

Abstract
CD30 is up-regulated in several human diseases and viral infections but its role in immune regulation is poorly understood. Here, we report the expression of a functional soluble CD30 homologue, viral CD30 (vCD30), encoded by ectromelia (mousepox) virus, a poxvirus that causes a severe disease related to human smallpox. We show that vCD30 is a 12-kD secreted protein that not only binds CD30L with high affinity and prevents its interaction with CD30, but it also induces reverse signaling in cells expressing CD30L. vCD30 blocked the generation of interferon γ–producing cells in vitro and was a potent inhibitor of T helper cell (Th)1- but not Th2-mediated inflammation in vivo. The finding of a CD30 homologue encoded by ectromelia virus suggests a role for CD30 in antiviral defense. Characterization of the immunological properties of vCD30 has uncovered a role of CD30–CD30L interactions in the generation of inflammatory responses.
Keywords: poxviruses, immunomodulation, cytokine, TNFR superfamily, Th-1
Introduction
CD30 and CD30L (CD153) are members of the TNFR and TNF superfamilies, respectively. CD30 was identified by the mAb Ki-1 against Hodgkin and Reed-Sternberg cells (1), the malignant component of Hodgkin's disease, and has been extensively used as a clinical disease marker. CD30 was subsequently found in resting CD8+ T cells, activated or virally transformed T and B cells, and at the surface of HIV-infected lymphocytes. CD30 is a type I membrane protein that can be cleaved by metalloproteases producing a soluble form (sCD30;*reference 2). The extracellular domain of CD30 shows cysteine-rich domains (CRDs) characteristic of the TNFR superfamily. CD30L is expressed as a type II membrane glycoprotein (2) in resting neutrophils and B cells, activated T cells and macrophages, and in neoplasic cells such as Burkitt-type lymphoma cells or B cells associated with lymphoproliferative disorders. The extracellular domain of CD30L shows homology to TNF, lymphotoxin, and CD40L (3). It is unclear whether CD30L also exists as a soluble form, as does TNF. Interaction of CD30L with cells expressing CD30 induces signals mediated by nuclear factor κB and TNFR-associated factor 2 that cause cell proliferation or cell death. Interestingly, upon binding to CD30, CD30L is also able to signal. One of the consequences of this reverse signaling is cell proliferation (4).
The role of CD30–CD30L interaction in health and disease is still not totally understood, in part due to the pleiotropic nature of CD30 signals. Mice lacking a functional CD30 gene show defective negative thymocyte selection (5), whereas transgenic mice expressing CD30 in the thymus have enhanced thymic negative selection (6). A recent study of genes targeted by CD30 suggests that Fas, TRAIL, CCR7, TNFR-associated factor 1, and cIAP2 are up-regulated whereas FasL, perforin, granzyme B, and c-myc seem to be down-regulated (7). Finally, the reasons for increased levels of sCD30 in malignant lymphomas, viral infection (HIV, human T cell leukemia virus, and EBV), and several immunological disorders such as systemic lupus erythematosus or rheumatoid arthritis are not known (8).
Poxviruses are a family of complex DNA viruses that encode up to 200 genes and infect a wide variety of hosts (9). Smallpox was a devastating disease caused by variola virus (VaV), one of the most virulent human pathogens. Vaccinia virus (VV) is the best characterized poxvirus and the vaccine used to achieve the global eradication of smallpox by 1977, but its origin and natural host are unknown. Cowpox virus (CPV) is probably a rodent virus that sporadically infects other animal species. Ectromelia virus (EV) is a highly virulent natural pathogen of mice that causes mousepox and has been isolated from outbreaks in laboratory mouse colonies (10). Like VaV, EV has a restricted host range, causes severe disease with high mortality rate and skin lesions in the later stages of infection. These similarities with smallpox make EV an interesting experimental model for virus–host interactions.
Poxviruses encode a unique collection of genes that evade host immune responses. These molecules are often secreted and include cytokine homologues and soluble cytokine receptors or binding proteins (11–13). Some of these viral genes seem to have been acquired from the host and modified during virus evolution to confer an advantage for virus replication, survival, or transmission. EV encodes receptors or binding proteins for TNF (14), IL-1β (15), IFN-γ (16, 17), IFN-α/β (17, 18), IL-18 (19, 20), and chemokines (15, 21). EV also encodes antiapoptotic proteins (22, 23) and an intracellular protein that confers IFN resistance (17).
The TNF binding activity encoded by orthopoxviruses is particularly interesting because there are four distinct viral TNFRs (vTNFRs): cytokine response modifier (Crm) B (24), CrmC (25), CrmD (14), and CrmE (26). These molecules show different ligand specificity and are expressed at different times after infection, but their relative contribution to viral pathogenesis is not well understood. Maybe the vTNFRs bind other members of the growing TNF family and protect the virus from the action of other ligands.
We report the identification and characterization of a novel member of the TNFR superfamily, a homologue of CD30 encoded by EV that blocks the binding of CD30L to its receptor and induces reverse signaling in cells expressing CD30L. Moreover, the viral CD30 (vCD30) abrogates T cell proliferation in vitro and in vivo it blocks type 1 but not type 2 cytokine–mediated T cell responses. These studies not only describe a novel immunomodulatory strategy of poxviruses, but also pave the way to the role of CD30–CD30L in viral infections and in type I cytokine–mediated inflammatory diseases.
Materials and Methods
Reagents
Recombinant mouse CD30L (ED50 = 50 − 150 μg/ml), human CD30 (ED50 = 0.03 − 0.1 μg/ml), and mouse CD30 (ED50 = 0.03 − 0.1 μg/ml) were purchased from R&D Systems. The iodine-125 (103.7 mCi/ml) and iodogen reagent used to radioiodinate mouse CD30L were purchased from Amersham Biosciences and Pierce Chemical Co., respectively. The protein A–coated FlashPlates used for binding and affinity studies were purchased from PerkinElmer. The mouse mAb specific for the histidine tag and the FITC-conjugated goat anti–mouse Igs used in flow cytometry were from CLONTECH Laboratories, Inc. and Dako, respectively. Mycobacterium tuberculosis whole cell lysates (H37Rv strain) and protein-purified derivative (PPD) were obtained from Mycos Research. Schistosome eggs and soluble egg Ags (SEA) were prepared as previously described (27).
Cells, Viruses, and Viral DNA Preparations
The growth of BSC-I, TK−143B, and K562 cells, and the sources of VV Western Reserve (WR) strain and EV isolates Hampstead and Naval have been described (15). VV and EV were propagated in BSC-I cells and viral genomic DNA was prepared as previously described (28). The growth of Autographa californica nuclear polyhedrosis virus in Spodoptera frugiperda 21 insect cells has been described (29). Tn5 B1-4 (Hi5) insect cells were cultured in EX-CELL serum-free medium as suggested by the supplier (European Collection of Cell Cultures).
DNA Sequencing
Specific oligonucleotides, CD30-1 (5′ GTTCTGGATACATGCACAAAG 3′) and CD30-2 (5′ GGAGGATAATCATTTGCAAACG 3′), were designed based on the sequence of CPV strain GRI90 open reading frame (ORF) D13L (30), and used to amplify the cognate genes from viral DNA preparations from VV WR and EV Hampstead and Naval by PCR using Taq DNA polymerase. PCR products were sequenced by the DNA Sequencing Service of the Department of Biochemistry, Cambridge University, Cambridge, United Kingdom. The sequence data were analyzed using Genetics Computer Group computer programs (31).
Construction of Recombinant Baculovirus Expressing the EV Hampstead vCD30 Gene
The EV Hampstead vCD30 gene was amplified by PCR using Pfu DNA polymerase, virus DNA as template, and oligonucleotides CD30-3 (5′ CGCAAGCTTGGATCCATGAAGATGAATACTATC TTTTTATC 3′) and CD30-4 (5′ CGCGCGGCCGCTGATGAGTATTTATGATAACAAAG 3′), which correspond to the 5′ and 3′ ends of the ORF and provide HindIII/BamHI and NotI sites, respectively. The resultant product was cloned into HindIII- and NotI-digested pBac1 (Novagen), creating plasmid pMS2 (EV Hampstead vCD30). The DNA sequence of the insert was confirmed to not contain mutations. The Fc fragment of the human IgG1 was cut from pIGplus (R&D Systems) and subcloned into NotI/SphI sites of pMS2, which created plasmid pMS18 (EV Hampstead vCD30-Fc). Recombinant baculovirus was produced as previously described (29) and termed AcCD30-Fc (EV Hampstead vCD30-Fc, AcMS18). Control recombinant baculovirus expressing EV Hampstead–truncated CrmD (AcCrmD-CRD1,2-Fc) was constructed as AcCD30-Fc (unpublished data).
Purification of the Baculovirus Recombinant vCD30-Fc Protein
Hi5 cultures were infected with recombinant baculoviruses at 10 pfu/cell and supernatants were harvested 3–4 d later when full infection was observed. The recombinant Fc fusion proteins were subsequently purified using Protein A HiTrap columns (Amersham Biosciences). The purified protein was then analyzed by SDS-PAGE in 12% acrylamide gels and stained with Coomassie blue. Protein concentration was determined using the Bio-Rad protein assay reagent.
Construction of Recombinant VV Expressing the EV Hampstead vCD30 Gene
The EV Hampstead vCD30 gene was amplified by PCR with virus DNA as template, Pfu DNA polymerase, and oligonucleotides CD30-3 and CD30-5 (5′ CGCGGTACCTCATGATGAGTATTTATGATAACAAAG 3′) containing KpnI restriction site. The DNA fragment was cloned into BamHI- and KpnI-digested pMJ601 (provided by B. Moss, National Institutes of Health, Bethesda, MD; reference 32), creating plasmid pMS12 (EV Hampstead vCD30). The DNA sequence of the insert was confirmed to not contain mutations. The recombinant VV was produced as previously described (29) and termed VVCD30 (EV Hampstead vCD30, vMS12).
Metabolic Labeling of VVCD30 and Electrophoretic Analysis
BSC-I cells were infected with VV WR or VVCD30 at 10 pfu/cell. Cultures were pulse labeled with 150 μCi/ml [35S]methionine (1,200 Ci/mmol; Amersham Biosciences) and 150 μCi/ml [35S]cysteine (600 Ci/mmol; NEN Life Science Products) in methionine- and cysteine-free medium in the absence of serum. Cells or media were dissociated in sample buffer and analyzed by SDS-PAGE in 12% acrylamide gels and visualized by fluorography with Amplify (Amersham Biosciences).
Preparation of VV and EV Supernatants
BSC-I cells were mock infected or infected with VV-WR, VVCD30, EV Hampstead, and EV Naval at 10 pfu/cell in phenol red– and serum-free medium. Supernatants were harvested at 2 (for the VV infections) or 3 (for the EV and mock infections) d after infection and prepared and inactivated as previously described (33).
CD30L Binding Assay
Recombinant mouse CD30L was radioiodinated to a specific activity of 106 cpm/μg using the Iodogen method (34). Approximately 150 pM of 125I-CD30L was incubated for 12 h with 5 ng purified vCD30-Fc or recombinant mouse CD30 in a protein A–coated FlashPlate. The binding medium was phenol red–free MEM, 0.1% BSA, 20mM Hepes, pH 7.5. The amount of CD30L bound to the viral receptor was measured in a Packard Topcount microplate counter. Nonspecific binding was determined by incubating 125I-CD30L with binding medium only. For the competition studies, a 500-fold molar excess of cold mouse CD30L was added to the recombinant mouse or viral receptors before the addition of 125I-CD30L. To test the CD30 binding activity in supernatants of EV Hampstead and Naval (VV WR or VVCD30), 50 μl supernatant equivalent to 1.5 × 104 cells were preincubated with 125I-CD30L before being added to the recombinant mouse or viral receptors. For the determination of the affinity constant of both mouse and vCD30 to the CD30L, binding assays with increasing amounts of 125I-CD30L against a fixed amount of recombinant CD30 (2 and 0.5 ng mouse or viral protein, respectively) were performed. The results were analyzed with the LIGAND software (35). For the determination of membrane-bound activity of vCD30, BSC-I cells were mock infected or infected with VV WR, VVCD30, and EV Hampstead at 10 pfu/cell. 24 h later, human 125I-CD30L was added and bound 125I-CD30L was determined by phthalate oil centrifugation (29).
Kinetics of vCD30 Production During EV Infection
BSC-I cells were mock infected or infected with 10 pfu EV Hampstead per cell in the absence or presence of 40 μg/ml cytosine arabinoside (AraC), an inhibitor of DNA replication, and harvested at different times after infection. Supernatants were inactivated and the CD30 binding activity was tested as described above. Total RNA was extracted using the guanidine thiocyanate–based DNA/RNA Isolation Kit (Promega) according to the manufacturer's instructions. Total RNA (from 7 × 104 cells) was then analyzed by RT and then by PCR. RT was performed in the presence of oligo(dT)15 (Promega), RNAsin (Amersham Biosciences), and avian myeloblastosis virus RT (Boehringer). The cDNA (2.5 μl of 40 μl; provided by K. Shair, University of Cambridge, Cambridge, United Kingdom) was amplified by PCR using Taq polymerase and oligonucleotides specific for vCD30, CD30-3, and CD30-4. DNA from BSC-I cells was included as a negative control.
Biological Activity of the vCD30
In Vitro Studies.
1 μg/ml recombinant soluble mouse CD30L preincubated with RPMI, a 25-fold excess of vCD30, or human IgG1 for 1.5 h at 4°C was added to 106 K562 cells and incubated for 2 h at 4°C. After this period, cells were incubated for 40 min at 4°C with a mouse mAb specific for the histidine tag (1 μg/ml in 0.1% BSA in PBS) that would recognize CD30L bound to the cell membrane. This Ab was subsequently developed with an FITC-labeled goat anti–mouse Ig Ab for 30 min at 4°C. Cells binding CD30L were then detected by FACS® analysis. Unstained cells and cells stained in the absence of CD30L were included as a control.
To test the ability of vCD30 to induce reverse signaling via membrane bound CD30L, 5 × 104 freshly isolated human neutrophils were incubated in a volume of 100 μl for 5 h at 37°C in 96-well plates precoated with 10 μg/ml of mouse, human vCD30, human IgG1, CD30, or PBS as previously described (4). After this period, supernatants were harvested and the production of IL-8 was measured by ELISA (Diaclone). To address the possible interference of vCD30 in the development of CTL responses, 4 × 105 freshly isolated splenocytes from BALB/c mice were mixed in 96-well plates with 2 × 104 L929 cells in a final volume of 200 μl RPMI in the presence or absence of 10 μg/ml vCD30-Fc or IgG1 supplemented with 10% FCS, sodium pyruvate, and nonessential amino acids for 5 d at 37°C, 5% CO2. After this period, IL-2 (Roche) was added to a final concentration of 50 U/ml and the incubation was held for an additional 2 d. Finally, cells were harvested and the viable cells counted by trypan blue exclusion. 105 activated splenocytes were mixed again with 104 L929 in the presence or absence of 10 μg/ml vCD30-Fc or IgG1 and the number of cells producing IFN-γ were measured using an ELISPOT assay (R&D Systems).
In Vivo Studies.
Type 1 and 2 cytokine–dominated pulmonary granulomas were induced by mycobacterial or SEA, respectively, as previously described (36). Female BALB/c mice were obtained from Harlan. vCD30-Fc or controls CrmD-CRD1,2-Fc and IgG1 (Sigma-Aldrich) were injected intraperitoneally (10 μg per injection) throughout sensitization and elicitation of bead granulomas, i.e., on days 0, 7, 14, and 16, or were injected only during elicitation of granulomas on days 14 and 16. On day 0, mice were sensitized by intraperitoneal injection of 20 μg M. tuberculosis whole cell lysates in complete Freund's adjuvant (type 1 cytokine sensitization) or 5,000 Schistosoma mansoni eggs (type 2 cytokine sensitization). 14 d later mice were injected intravenously with 5,000 Sepharose 4B beads covalently coupled with PPD or SEA. Mice were killed 4 d after bead injection on day 18. The left lung lobe was snap frozen and used for cytokine analysis. The remainder of the lung was fixed for histological studies. The diameters of the granuloma surrounding at least 50 individual beads per mouse were measured. Group mean granuloma volumes from four or five mice per group are presented. The statistical differences between groups were determined using Student's t test.
Lung tissue cytokines were determined as previously described using ELISA protocols (37). Control naive mouse lungs were processed to determine basal lung cytokine levels. Data were expressed as nanogram cytokine per milligram lung protein. The spleen and draining mediastinal LN were removed on the day the mice were killed and used for cell culture and intracellular cytokine staining (27). In brief, spleen cell suspensions were cultured for 6 h in media alone or in the presence of 6 μg/ml Con A (Sigma-Aldrich) and 10 μg/ml Brefeldin A (Sigma-Aldrich) added during the last 4 h. All intracellular detection reagents were from Caltag. Cells were surface stained with tri-color–conjugated anti-CD4 or CD8 mAbs. After cell permeabilization, cells were incubated with an FITC-conjugated anti–IFN-γ–mAb, PE-conjugated anti–IL-4 mAb, or FITC- or PE-conjugated isotype control mAbs. For FACS® analysis CD4+ or CD8+ lymphocytes were gated and quadrants were set using isotype control mAbs. The frequencies of IFN-γ– and IL-4–stained cells are expressed as percentages.
Nucleotide Sequence Accession Number
The sequence data for the EV Hampstead– and Naval-encoded CD30 homologue are available from GenBank/EMBL/DDBJ under accession numbers AJ507059 and AJ507060, respectively.
Results
Identification of a Novel Member of the TNFR Superfamily Encoded by EV.
Analysis of the CPV strain GRI90 sequence (30) revealed the presence of an ORF (D13L) with sequence similarity to host CD30, a TNFR superfamily member, and distinct from the previously identified poxvirus TNFRs (CrmB, CrmC, CrmD, and CrmE). PCR and sequence analysis of the cognate gene in EV isolates Hampstead and Naval showed the existence of an intact vCD30 gene (Fig. 1 a). The predicted viral molecule lacked N-glycosylation sites, was considerably smaller (12 kD) than the mouse or human counterparts (52 and 120 kD, respectively), and aligned with the second and third CRDs found in the extracellular domain of the host CD30, with some motifs highly conserved (Fig. 1 b). Interestingly, vCD30 showed a higher similarity to mouse (63.7%) than to human (56.7%) CD30. The existence of a signal peptide and the lack of transmembrane domain suggested that vCD30 may act as a soluble decoy receptor for host CD30L.
Figure 1.

Sequence of vCD30. Pairwise alignment of the predicted amino acid sequence of vCD30 in (a) three distinct orthopoxviruses and (b) of human, mouse, and vCD30. Dark shadows represent (a) differences or (b) similarities. Gray boxes, regions of high similarity; ·, deletions; *, stop codons. The predicted signal peptide (SP), CRDs, and transmembrane domain (TM) are indicated. These sequence data are available from GenBank/EMBL/DDBJ under accession numbers: Y11842 (CPV-GRI90 vCD30), M83554 (human CD30), U25416 (mouse CD30), AJ507059 (EV Hampstead vCD30), and AJ507060 (EV Naval vCD30).
Characterization of the vCD30 Protein.
A recombinant VV WR expressing the EV vCD30 under a strong promoter was constructed. PCR analysis indicated the absence of the vCD30 gene in VV WR (not depicted). Pulse labeling experiments with [35S]methionine and cysteine showed that vCD30 was efficiently secreted from infected cells as a 12-kD protein (Fig. 2 a). The EV vCD30 gene was also expressed in the baculovirus system fused to the Fc region of human IgG1. The recombinant product (vCD30-Fc) was purified by affinity chromatography on protein A Sepharose and its molecular size was consistent with vCD30 encoding a 12-kD protein (Fig. 2 b). In addition, matrix-assisted laser desorption ionization time-of-flight mass spectrometry analysis of the purified vCD30-Fc (performed by S. Mas and B. Aguado, Medical Research Council Human Genome Mapping Project, Cambridge, United Kingdom) and the minor contaminant band only identified peptides corresponding to vCD30 or the Fc region of human IgG1, demonstrating the high purity of the vCD30-Fc preparation and indicating that the minor contaminant band results from proteolytic degradation of vCD30-Fc (not depicted).
Figure 2.


Expression of vCD30 in the VV expression system and as an Fc fusion protein. (a) BSC-I cells were infected with VV-WR or VVCD30 and pulse labeled with [35S]cysteine and [35S]methionine from 4 to 8 h after infection. Proteins present in cells and media were analyzed by SDS-PAGE in the presence of 2-ME and visualized by fluorography. The position of the expressed protein in supernatants and cell extracts is indicated. (b) Hi5 insect cells were infected with the recombinant baculovirus expressing vCD30-Fc, harvested 3 d after infection, and the recombinant protein was purified by affinity chromatography in a protein A column. The fractions containing the purified vCD30-Fc were then concentrated and analyzed by SDS-PAGE. The position of vCD30-Fc is indicated. In both a and b, molecular masses in kD are shown.
CD30L Binding Activity, Specificity, and Affinity of vCD30.
CD30L binding activity was determined using a scintillation proximity assay. Purified recombinant vCD30-Fc or mouse CD30–Fc were incubated for 12 h with radiolabeled mouse CD30L in protein A–coated FlashPlates containing a thin layer of scintillant in the interior of each well. In this assay, recombinant CD30 proteins are immobilized by the Fc portion and bound 125I-CD30L induces a signal detectable in a scintillation counter. Free 125I-CD30L is not detected and there is no need to remove it. As shown in Fig. 3 a, both vCD30-Fc and mouse CD30–Fc specifically bound 125I-CD30L. Interestingly, the viral protein seemed to have a better binding capacity than the mouse counterpart and this was observed over a range of protein concentrations (not depicted). This difference may reflect a slightly higher affinity of vCD30 or the loss of some biological activity of the mouse protein during the purification procedure. Control IgG1 did not show binding activity in this assay (not depicted). vCD30 did not bind iodinated human TNF in similar binding assays (not depicted).
Figure 3.

(a) CD30L binding activity of vCD30-Fc and (b) recombinant vCD30 expressed from VVCD30. (a) 5 ng vCD30-Fc or mouse CD30–Fc were mixed with 150 pM mouse 125I-CD30L in the presence or absence of unlabeled CD30L and added without preincubation to protein A–coated FlashPlates. (b) 75 μl supernatants, equivalent to 1.5 × 104 cells, from cultures mock infected or infected with VVCD30 or WR were preincubated with 200 pM mouse 125I-CD30L, mixed with 5 ng vCD30-Fc, and then added to the protein A–coated FlashPlates. Bound 125I-CD30L was determined in a Packard Topcount microplate counter. The background radioactivity in the absence of recombinant protein has been subtracted. 125I-CD30L binding of duplicate samples (mean ± SD) is shown.
The activity of the recombinant vCD30 expressed from VV (VVCD30) was determined by measuring its ability to block the binding of 125I-CD30L to vCD30-Fc. Radiolabeled CD30L was incubated with supernatants from cells uninfected or infected with VVCD30 or VV WR before its addition to vCD30-Fc in the FlashPlate binding assay. Only the VVCD30 supernatant blocked the binding of 125I-CD30L to the vCD30-Fc. This confirmed the absence of CD30L binding activity in VV WR and demonstrated that the ORF cloned into the recombinant virus VVCD30 was expressed as a secreted protein that binds CD30L (Fig. 3 b).
TNF binding activity at the surface of cells infected with VV strain Lister has been reported (29). However, binding assays of 125I-CD30L to cells infected with VVCD30 failed to detect CD30L binding activity at the cell surface (not depicted).
The binding affinity of mouse and vCD30 for mouse CD30L was determined in binding assays using the protein A–coated FlashPlates with increased doses of labeled ligand. Scatchard analyses showed an affinity of 0.66 ± 0.30 nM (mean of three independent experiments, one of which is represented in Fig. 4) for vCD30. The affinity determined for mouse CD30 was 0.93 ± 0.11 nM (mean of three independent experiments, not depicted), comparable to that determined by other methods for the interaction of human CD30 with mouse or human CD30L (2.5 ± 0.3 nM; reference 3).
Figure 4.

Affinity of vCD30 for mouse CD30L. Half a nanogram of vCD30 was incubated in the protein A–coated FlashPlates with different amounts of mouse 125I-CD30L, and the radioactivity bound was determined in a Packard Topcount microplate counter. Data were converted to the Scatchard coordinate system and analyzed with the LIGAND software. Specific 125I-CD30L binding of duplicate samples (mean ± SD) is shown.
The EV-encoded CD30 Is Expressed at Late Times after Infection and Binds CD30L.
To investigate the binding activity of natural vCD30, supernatants from cells uninfected or infected with EV isolates Hampstead or Naval were tested in binding assays. The naturally produced EV protein efficiently blocked the binding of mouse 125I-CD30L to vCD30-Fc (Fig. 5 a). Moreover, vCD30 was expressed at late times after infection because supernatants prepared in the presence of AraC, an inhibitor of DNA replication that allows early protein expression but prevents synthesis of late viral proteins, did not show binding activity (Fig. 5 b). Failure to detect by RT-PCR vCD30-specific transcripts in cell extracts at early times after infection in the presence of AraC confirmed this result (not depicted). As a positive control for the RT-PCR, we detected transcripts specific for the early gene encoding the viral epidermal growth factor (not depicted). There is no consensus poxvirus late promoter sequence upstream of the vCD30 gene (9).
Figure 5.

CD30L binding activity and kinetics of production of the natural EV vCD30. (a) EV sCD30L binding activity. 200 pM mouse 125I-CD30L was preincubated with supernatant, equivalent to 1.5 × 104 cells, from BSC-I cells mock infected or infected with EV strains Hampstead or Naval, and then incubated with 5ng vCD30-Fc in a protein A–coated FlashPlate. The binding of 125I-CD30L was determined in a Packard Topcount microplate counter. (b) BSC-I cells were mock infected or infected with EV strain Hampstead in the absence or presence of AraC. 6 (Early) or 24 h after infection (Late), supernatants were harvested and an aliquot, equivalent to 5 × 104 cells, was tested for its ability to block the binding of 200 pM 125I-CD30L to 5 ng vCD30-Fc. Bound 125I-CD30L was determined as described above. The background radioactivity corresponding to the binding medium has been subtracted. Specific 125I-CD30L binding of duplicate samples (mean ± SD) is shown.
Biological Activity of vCD30.
The ability of vCD30 to block CD30L binding to cell surface receptors was investigated. First, we screened by flow cytometry human and mouse cell lines for CD30 expression using soluble recombinant mouse CD30L that cross reacts with human receptors (3). Staining profiles indicated that human monocyte K562 cells expressed high levels of CD30 (Fig. 6 a). The addition of a 25-fold excess of vCD30-Fc, but not of IgG1, efficiently blocked CD30L binding to K562 cells (Fig. 6 a). As little as a 10-fold excess of vCD30-Fc was sufficient to interfere with the binding of CD30L to its cellular receptors (not depicted).
Figure 6.
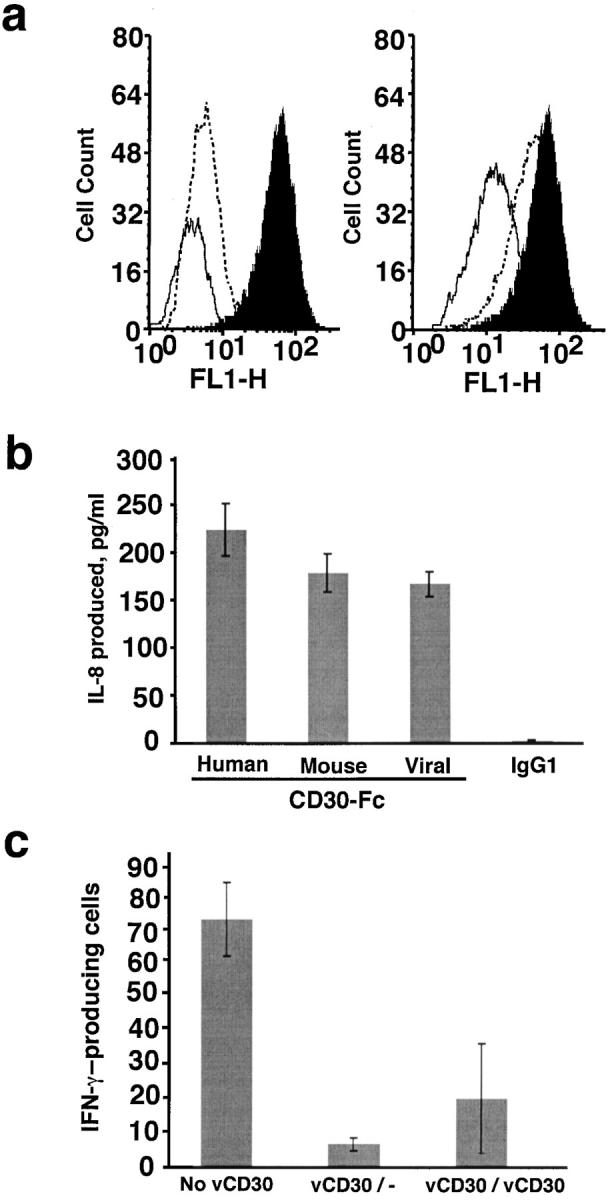
Biological activities of vCD30. (a) Blockade of the binding of soluble CD30L to CD30 expressed at the cell membrane. K562 cells were incubated with a 6× histidine-tagged CD30L and binding was detected with the mouse Ab specific for the histidine tag on the CD30L molecule followed by an FITC-goat anti–mouse Ab (filled histogram). This binding was competed by a 25-fold excess of vCD30-Fc (right, solid line), but not by the same excess of IgG1 (right, dashed line). The profile of unstained cells (left, dashed line) and of cells stained in the absence of CD30L (left, solid line) is also shown. (b) IL-8 production by neutrophils induced by human, mouse, or vCD30-Fc. Freshly isolated neutrophils were incubated for 5 h in the presence of the indicated immobilized Fc fusion proteins or IgG1. Supernatants were harvested and assayed for IL-8 production by ELISA. IL-8 secretion of duplicate samples (mean ± SD) is shown. The figure shows one representative experiment of three done with neutrophils from different donors. (c) Role of vCD30 in in vitro T cell responses. Freshly isolated splenocytes from BALB/c mice were mixed with irradiated L929 cells in the absence or presence of vCD30. 5 d later, IL-2 was added and the incubation was held for an additional 2 d. After this priming phase, the viable cells were harvested and incubated with irradiated L929 for 20 h in the presence of vCD30. Cell activation was measured by their ability to produce IFN-γ. The number of cells secreting IFNγ was assayed by ELISPOT. The result of duplicate samples (mean ± SD) is shown. vCD30/−, vCD30 added only in the priming phase; vCD30/vCD30, vCD30 added in both phases.
In addition to blocking the CD30–CD30L interaction, vCD30 had the potential to bind CD30L expressed at the cell surface and trigger intracellular signals. Neutrophils constitutively express CD30L, but not CD30, and rapidly produce IL-8 when stimulated by CD30 (4). Therefore, we analyzed the production of IL-8 by freshly isolated human neutrophils in response to immobilized human, mouse, or vCD30-Fc proteins or IgG1. As shown in Fig. 6 b, the viral homologue induced a response comparable to that of the mammalian receptors, whereas human IgG1 had no effect on IL-8 production. The latter indicated that in this experimental system, CD30–CD30L interactions leading to IL-8 production do not occur in the absence of vCD30. Therefore, the effect observed is due to signaling through CD30L after interaction with vCD30 and not to the inhibition of CD30–CD30L interactions by vCD30 acting as a soluble decoy receptor.
Finally, we investigated a possible role of vCD30, and indirectly of the CD30–CD30L pair, in the development of T cell responses in vitro. We determined the influence of vCD30 on the activation of IFN-γ–producing cells in an MLR. As shown in Fig. 6 c, vCD30 almost completely abrogated the production of IFN-γ by splenocytes from BALB/c mice exposed to L929 cells of different haplotype. The presence of vCD30 in the priming phase was sufficient to cause this effect. This result suggested an important role of the CD30–CD30L interaction for the establishment of T cell responses, particularly at early stages of activation. Moreover, the viral protein might be targeting this interaction to protect the virus against host T cell responses. The presence of IgG1 had no effect on the development of the T cell response (not depicted).
Biological Role of vCD30 In Vivo.
To address the potential immunomodulatory activity of vCD30 in vivo, the effects of vCD30-Fc treatment on inflammation in a pulmonary granuloma model were investigated. In this model, type 1 and 2 cytokine–mediated granulomas are induced in mouse lungs by bead-immobilized mycobacterial PPD or SEA, respectively. Mice were treated with vCD30-Fc or control IgG1 during Ag sensitization and elicitation (four treatments over 18 d), or only during elicitation (two treatments over 4 d). The administration of vCD30-Fc to mice caused a significant impaired type 1 cytokine–mediated inflammatory response with pulmonary granuloma size reduced >80% compared with IgG1-treated mice (Fig. 7 , a and b). This effect was observed when vCD30 was administered throughout both the sensitization and elicitation response or only during elicitation (Fig. 7 b). In contrast, type 2 cytokine–mediated pulmonary inflammation was not modified by vCD30 administration, as vCD30-Fc–treated mice had comparable or marginally larger granulomas than obtained in control animals (Fig. 7, a and b). The composition of the cellular infiltrate surrounding the type 1 or 2 granulomas was not altered by vCD30 treatment (not depicted).
Figure 7.

Role of vCD30 during in vivo type 1 or 2 cytokine–mediated pulmonary inflammation. BALB/c mice were type 1 or 2 sensitized and pulmonary granulomas were elicited as described in Materials and Methods. Mice were treated with vCD30-Fc or control IgG1 throughout the sensitization (S) and elicitation (E; four treatments) or only during elicitation (two treatments). (a) Photomicrographs of hematoxylin and eosin–stained sections of representative type 1 (lymphocytes-, monocytes-, and neutrophils-rich) or 2 (eosinophil-dominated) pulmonary granulomas surrounding Ag-coated beads in mice treated with vCD30-Fc or IgG1. (b) Quantification of the granuloma volume (mean ± SD) from four or five mice per group. Significantly smaller type 1 granulomas (*, P < 0.01; Student's t test) were found in mice treated two or four times with vCD30 compared with IgG1-treated mice. (c) vCD30-Fc treatment reduced lung tissue IFN-γ and IL-12 in type 1–sensitized mice. The levels of IFN-γ, IL-12, IL-4, and IL-5 in lung tissue homogenates were tested in ELISA and are expressed as nanogram cytokine per milligram lung protein. (d) Intracellular detection of alterations in the frequencies of IFN-γ– and IL-4–producing CD4+ and CD8+ T cells in type 1– or 2–sensitized mice. Data in quadrants represent the percentage of positively stained cells. All data are representative from two separate experiments (n = four or five mice per group). Cytokine and FACS® data presented are from mice treated four times with vCD30-Fc. Comparable data was obtained in two separate experiments when mice were only treated two times.
The effect of vCD30 on pulmonary cytokine responses was measured in lung tissue homogenates. Levels of type 1 or 2 cytokines in the lungs of control IgG1–treated mice were biased in the respective type 1– (elevated IFN-γ and IL-12) or 2– (IL-4 and IL-5) mediated pulmonary inflammatory responses (Fig. 7 c). vCD30 treatment of mice with type 1 granulomas caused a substantial reduction in lung IFN-γ and albeit to a lesser degree, IL-12 levels (Fig. 7 c) with no alterations in IL-4 and IL-5 levels. Cytokine levels in mice with type 2 granulomas were not altered by vCD30, although IFN-γ and IL-12 levels were lower in vCD30-treated mice (Fig. 7 c). Changes in pulmonary type 1 cytokine levels occurred when vCD30 was administered throughout the experiment (Fig. 7 c) or only during elicitation (not depicted). These results demonstrate that the formation of type 1, but not type 2, cytokine–mediated pulmonary inflammation was impaired by vCD30 in vivo. To address if the reduced IFN-γ production in vCD30-treated mice was associated with alterations in the production of type 1 (IFN-γ) or type 2 (IL-4) cytokines by T cells, we performed intracellular cytokine staining on CD4+ or CD8+ T cells from spleens. Cells from the spleens of type 1 granuloma–sensitized mice treated with vCD30 had two- to threefold lower frequencies of both Th1 and Tc1 cells compared with control IgG1–treated mice (Fig. 7 d). Similarly, vCD30 treatment also reduced the numbers of IFN-γ–secreting T cells in type 2 granuloma–sensitized mice (Fig. 7 d). These results support a preferential effect of vCD30 on type 1 T cells.
In addition, the role of vCD30 in the development of Th1-like responses was controlled with a truncated version of another EV vTNFR. A recombinant inactive form of CrmD consisting of the first two CRDs (CrmD-CRD1,2-Fc) and unable to protect cells from TNF–induced necrosis was produced in the baculovirus system and purified according to the same protocols used for vCD30-Fc (unpublished data). Mice exposed to mycobacterium or schistosoma antigens as described above were treated with vCD30-Fc, CrmD-CRD1,2-Fc, or PBS, and the levels of cytokines in the lung were determined. Table I shows that vCD30-Fc, but not CrmD-CRD1,2-Fc, specifically down-regulated Th1 cytokine production, confirming the previous result and demonstrating that the role attributed to vCD30-Fc was specific.
Table I.
Modulation of Cytokine Production by vCD30 in a Mouse Model of Inflammation
| Th1 response
|
Th2 response
|
|||||
|---|---|---|---|---|---|---|
| vCD30-Fc | CrmD-CRD1,2-Fc | PBS | vCD30-Fc | CrmD-CRD1,2-Fc | PBS | |
| IFN-γ | 0.33 ± 0.2 | 3.31 ± 0.57 | 3.64 ± 0.47 | 0.29 ± 0.16 | 0.94 ± 0.19 | 0.81 ± 0.26 |
| IL-12 | 0.82 ± 0.28 | 3.35 ± 0.6 | 3.20 ± 0.73 | 0.17 ± 0.08 | 0.39 ± 0.15 | 0.43 ± 0.16 |
| IL-4 | 0.34 ± 0.09 | 0.05 ± 0.04 | 0.09 ± 0.05 | 3.12 ± 0.49 | 3.81 ± 0.44 | 2.93 ± 0.63 |
| IL-5 | 0.88 ± 0.2 | 0.38 ± 0.12 | 0.32 ± 0.21 | 5.93 ± 0.89 | 5.72 ± 0.67 | 5.75 ± 0.98 |
Cytokine responses in the lungs of mice exposed to mycobacterium (Th1 response) or schistosoma antigens (Th2 response) and treated on days 14 and 16 with 10 μg vCD30-Fc, CrmD-CRD1,2-Fc, or PBS. Levels of IFN-γ, IL-12, IL-4, and IL-5 in lung tissue homogenates as tested by ELISA and expressed as nanogram cytokine per milligram lung protein (mean ± SD) from five mice per group.
Discussion
We report the identification and characterization of a homologue of mammalian CD30, designated vCD30 and encoded by EV. vCD30 is a new member of the TNFR superfamily that binds CD30L. Comparative studies in 10 other EV isolates showed that all of them encode an active vCD30 (unpublished data). The complete genome sequence of EV has been recently determined (available at http://www.sanger.ac.uk) and shows that the vCD30 gene described here is the only CD30 homologue encoded by the virus (unpublished data). Thus it is most likely that the gene we have characterized is responsible for the activity expressed in EV infections. The presence of an active vCD30 seems restricted to a few poxviruses. An intact vCD30 gene has been identified in EV isolates and CPV GRI-90 (30), whereas it is not found in VV strain WR (this paper) or in 18 other poxviruses including 3 strains of VaV, whose complete genome has been sequenced and is available in public databases. Maybe different immunomodulatory genes compensate for the lack of vCD30 in other poxviruses.
Amino acid sequence similarity of vCD30 with the human and mouse counterparts is confined to a region of the extracellular domain because the viral protein lacks the transmembrane or cytoplasmic domains. The extracellular domain of human CD30 consists of a duplicate structure of three CRDs, whereas mouse CD30 only has the first cluster and the viral counterpart have the second and third CRDs. This demonstrates direct involvement of the second and third CRDs in ligand binding. Moreover, the conservation of some motifs in the three molecules may point at specific residues involved in CD30L binding. Both mouse and human CD30 can be proteolytically cleaved by a zinc metalloprotease to produce a soluble form (sCD30) that is larger than vCD30. The viral protein appears to retain the minimal structure necessary for efficient CD30L binding activity. The higher similarity of vCD30 to mouse CD30 is consistent with mice being the natural host for EV.
vCD30 is the fifth member of the TNFR superfamily identified in poxviruses and the only one that does not bind TNF, which suggests a distinct role during viral infection. The sequence similarity between vCD30 and the other vTNFRs (CrmB, CrmC, CrmD, and CrmE) is confined to the CRDs. CrmB and CrmD show an extended COOH-terminal region with no sequence similarities in databases, which is not required for TNF binding (unpublished data). Interestingly, the ORF downstream vCD30 in the EV strain Naval genome has similarity to the COOH-terminal extension found in some vTNFRs and no intergenic region is found (unpublished data). We propose that the ancestral vCD30 gene may have contained the COOH-terminal extension that has been lost in the gene presently found in EV.
vCD30 is a 12-kD protein secreted at late times during EV infection that binds CD30L with high affinity and inhibits its binding to cell surface CD30. This function of vCD30 is similar to that of the poxvirus soluble cytokine decoy receptors, including vTNFRs. However, vCD30 has an additional unique property. It induces reverse signaling in cells expressing CD30L. Therefore, vCD30 may not be classified as a genuine decoy receptor and we propose that the mechanism of action of vCD30 is totally different from that adopted by vTNFRs. Because secreted CD30L has not been identified, vCD30 is acting at the cell surface, both mimicking signal transduction mediated by CD30 and inhibiting the effect of CD30L in cells expressing CD30. We also demonstrate that vCD30 is a soluble molecule with no membrane-associated binding activity, as it has been described for TNF binding activity in VV Lister (29).
The expression of CD30 is associated with the activated status of T cells. In vitro, it has been mainly associated with a Th2/Th0 phenotype (38), although in vivo studies suggest that the relationship between CD30+ T cells and Th1 or Th2 profiles is very complex. Recent studies support a novel regulatory mechanism for CD30 in Th1 polarized responses such as rheumatoid arthritis (39). We show that the blockade of the binding of CD30L to CD30 by the viral protein and/or the activation of CD30L by vCD30 is responsible for the inhibition of IFN-γ production by activated splenocytes in MLR. The potent in vivo inhibition of pulmonary granuloma formation by vCD30 in type 1, but not 2, cytokine–sensitized mice supports a preferential role for CD30–CD30L in type 1 cytokine–mediated responses. Consistent with our in vitro data, a potential direct affect of vCD30 on type 1 T cells was shown by the diminished frequencies of IFN-γ–producing T cells detected in vCD30-treated mice. These data support increasing evidence for a key role of CD30 in type 1 responses in vivo. Interestingly, vCD30 treatment was effective in suppressing type 1 cytokine–mediated inflammation when administered either throughout the Ag sensitization and elicitation phase or only during elicitation. In the context of viral infection, this is probably compromising a protective antiviral immune response, which is known to be Th1-like T cell mediated.
CD30 is also known to be expressed in activated B cells (2). CD30L was found to be a potent mediator of mouse B cell growth and differentiation in vitro, although different results were found with human B cells (2). Therefore, vCD30 may also interfere with B cell responses to the virus. Finally, vCD30 may also modulate signaling between B and T cells, a process in which CD30 has been implicated.
Viral mechanisms of immune modulation may point at those host molecules that contribute to antiviral responses. The remarkable finding of a virus-encoded CD30 homologue uncovers a potential role of the CD30–CD30L system in antiviral immune responses. Increased levels of sCD30 have been reported in pathological conditions and after infection with HIV, EBV, hepatitis virus B and C, measles virus, and varicella-zoster virus, but the biological significance is not known. Viral proteins have been optimized during virus–host coevolution to become potent inhibitors of host immune responses. Maybe the biological properties of vCD30 differ from those of the host sCD30.
Characterization of viral immunomodulatory proteins may also shed light into the function of the host counterparts. By using vCD30, we demonstrate a role of CD30–CD30L interactions in the generation of Th1 inflammatory responses. vCD30 may also provide alternative strategies to effectively block the activity of CD30 in vivo, which might be applied to modulate an overreactive immune response in a number of human disease conditions (8). vCD30 is another example of proteins from virulent viruses that have promise as therapeutic reagents.
Acknowledgments
We thank Sebastian Mas and Begonia Aguado for the mass spectrometry analysis of vCD30-Fc.
This work was funded by the Wellcome Trust (grants 051087/Z/97/Z and 057381). M. Saraiva is funded by Fundacao para a Ciencia e Tecnologia-Praxis XXI (grant BD-18081/98). P.G. Fallon was supported by a Wellcome Trust Career Development Fellowship and A. Alcami is a Wellcome Trust Senior Research Fellow.
P. Smith's and P.G. Fallon's present address is Department of Biochemistry, Trinity College, Dublin 2, Ireland.
Footnotes
Abbreviations used in this paper: AraC, cytosine arabinoside; CPV, cowpox virus; CRD, cysteine-rich domain; Crm, cytokine response modifier; EV, ectromelia virus; ORF, open reading frame; PPD, protein-purified derivative; sCD30, soluble CD30; SEA, soluble egg Ags; VaV, variola virus; vCD30, viral CD30; vTNFR, viral TNFR; VV, vaccinia virus; WR, Western Reserve.
References
- 1.Schwab, U., H. Stein, J. Gerdes, H. Lemke, H. Kirchner, M. Schaadt, and V. Diehl. 1982. Production of a monoclonal antibody specific for Hodgkin and Sternberg Reed cells of Hodgkin's disease and a subset of normal lymphoid cells. Nature. 299:65–67. [DOI] [PubMed] [Google Scholar]
- 2.Horie, R., and T. Watanabe. 1998. CD30: expression and function in health and disease. Semin. Immunol. 10:457–470. [DOI] [PubMed] [Google Scholar]
- 3.Smith, C.A., H.J. Gruss, T. Davis, D. Anderson, T. Farrah, E. Baker, G.R. Sutherland, C.I. Brannan, N.G. Copeland, N.A. Jenkins, et al. 1993. CD30 antigen, a marker for Hodgkin's lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell. 73:1349–1360. [DOI] [PubMed] [Google Scholar]
- 4.Wiley, S.R., R.G. Goodwin, and C.A. Smith. 1996. Reverse signaling via CD30 ligand. J. Immunol. 157:3635–3639. [PubMed] [Google Scholar]
- 5.Amakawa, R., A. Hakem, T.M. Kundig, T. Matsuyama, J.J. Simard, E. Timms, A. Wakeham, H.W. Mittruecker, H. Griesser, H. Takimoto, et al. 1996. Impaired negative selection of T cells in Hodgkin's disease antigen CD30-deficient mice. Cell. 84:551–562. [DOI] [PubMed] [Google Scholar]
- 6.Chiarle, R., A. Podda, G. Prolla, E.R. Podack, G.J. Thorbeecke, and G. Inggirami. 1999. CD30 overexpression enhances negative selection in the thymus and mediates programmed cell death via Bcl-2-sensitive pathway. J. Immunol. 163:194–205. [PubMed] [Google Scholar]
- 7.Muta, H., L.H. Boise, L. Fang, and E.R. Podack. 2000. CD30 signals integrate expression of cytotoxic effector molecules, lymphocyte trafficking signals, and signals for proliferation and apoptosis. J. Immunol. 165:5105–5111. [DOI] [PubMed] [Google Scholar]
- 8.Gruss, H.J., A. Pinto, J. Duyster, S. Poppema, and F. Herrmann. 1997. Hodgkin's disease: a tumor with disturbed immunological pathways. Immunol. Today. 18:156–163. [DOI] [PubMed] [Google Scholar]
- 9.Moss, B. 1996. Poxviridae: the virus and their replication. In Virology. B.N. Fields, R.M. Chanock, D.M. Knipe, and P.M. Howley, editors. Lippincott-Raven Publishers, Philadelphia. 2637–2671.
- 10.Fenner, F., and R.M. Buller. 1997. Mousepox. In Viral Pathogenesis. N. Nathanson, F. Gonzalez-Scarano, R. Ahmed, H.L. Robinson, D.E. Griffin, K.V. Holmes, and F.A. Murphy, editors. Lippincott-Raven Publishers, Philadelphia. 535–553.
- 11.Alcami, A., and U.H. Koszinowski. 2000. Viral mechanisms of immune evasion. Immunol. Today. 21:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McFadden, G., and P.M. Murphy. 2000. Host-related immunomodulators encoded by poxviruses and herpesviruses. Curr. Opin. Microbiol. 3:371–378. [DOI] [PubMed] [Google Scholar]
- 13.Tortorella, D., B.E. Gewurz, M.H. Furman, D.J. Schust, and H.L. Ploegh. 2000. Viral subversion of the immune system. Annu. Rev. Immunol. 18:861–926. [DOI] [PubMed] [Google Scholar]
- 14.Loparev, V.N., J.M. Parsons, J.C. Knight, J.F. Panus, C.A. Ray, R.M. Buller, D.J. Pickup, and J.J. Esposito. 1998. A third distinct tumor necrosis factor receptor of orthopoxviruses. Proc. Natl. Acad. Sci. USA. 95:3786–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith, V.P., and A. Alcami. 2000. Expression of secreted cytokine and chemokine inhibitors by ectromelia virus. J. Virol. 74:8460–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mossman, K., C. Upton, R.M. Buller, and G. McFadden. 1995. Species specificity of ectromelia virus and vaccinia virus interferon-γ binding proteins. Virology. 208:762–769. [DOI] [PubMed] [Google Scholar]
- 17.Smith, V.P., and A. Alcami. 2002. Inhibition of interferons by ectromelia virus. J. Virol. 76:1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colamonici, O.R., P. Domanski, S.M. Sweitzer, A. Larner, and R.M. Buller. 1995. Vaccinia virus B18R gene encodes a type I interferon-binding protein that blocks interferon α transmembrane signaling. J. Biol. Chem. 270:15974–15978. [DOI] [PubMed] [Google Scholar]
- 19.Smith, V.P., N.A. Bryant, and A. Alcami. 2000. Ectromelia, vaccinia and cowpox viruses encode secreted interleukin-18-binding proteins. J. Gen. Virol. 81:1223–1230. [DOI] [PubMed] [Google Scholar]
- 20.Born, T.L., L.A. Morrison, D.J. Esteban, T. VandenBos, L.G. Thebeau, N. Chen, M.K. Spriggs, J.E. Sims, and R.M. Buller. 2000. A poxvirus protein that binds to and inactivates IL-18, and inhibits NK cell response. J. Immunol. 164:3246–3254. [DOI] [PubMed] [Google Scholar]
- 21.Graham, K.A., A.S. Lalani, J.L. Macen, T.L. Ness, M. Barry, L.Y. Liu, A. Lucas, I. Clark-Lewis, R.W. Moyer, and G. McFadden. 1997. The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology. 229:12–24. [DOI] [PubMed] [Google Scholar]
- 22.Turner, S.J., J. Silke, B. Kenshole, and J. Ruby. 2000. Characterization of the ectromelia virus serpin, SPI-2. J. Gen. Virol. 81:2425–2430. [DOI] [PubMed] [Google Scholar]
- 23.Brick, D.J., R.D. Burke, A.A. Minkley, and C. Upton. 2000. Ectromelia virus virulence factor p28 acts upstream caspase-3 in response to UV light-induced apoptosis. J. Gen. Virol. 81:1087–1097. [DOI] [PubMed] [Google Scholar]
- 24.Hu, F.Q., C.A. Smith, and D.J. Pickup. 1994. Cowpox virus contains two copies of an early gene encoding a soluble secreted form of the type II TNF receptor. Virology. 204:343–356. [DOI] [PubMed] [Google Scholar]
- 25.Smith, C.A., F.Q. Hu, T.D. Smith, C.L. Richards, P. Smolak, R.G. Goodwin, and D.J. Pickup. 1996. Cowpox virus genome encodes a second soluble homologue of cellular TNF receptors, distinct from CrmB, that binds TNF but not LT α. Virology. 223:132–147. [DOI] [PubMed] [Google Scholar]
- 26.Saraiva, M., and A. Alcami. 2001. CrmE, a novel soluble tumor necrosis factor receptor encoded by poxviruses. J. Virol. 75:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fallon, P.G., P. Smith, and D.W. Dunne. 1998. Type 1 and type 2 cytokine-producing mouse CD4+ and CD8+ T cells in acute Schistosoma mansoni infection. Eur. J. Immunol. 28:1408–1416. [DOI] [PubMed] [Google Scholar]
- 28.Esposito, J., R. Condit, and J. Obijeski. 1981. The preparation of orthopoxvirus DNA. J. Virol. Methods. 2:175–179. [DOI] [PubMed] [Google Scholar]
- 29.Alcami, A., A. Khanna, N.L. Paul, and G.L. Smith. 1999. Vaccinia virus strains Lister, USSR and Evans express soluble and cell-surface tumour necrosis factor receptors. J. Gen. Virol. 80:949–959. [DOI] [PubMed] [Google Scholar]
- 30.Shchelkunov, S.N., P.F. Safronov, A.V. Totmenin, N.A. Petrov, O.I. Ryazankina, V.V. Gutorov, and G.J. Kotwal. 1998. The genomic sequence analysis of the left and right species-specific terminal region of a cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology. 243:432–460. [DOI] [PubMed] [Google Scholar]
- 31.Genetics Computer Group. 1994. Program Manual for the Wisconsin Package.
- 32.Davison, A.J., and B. Moss. 1990. New vaccinia virus recombination plasmids incorporating a synthetic late promoter for high level expression of foreign proteins. Nucleic Acids Res. 18:4285–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alcami, A., J.A. Symons, P.D. Collins, T.J. Williams, and G.L. Smith. 1998. Blockade of chemokine activity by a soluble chemokine binding protein from vaccinia virus. J. Immunol. 160:624–633. [PubMed] [Google Scholar]
- 34.Markwell, M.A., and C.F. Fox. 1978. Surface-specific iodination of membrane proteins of viruses and eucaryotic cells using 1,3,4,5-tetrachloro-3α,6α-diphenylglycouril. Biochemistry. 17:4807–4817. [DOI] [PubMed] [Google Scholar]
- 35.Munson, P.J., and D. Rodbard. 1980. LIGAND: a versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 107:220–239. [DOI] [PubMed] [Google Scholar]
- 36.Chensue, S.W., K. Warmington, J.H. Ruth, N. Lukacs, and S.L. Kunkel. 1997. Mycobacterial and schistosomal antigen-elicited granuloma formation in IFN-γ and IL-4 knockout mice: analysis of local and regional cytokine and chemokine networks. J. Immunol. 159:3565–3573. (See published erratum 162:3106.) [PubMed] [Google Scholar]
- 37.Fallon, P.G., and D.W. Dunne. 1999. Tolerization of mice to Schistosoma mansoni egg antigens causes elevated type 1 and diminished type 2 cytokine responses and increased mortality in acute infection. J. Immunol. 162:4122–4132. [PubMed] [Google Scholar]
- 38.Del Prete, G., M. De Carli, F. Almerigogna, C.K. Daniel, M.M. D'Elios, G. Zancuoghi, F. Vinante, G. Pizzolo, and S. Romagnani. 1995. Preferential expression of CD30 by human CD4+ T cells producing Th2-type cytokines. FASEB J. 9:81–86. [PubMed] [Google Scholar]
- 39.Gerli, R., C. Lunardi, F. Vinante, O. Bistoni, G. Pizzolo, and C. Pitzalis. 2001. Role of CD30+ T cells in rheumatoid arthritis: a counter-regulatory paradigm for Th1-driven diseases. Trends Immunol. 22:72–77. [DOI] [PubMed] [Google Scholar]