walsh, R.L. Horswood, and R.M. Chanock. 1986. Enhancement of respiratory syncytial virus pulmonary pathology in cotton rats by prior intramuscular inoculation of FI virus. J. Virol. 57:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gershwin, L.J., E.S. Schelegle, R.A. Gunther, M.L. Anderson, A.R. Woolums, D.R. Larochelle, G.A. Boyle, K.E. Friebertshauser, and R.S. Singer. 1998. A bovine model of vaccine enhanced respiratory syncytial virus pathophysiology. Vaccine. 16:1225–1236. [DOI] [PubMed] [Google Scholar]
- 11.Prince, G.A., S.J. Curtis, K.C. Yim, and D.D. Porter. 2001. Vaccine-enhanced respiratory syncytial virus disease in cotton rats following immunization with Lot 100 or a newly prepared reference vaccine. J. Gen. Virol. 82:2881–2888. [DOI] [PubMed] [Google Scholar]
- 12.Murphy, B.R., A. Sotnikov, P.R. Paradiso, S.W. Hildreth, A.B. Jenson, R.B. Baggs, L. Lawrence, J.J. Zubak, R.M. Chanock, J.A. Beeler, et al. 1989. Immunization of cotton rats with the fusion (F) and large (G) glycoproteins of respiratory syncytial virus (RSV) protects against RSV challenge without potentiating RSV disease. Vaccine. 7:533–540. [DOI] [PubMed] [Google Scholar]
- 13.Connors, M., P.L. Collins, C.Y. Firestone, A.V. Sotnikov, A. Waitze, A.R. Davis, P.P. Hung, R.M. Chanock, and B.R. Murphy. 1992. Cotton rats previously immunized with a chimeric RSV FG glycoprotein develop enhanced pulmonary pathology when infected with RSV, a phenomenon not encountered following immunization with vaccinia-RSV recombinants or RSV. Vaccine. 10:475–484. [DOI] [PubMed] [Google Scholar]
- 14.Polack, F.P., P.G. Auwaerter, S.H. Lee, H.C. Nousari, A. Valsamakis, K.M. Leiferman, A. Diwan, R.J. Adams, and D.E. Griffin. 1999. Production of atypical measles in rhesus macaques: evidence for disease mediated by IC formation and eosinophils in the presence of fusion-inhibiting antibodies. Nat. Med. 5:629–634. [DOI] [PubMed] [Google Scholar]
- 15.Anderson, C.F., and D.M. Mosser. 2002. Cutting edge: biasing immune responses by directing antigen to macrophage fcgamma receptors. J. Immunol. 168:3697–3701. [DOI] [PubMed] [Google Scholar]
- 16.Teng, M.N., S.S. Whitehead, and P.L. Collins. 2001. Contribution of the respiratory syncytial virus G glycoprotein and its secreted and membrane-bound forms to virus replication in vitro and in vivo. Virology. 289:283–296. [DOI] [PubMed] [Google Scholar]
- 17.Peebles, R.S., Jr., J.R. Sheller, R.D. Collins, K. Jarzecka, D.B. Mitchell, and B.S. Graham. 2000. Respiratory syncytial virus (RSV)-induced AHR in allergically sensitized mice is inhibited by live RSV and exacerbated by FI RSV. J. Infect. Dis. 182:671–677. [DOI] [PubMed] [Google Scholar]
- 18.Regele, H., M. Exner, B. Watschinger, C. Wenter, M. Wahrmann, C. Osterreicher, M.D. Saemann, N. Mersich, W.H. Horl, G.J. Zlabinger, G.A. Bohmig. 2001. Endothelial C4d deposition is associated with inferior kidney allograft outcome independently of cellular rejection. Nephrol. Dial. Transplant. 16:2058–2066. [DOI] [PubMed] [Google Scholar]
- 19.Groothuis, J.R., E.A. Simoes, M.J. Levin, C.B. Hall, C.E. Long, W.J. Rodriguez, J. Arrobio, H.C. Meissner, D.R. Fulton, R.C. Welliver, et al. 1993. Prophylactic administration of respiratory syncytial virus immune globulin to high-risk infants and young children. The Respiratory Syncytial Virus Immune Globulin Study Group. N. Engl. J. Med. 329:1524–1530. [DOI] [PubMed] [Google Scholar]
- 20.Devey, M.E., and M.W. Steward. 1980. The induction of chronic antigen-antibody complex disease in selectively bred mice producing either high or low affinity antibody to protein antigens. Immunology. 41:303–311. [PMC free article] [PubMed] [Google Scholar]
- 21.Srikiatkhachorn, A., T. Braiciale. 1997. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J. Exp. Med. 186:421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hussell, T., C.J. Baldwin, A. O'Garra, and P.J. Openshaw. 1997. CD8+ T cells control Th2-driven pathology during pulmonary respiratory syncytial virus infection. Eur. J. Immunol. 27:3341–3349. [DOI] [PubMed] [Google Scholar]
- 23.Wills-Karp, M., J. Luyimbazi, X. Xu, B. Schofield, T.Y. Neben, C.L. Karp, and D.D. Donaldson. 1998. Interleukin-13: central mediator of allergic asthma. Science. 282:2258–2261. [DOI] [PubMed] [Google Scholar]
- 24.Connors, M., A.B. Kulkarni, C.Y. Firestone, K.L. Holmes, H.C. Morse III, A.V. Sotnikov, and B.R. Murphy. 1992. Pulmonary histopathology induced by respiratory syncytial virus (RSV) challenge of FI RSV-immunized BALB/c mice is abrogated by depletion of CD4+ T cells. J. Virol. 66:7444–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim, H.W., S.L. Leikin, J. Arrobio, C.D. Brandt, R.M. Chanock, and R.H. Parrott. 1976. Cell-mediated immunity to respiratory syncytial virus induced by inactivated vaccine or by infection. Pediatr. Res. 10:75–78. [DOI] [PubMed] [Google Scholar]
- 26.Kraus, P.J., J.D. Cherry, J.M. Carney, M.J. Naiditch, and K. O'Connor. 1980. Measles-specific lymphocyte reactivity and serum antibody in subjects with different measles histories. Am. J. Dis. Child. 134:567–571. [DOI] [PubMed] [Google Scholar]
- 27.Humbles, A.A., B. Lu, C.A. Nilsson, C. Lilly, E. Israel, Y. Fujiwara, N.P. Gerard, and C. Gerard. 2000. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature. 406:998–1001. [DOI] [PubMed] [Google Scholar]
- 28.Karp, C.L., A. Grupe, E. Schadt, S.L. Ewart, M. Keane-Moore, P.J. Cuomo, J. Kohl, L. Wahl, D. Kuperman, S. Germer, et al. 2000. Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nat. Immunol. 1:221–226. [DOI] [PubMed] [Google Scholar]
- 29.Hopken, U.E., B. Lu, N.P. Gerard, and C. Gerard. 1997. Impaired inflammatory responses in the reverse arthus reaction through genetic deletion of the C5a receptor. J. Exp. Med. 186:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang, Y., B.F. Ramos, and B.A. Jakschik. 1992. Neutrophil recruitment by tumor necrosis factor from mast cells in IC peritonitis. Science. 258:1957–1959. [DOI] [PubMed] [Google Scholar]