

Abstract
The failure of CD25+ regulatory T cells (Tregs) to proliferate after T cell receptor (TCR) stimulation in vitro has lead to their classification as naturally anergic. Here we use Tregs expressing a transgenic TCR to show that despite anergy in vitro, Tregs proliferate in response to immunization in vivo. Tregs also proliferate and accumulate locally in response to transgenically expressed tissue antigen whereas their CD25− counterparts are depleted at such sites. Collectively, these data suggest that the anergic state that characterizes CD25+ Tregs in vitro may not accurately reflect their responsiveness in vivo. These observations support a model in which Treg population dynamics are shaped by the local antigenic environment.
Keywords: CD4+ T lymphocytes, peripheral tolerance, autoantigen, regulatory T cells, autoimmunity
Introduction
The critical ability of regulatory T cells (Tregs)*to control diseases, particularly autoimmunity, has sparked much interest in how such cells develop and function. Thymic-derived CD4+ CD25+ T cells constitute a major population of Tregs that are able to inhibit T cell responses both in vitro (1–3) and in vivo (4, 5). Tregs are widely believed to recognize self-antigens. In fact, the number of CD4+ CD25+ cells that is selected in the thymus has been shown to be proportional to the diversity of self-peptides presented in the context of MHC class II molecules on thymic epithelium (6). Elegant studies using PVG rats have suggested that Tregs emerging from the thymus require access to their specific autoantigen in the periphery to survive as a functional population, and this may reflect a requirement for self-antigen–driven expansion (7). On face value, such a scenario is hard to reconcile with the profound anergy exhibited by Tregs in response to TCR engagement in vitro (1, 2, 8).
To study the response of Tregs to antigen in vivo, we have taken advantage of a murine model that allows the production of a large number of CD25+ T cells with regulatory function that bear a transgenic TCR. Using the clonotypic antibody to identify these cells after adoptive transfer to nontransgenic recipients, we reveal a key difference between the responsiveness of Tregs to encounter with antigen in vivo versus in vitro. The cells are anergic to antigen stimulation in vitro, but undergo proliferation if presented with immunizing antigen in an in vivo context. Furthermore, after adoptive transfer of TCR transgenic Tregs to mice transgenically expressing the relevant antigen (OVA) as a self-protein in a peripheral tissue, proliferation of a fraction of the Tregs can be detected in the lymphoid tissue draining the site of antigen expression. Although Tregs have been shown to be capable of homeostatic proliferation in lymphopenic hosts (9, 10), they are believed to be refractory to antigen-driven proliferation based on in vitro studies. Our results indicate that the constraints on antigen-driven Treg proliferation documented in vitro are not apparent in vivo and provide new insight into the biology of this critical T cell subset.
Materials and Methods
Mice.
DO11.10 TCR transgenic mice and BALB/c mice were purchased from The Jackson Laboratory. RAG2−/− mice were purchased from Taconic Laboratories. Rat insulin promoter (RIP)-mOVA mice on a BALB/c background expressing a membrane-bound form of OVA under the control of the RIP (from line 296-1B) were provided by W. Heath (The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia). Mice were housed in the University of California San Francisco animal facility and used according to the guidelines of the Institutional Committee on Animal Research. Mice were genotyped using PCR and flow cytometry and were between 6 and 12 wk of age at the start of each experiment.
T Cell Transfers.
Combined LN (axillary, inguinal, brachial, popliteal, and mesenteric) cells from DO11 × RIP-mOVA double transgenic mice (on a RAG−/− background where indicated) were stained with the clonotypic antibody KJ-126-APC (Caltag) and CD25-PE and purified using high speed cell sorting (MoFlo®; DakoCytomation). 0.5–1 × 106 cells were transferred into recipient mice by tail vein injection. Where indicated, cells were incubated before transfer with 1 μM carboxy-fluorescein diacetate succinimidyl ester (CFSE; Molecular Probes) for 10 min at room temperature followed by two washes with RPMI supplemented as described below. Absolute cell numbers were calculated based on percentage of CD4+ KJ+ cells and total cell counts. Fold expansion is calculated using the average values from unimmunized mice within each experiment.
Immunization.
OVA protein (Sigma-Aldrich) was prepared emulsified in IFA (Difco) and 200 μg was administered s.c. in the flank where indicated. Axillary and inguinal LNs were taken as draining LNs and cervical LNs were taken as nondraining LNs.
Flow Cytometry.
Antibodies used for staining were KJ-126-biotin/APC, CD25-FITC/PE (PC61), CD62L-FITC (MEL-14), CD69-PE (H1.2F3), cytotoxic T lymphocyte–associated antigen 4 (CTLA-4)-PE (UC10-4F10-11), CD4-FITC/PERCP (L3T4), TCR-Vα2-PE (B20.1), IL-2-PE (JES6-544), OX40-biotin (OX-86), streptavidin-PE/PERCP, IL-7Rα-PE (SB/14), and CD40 ligand (CD40L)-PE (MR1). All antibodies were purchased from BD Biosciences unless otherwise indicated. In some experiments, labeling with CFSE was used to identify adoptively transferred KJ+ CD25+ or KJ+ CD25− cells from cotransferred responder DO11 cells. For intracellular cytokine staining, cells were restimulated for 4 h with 1 μg/ml OVA peptide in the presence of 10 μg/ml brefeldin A for the final 3 h. Cells were fixed for 10 min with 4% paraformaldehyde after surface staining and then permeabilized with 0.5% saponin (Sigma-Aldrich) and stained with antibodies against intracellular markers for 15 min at room temperature. Stained cells were washed once with 0.5% saponin and once with 1% FBS in PBS before analysis. Gates were set using isotype-matched control antibodies.
In Vitro Proliferation.
2.5 × 104 T cells purified by MoFlo® sorting were cultured with 1.25 × 105 spleen cells from nontransgenic mice in 0.2 ml RPMI 1640 supplemented with 1 mM l-glutamine, penicillin, streptomycin, nonessential amino acids, sodium pyruvate, Hepes (all from Life Technologies), 5 × 10−5 M 2-ME, and 10% FBS (Sigma-Aldrich) containing the indicated concentration of OVA323–339 peptide. For restimulation of draining LN cells, populations that contained an equivalent percentage of KJ+ cells were cultured at a concentration of 5 × 106 total cells per ml in RPMI 1640 supplemented as described above. Proliferation assays were pulsed with 1 μCi [3H]thymidine (New England Nuclear) for the final 7–8 h of the 72-h period and incorporated radioactivity was measured in a Betaplate scintillation counter (LBK Pharmacia).
Results
Clonotype+ CD25+ Cells in DO11 × RIP-mOVA Mice.
We observed that in double transgenic mice expressing a membrane-bound form of OVA under the control of the RIP (RIP-mOVA) and also expressing the DO11 TCR, there was a large population of clonotype+ cells that expressed CD25 (Fig. 1, A and B) . The KJ-126+ CD25+ cells (from hereon referred to as KJ+ CD25+) were present in the secondary lymphoid tissues and enriched in the pancreatic LN that drains the site of peripheral OVA expression (Fig. 1 A). Although CD25 is transiently up-regulated during T cell activation, the surface expression profile of the KJ+ CD25+ cells did not indicate activation because they were almost exclusively CD62Lhi and did not exhibit elevated levels of CD69 despite expressing high levels of intracellular CTLA-4 when compared with KJ+ CD25− cells from the same mouse (Fig. 1 C). Instead, the phenotype of the KJ+ CD25+ cells suggested that they were Tregs. The KJ+ CD25+ cells expressed higher OX40 and lower IL-7 receptor α than the KJ+ CD25− cells (Fig. 1 C). This is consistent with microarray analysis of nontransgenic CD4+ CD25+ cells that showed increased mRNA for OX40 (11) and decreased mRNA for IL-7 receptor α (9) compared with CD4+ CD25− cells.
Figure 1.

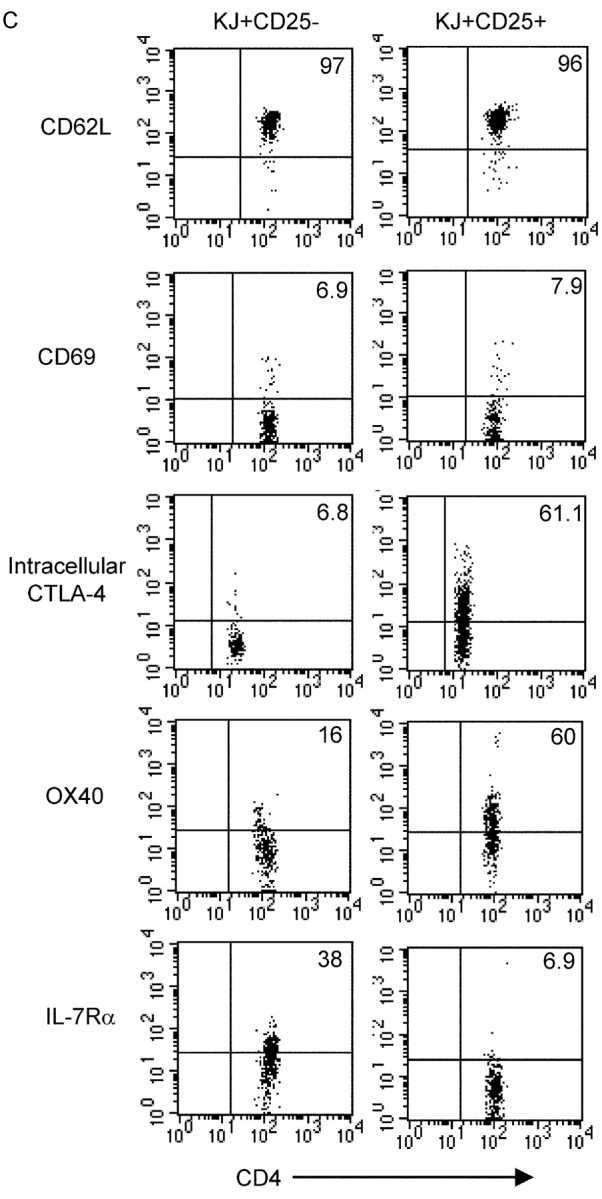
Abundant clonotype+ CD25+ cells in DO11 × RIP-mOVA double transgenic mice. (A) FACS® profiles of DO11 single positive versus DO11 × RIP-mOVA double transgenic littermates at 7 wk of age. Cells were isolated from the indicated lymphoid tissues and stained with CD4-PERCP, CD25-PE, and KJ-126-APC. Plots are gated on CD4+ KJ+ cells (except for thymus plots, which are gated on CD4+ CD8− KJ+ cells) and show the percentage of CD25+ cells. (B) Absolute number of CD4+ KJ+ CD25+ cells in the lymphoid tissues of DO11 single positive versus DO11 × RIP-mOVA double transgenic littermates. Data show mean and standard deviation from three mice of each genotype aged 6–7 wk. (C) Peripheral LN cells from DO11 × RIP-mOVA double transgenic mice were stained with various combinations of KJ-126-APC, CD62L-FITC, CD69PE, CD4-PERCP, OX40- biotin, streptavidin-PE, IL-7Rα-PE, and CD25-FITC/PE. For intracellular staining cells were surface stained and then permeabilized and stained with CTLA-4-PE. Expression profiles of each marker are shown for gated CD4+ KJ+ CD25− versus CD4+ KJ+ CD25+ cells.
We hypothesized that the KJ+ CD25+ cells were Tregs that had arisen as a result of the intrathymic activity of the insulin promoter (12). Indeed, functionally significant levels of OVA are expressed in the thymus of RIP-mOVA mice because the thymic deletion of OT-1 cells in double transgenic (OT-1 × RIP-mOVA) mice (13) is abolished in thymectomized RIP-mOVA mice grafted with a nontransgenic thymus and OT-1 bone marrow (14). Consistent with a thymic origin, KJ+ CD25+ cells were present in the CD4+ CD8− fraction of thymocytes from DO11 × RIP-mOVA mice (Fig. 1, A and B). The total number of DO11 T cells in the thymus in DO11 × RIP-mOVA double transgenic mice was reduced compared with single positive DO11 mice (7.5 ± 2.4 × 106 compared with 21.5 ± 5.9 × 106, respectively), suggesting that thymic OVA expression also induced negative selection of a proportion of the clonotype+ cells.
The KJ+ CD25+ cells were not enriched for cells expressing endogenous TCRα chains because the proportion of cells expressing Vα2 (an endogenous TCRα chain) was slightly lower in the KJ+ CD25+ population than in the KJ+ CD25− population (Fig. 2 A). Levels of the transgenic TCR were equivalent between KJ+ CD25+ cells and KJ+ CD25− cells as assessed by KJ-126 staining (Fig. 2 A). The lack of increased endogenous TCRα chain usage argued against a requirement for the rearrangement of additional TCRs for the development of the KJ+ CD25+ cells. Consistent with this, the development of KJ+ CD25+ cells was intact in DO11 × RIP-mOVA mice bred to a RAG-deficient background (Fig. 2 B). In line with previous reports (15), the small number of CD25+ cells that develop in conventional DO11 mice require endogenous TCRα chains and do not arise in DO11 RAG−/− mice (Fig. 2 B). Thymic deletion of DO11 cells was also evident in the DO11 × RIP-mOVA RAG−/− mice (8.7 ± 5.2 × 106 KJ+ CD4+ CD8− cells in the thymus of DO11 × RIP-mOVA RAG−/− mice compared with 20.7 ± 6.7 × 106 in DO11 RAG−/− mice). Therefore, both deletion and Treg differentiation of DO11 cells were evident in antigen-bearing mice in a manner that was independent of endogenous TCRα chain usage.
Figure 2.

The generation of KJ+ CD25+ cells does not require endogenous TCRα chains. (A) Frequency of TCR Vα2 usage and expression levels of the transgenic TCR on peripheral LN cells from DO11 × RIP-mOVA mice. Histogram labels depict the median fluorescence channel. (B) Proportion of CD4+ KJ+ cells expressing CD25 in DO11 versus DO11 × RIP-mOVA mice on a RAG−/− background. Cells were isolated and stained as described in Fig. 1.
KJ+ CD25+ Cells Suppress T Cell Responses In Vitro and In Vivo.
To confirm that the KJ+ CD25+ cells were Tregs we analyzed their suppressive function in vitro. For these experiments, we sorted highly pure populations of KJ+ CD25− and KJ+ CD25+ cells from pooled peripheral LNs of DO11 × RIP-mOVA double transgenic mice. KJ+ CD25+ cells did not proliferate in response to OVA323–339 peptide and APCs, whereas KJ+ CD25− cells purified from the same mice showed a robust proliferative response (Fig. 3 A). Furthermore, in cocultures, KJ+ CD25+ cells potently suppressed the proliferation of KJ+ CD25− cells (Fig. 3 A). KJ+ CD25+ cells isolated from DO11 × RIP-mOVA mice appeared to suppress with equivalent potency to the much less abundant KJ+ CD25+ cells isolated from conventional DO11 mice (suppression observed at 0.1 μg/ml OVA using a 1:1 ratio of suppressors/responders was 96 versus 93%, respectively, and using a ratio of 0.5:1 was 69 versus 74%, respectively). KJ+ CD25+ cells from conventional DO11 mice can presumably still be induced to suppress via their transgenic TCR despite undergoing thymic selection on the basis of an alternative TCR.
Figure 3.

KJ+ CD25+ cells are suppressive in vitro and in vivo. (A) KJ+ CD25− and KJ+ CD25+ populations were purified from pooled peripheral LNs of DO11 × RIP-mOVA double transgenic mice by high speed cell sorting and stimulated in 96-well plates, either alone or in combination at a 1:1 ratio, with BALB/c splenocytes and the indicated concentration of OVA323–339 peptide. Data show one experiment that is representative of three. (B) 2 × 106 DO11 T cells (from a single positive DO11 mouse) were injected into BALB/c recipients either alone or in combination with 106 CFSE-labeled KJ+ CD25+ cells or 106 CFSE-labeled KJ+ CD25− cells. Recipient mice were immunized s.c. with 200 μg OVA/IFA where indicated. Draining LNs were isolated at day 3 and the absolute number of responder DO11 cells (CFSE−) is shown. Data show one experiment (two mice per group) and are representative of two separate experiments.
One advantage of using a TCR transgenic system to study Treg function is the ability to perform in vivo experiments in which “responder” and “suppressor” T cells of the same specificity can be adoptively transferred into nontransgenic hosts and tracked using the clonotypic antibody KJ-126. We were therefore able to assess whether KJ+ CD25+ cells were capable of suppressing the response of naive DO11 cells to OVA emulsified in IFA. To distinguish between the two populations, we used CFSE labeling to mark the suppressor cells (KJ+ CD25+ cells or control transfers of KJ+ CD25− cells). Fig. 3 B shows that the accumulation of responder (CFSE−) DO11 cells in response to immunization was decreased if CFSE-labeled KJ+ CD25+ cells were cotransferred before immunization. Control transfers of CFSE-labeled KJ+ CD25− cells did not decrease the accumulation of DO11 cells after immunization, indicating that the inhibition observed with KJ+ CD25+ cells was not simply a consequence of competition for peptide. No accumulation of DO11 cells was observed in nondraining LNs in response to immunization (not depicted).
KJ+ CD25+ Cells Proliferate in Response to Immunization.
During the course of these experiments, we made the unexpected observation that the KJ+ CD25+ cells appeared to divide after immunization as assessed by loss of CFSE dye. To characterize directly the response of KJ+ CD25+ cells to encounter with antigen in vivo, we adoptively transferred CFSE-labeled KJ+ CD25− or KJ+ CD25+ cells into BALB/c recipients immunized with OVA protein in IFA, and examined the CFSE profiles of the KJ+ cells isolated from the draining LNs 3, 6, or 8 d later. As expected, KJ+ CD25− cells showed a strong proliferative response to immunization (Fig. 4 A). Strikingly, despite being anergic to stimulation in vitro (Fig. 3 A), the adoptively transferred KJ+ CD25+ cells proliferated in response to immunization in vivo (Fig. 4 A). KJ+ CD25− and KJ+ CD25+ cells isolated from nondraining LNs remained largely CFSE high (Fig. 4 A, day 3 data shown). Table I shows the percentage of KJ+ cells that were CFSE low, the median fluorescence channel of the divided fraction, and the absolute number of KJ+ cells for each of the conditions shown in Fig. 4 A.
Figure 4.

KJ+ CD25+ cells proliferate in vivo in response to immunization. 0.5 × 106 CFSE-labeled KJ+ CD25− or KJ+ CD25+ cells were adoptively transferred into BALB/c recipients that were immunized s.c. 24 h later with 200 μg OVA/IFA where indicated. Draining and nondraining LNs were isolated at the indicating time points after immunization and a fraction were stained with KJ-126-APC, CD4-PERCP, and CD25-PE whereas the rest were reserved for restimulation. (A) CFSE profile of gated CD4+ KJ+ cells harvested at the indicated time points after immunization. (B) Absolute number of CD4+ KJ+ cells at day 3 after immunization (symbols depict separate experiments). (C) Draining LN cells from two recipients of KJ+ CD25− cells and two recipients of KJ+ CD25+ cells were isolated 3 d after immunization and restimulated in vitro with OVA323–339 peptide. Proliferation was assessed 72 h later. Similar data were obtained in three independent experiments. (D) KJ+ CD25+ cells were sorted from conventional DO11 mice, CFSE labeled, and 106 cells were adoptively transferred into BALB/c recipients that were immunized where indicated 24 h later as described above. CFSE profiles of gated CD4+ KJ+ cells 3 d after immunization are shown.
Table I.
Proliferation of KJ+ CD25+ and KJ+ CD25− Cells in Response to Immunization
| Percent M2 (CFSE low) | Median fluorescence channel (within M2) | Number KJ+ cells (×10-3) | |
|---|---|---|---|
| KJ+ CD25− | |||
| No immunization (day 3) | 6.34 | 37.8 | 6.32 |
| Day 3 after immunization | 71.34 | 54.25 | 22.5 |
| Day 6 after immunization | 85.8 | 59.64 | 93.0 |
| Day 8 after immunization | 98.56 | 33.76 | 111.36 |
| Nondraining LN (day 3) | 11.73 | 32.78 | 1.64 |
| KJ+ CD25+ | |||
| No immunization (day 3) | 2.54 | 39.24 | 4.35 |
| Day 3 after immunization | 63.21 | 77.74 | 12.7 |
| Day 6 after immunization | 89.52 | 99.65 | 31.0 |
| Day 8 after immunization | 93.63 | 87.32 | 39.9 |
| Nondraining LN (day 3) | 10.36 | 42.17 | 2.05 |
The data depicted in Fig. 4 A were quantitated in terms of the percentage of KJ+ cells that were CFSE low, the median fluorescence channel within the divided population, and the absolute number of CD4+ KJ+ cells at each time point.
The absolute number of KJ+ CD25+ cells in the draining LNs increased in response to immunization (Fig. 4 B). The fold expansion observed in the recipients of KJ+ CD25+ cells 3 d after immunization in the experiments shown in Fig. 4 B (3.9-, 2.7-, 2.5-, and 2.1-fold) was similar to that observed for the KJ+ CD25− cells (3.5-, 2.6-, and 3.0-fold). Strikingly, when draining LN cells were isolated from recipient mice 3 d after immunization and subjected to restimulation in vitro, LN cells from recipients of KJ+ CD25− cells showed a strong proliferative response to OVA323–339 peptide whereas LN cells from recipients of KJ+ CD25+ cells did not proliferate (Fig. 4 C). The percentage of KJ+ cells in the starting populations isolated from recipient mice was equivalent in both cases (0.08 and 0.06% for recipients of KJ+ CD25− cells and 0.08 and 0.05% for recipients of KJ+ CD25+ cells in the experiment shown in Fig. 4 C). Collectively, these data suggest that although Tregs are anergic to simulation in vitro, they are able to proliferate in response to antigen in vivo. The observations made using KJ+ CD25+ cells from DO11 × RIP-mOVA mice were confirmed using the much rarer KJ+ CD25+ cells isolated from conventional DO11 mice that also proliferated in response to immunization (Fig. 4 D). Antigen-driven proliferation did not block the ability of Tregs to suppress immune responses because the KJ+ CD25+ cells could suppress T cell responses under the same conditions that induced their own proliferation (Fig. 3 B).
Defective IL-2 Induction in KJ+ CD25+ Cells.
To assess whether the proliferation of KJ+ CD25+ cells reflected an equivalent program of activation to that exhibited by the KJ+ CD25− cells, we examined cytokine expression by intracellular staining. Although the KJ+ CD25+ cells entered cell cycle, there was a striking lack of IL-2 production in these cells compared with the KJ+ CD25− cells at all the time points examined (Fig. 5 A). It is intriguing to speculate that this might be associated with the preferential expression of the transcription factor Foxp3 in CD25+ Tregs (16, 17) that has been shown to inhibit transcription of IL-2 in a Jurkat transfection system (18). At day 3 after immunization, a proportion of the KJ+ CD25− cells produced IFNγ and a smaller fraction stained positive for IL-4 (Fig. 5 B). However, neither of these effector cytokines could be detected in KJ+ CD25+ cells responding to immunization (Fig. 5 B). We were also unable to detect IL-10 protein in the KJ+ CD25+ cells either before or after immunization (Fig. 5 B and unpublished data). In addition to supplying cytokines, helper T cells can also modulate immune responses by the activation-induced provision of CD40L. To assess the capacity of CD25+ Tregs to fulfill this function, we examined CD40L expression in the KJ+ CD25+ cells responding to immunization. Levels of CD40L were greatly reduced in KJ+ CD25+ cells compared with KJ+ CD25− cells. Collectively, these data suggest that CD25+ Tregs make a qualitatively different response to immunization compared with CD25− cells.
Figure 5.

Cytokine profiles of KJ+ CD25+ cells responding to immunization. 0.5 × 106 CFSE-labeled KJ+ CD25− or KJ+ CD25+ cells were adoptively transferred into BALB/c recipients that were immunized s.c. 24 h later with 200 μg OVA/IFA. Draining LNs were isolated at the indicated time point after immunization, restimulated for 4 h with 1 μg/ml OVA323–339 peptide, fixed, permeabilized, and stained for intracellular proteins as described in Materials and Methods. Plots are gated on CD4+ KJ+ cells. (A) Kinetic analysis of IL-2 induction in response to immunization. (B) Analysis of IFNγ, IL-4, IL-10, and CD40L at day 3 after immunization. For IL-10 detection, CD4 was detected with PERCP rather than FITC accounting for the slightly lower fluorescence observed. Data are representative of at least three independent experiments.
Response of KJ+ CD25+ Cells to Tissue-expressed Antigen.
The observation that KJ+ CD25+ cells could proliferate in response to immunogenic antigen raised the intriguing possibility that Tregs are not anergic in vivo but instead might be capable of responding to the self-antigens that they recognize. To address this possibility, we examined the response of KJ+ CD25+ cells to OVA expressed transgenically as a self-antigen on pancreatic β cells. It has previously been shown that naive CD4 T cells specific for a pancreas-expressed protein undergo proliferation in the pancreatic LN (19–21). We therefore compared the proliferative response of CFSE-labeled KJ+ CD25+ cells with that of CFSE-labeled KJ+ CD25− cells after adoptive transfer into mice bearing the cognate antigen, OVA, in the pancreas. 6 d after adoptive transfer to RIP-mOVA mice, a proportion of the KJ+ CD25− had undergone proliferation in response to tissue-derived antigen as judged by loss of CFSE dye (Fig. 6 A). The proliferation of KJ+ CD25+ cells was less marked than that of KJ+ CD25− cells. However, the CFSE profiles suggested that a small fraction of KJ+ CD25+ cells had entered cell cycle in the pancreatic LN in a manner that was not observed in nondraining LNs isolated from the same mice (Fig. 6, A and B). KJ+ CD25− or KJ+ CD25+ cells adoptively transferred into nontransgenic recipients did not proliferate in either the inguinal LN or pancreatic LN (not depicted).
Figure 6.

Proliferative response of KJ+ CD25− and KJ+ CD25+ cells to tissue-expressed antigen. 106 CFSE-labeled KJ+ CD25− or KJ+ CD25+ cells were adoptively transferred into RIP-mOVA recipients. 6 d later pancreatic LNs and inguinal LNs were isolated from recipient mice and stained with KJ-126-APC, CD4-PERCP, and CD25-PE. (A) CFSE profiles of gated CD4+ KJ+ cells in one experiment. MFI shows the median fluorescence channel for the CFSE low fraction. (B) Percentage of CD4+ KJ+ cells that were CFSE low in four separate experiments.
Reciprocal Homeostasis of KJ+ CD25+ and KJ+ CD25− Cells
Proliferation of CD25− T cells in response to tissue-derived antigen is not associated with an increase in cell number. In fact, we have previously noted that the number of DO11 cells decreases after transfer to RIP-mOVA mice compared with nontransgenic recipients (unpublished data). This phenomenon has also been observed for CD8 T cells and is consistent with the induction of apoptosis in the cells responding to tissue antigen (14). To directly compare the effect of encounter with tissue antigen on KJ+ CD25− cells versus KJ+ CD25+ cells, we adoptively transferred purified populations into either RIP-mOVA mice or their transgene-negative littermates and assessed the absolute number of KJ+ cells in the pancreatic LNs 8 d later. As we have previously observed, the number of KJ+ CD25− cells recovered from the pancreatic LNs of RIP-mOVA mice was lower than that recovered from nontransgenic hosts (Fig. 7 A). The number of KJ+ CD25+ cells recovered from the LNs of nontransgenic mice tended to be lower than that of KJ+ CD25− cells, likely reflecting the hyporesponsiveness of Tregs to lymphoid chemokines that may limit their ability to enter lymphoid tissues (9). Despite this, the number of KJ+ CD25+ cells recovered from the pancreatic LN of RIP-mOVA recipients was higher than that recovered from transgene-negative littermates (Fig. 7 A). In line with preferential accumulation at the site of self-antigen, the percentage of KJ+ CD25+ cells in pancreatic LNs was consistently greater than that seen in nondraining nodes within the RIP-mOVA recipients (Fig. 7 B). When the rearrangement of additional TCR specificities in the KJ+ cells was precluded by RAG deficiency, the same trends were observed both at day 8 (Fig. 7, A and B) and day 14 (unpublished data). Collectively, these experiments indicate that CD25− and CD25+ cells bearing the same antigen specificity respond differentially to encounter with antigen draining from a peripheral tissue. CD25− cells proliferate yet ultimately decrease in number, consistent with the induction of apoptosis. In contrast, CD25+ cells show a small proliferative response to tissue antigen and their representation is increased locally, both proportionally and in terms of absolute cell number.
Figure 7.

Differential regulation of KJ+ CD25− and KJ+ CD25+ cell numbers in response to self-antigen. 0.5 × 106 KJ+ CD25−, KJ+ CD25− RAG−/−, KJ+ CD25+, or KJ+ CD25+ RAG−/− cells were adoptively transferred into RIP-mOVA recipients or their transgene-negative littermates. 8 d later recipient mice were killed and pancreatic LN cells and inguinal LN cells were stained with KJ-126-APC, CD4-PERCP, and CD25-PE. (A) Absolute number of CD4+ KJ+ cells in the pancreatic LNs of RIP-mOVA or transgene-negative recipients. (B) Percentage of KJ+ cells (within the CD4+ population) in the pancreatic LNs and inguinal LNs of RIP-mOVA recipients. Lines join data points from the same experiment.
Discussion
In vitro characterization of CD4+ CD25+ Tregs has led to the view that this subset constitutes a “naturally anergic” population (1, 15). In fact, treatments that break Treg anergy, such as the addition of exogenous IL-2 to cultures, tend to abrogate the ability of Tregs to suppress (2, 8). The diverse antigen specificity of naturally occurring CD4+ CD25+ cells has so far precluded study of the impact of antigen receptor signaling on peripheral Treg responses in vivo. We have circumvented this problem by using Tregs whose specificity is dictated by a transgenic TCR. Although refractory to stimulation in vitro, we demonstrate that Tregs proliferate in vivo in response to immunization and this does not abrogate their ability to suppress CD25− T cells. Although the number of Tregs that enter cell cycle is similar to that seen in the control population of TCR transgenic CD25− cells, the qualitative aspects of the response are markedly different as exemplified by defective induction of IL-2, effector cytokines, and CD40L in the Tregs. Thus, despite proliferation, Tregs do not adopt a phenotype consistent with the provision of T cell help to CD8 cells or B cells.
The proliferation of Tregs in vivo was somewhat surprising given their nonresponsiveness in vitro. However, Tregs are known to be highly sensitive to TCR signaling and can be induced to exert inhibition at peptide concentrations up to 100-fold lower than those required for the proliferation of CD25− cells (1). Furthermore, the finding that CD4+ CD25+ cells exhibit robust MHC class II–dependent proliferation in lymphopenic recipients (9, 10) suggests that their TCR is clearly capable of transmitting a proliferative signal. The reversion to an anergic phenotype upon removal of Tregs to an in vitro setting in the former study is intriguingly similar to the findings reported here. One possibility is that the apparently anergic phenotype of Tregs in vitro might be a feature of the high cell density in culture that allows inhibition by cell–cell contact in a manner that does not occur within secondary lymphoid tissues. Alternatively, Tregs stimulated in culture may secrete inhibitory cytokines that accumulate and suppress their proliferation. Another plausible explanation for the differential responsiveness observed in this study is that additional growth factors are available in vivo to support Treg proliferation and that these are lacking in minimalist in vitro setups.
There is some evidence to suggest that CD25+ Tregs isolated from different anatomical sites are not functionally equivalent. In an inflammation-induced diabetes model, Tregs had to be derived from the pancreatic LN to exert disease protection (22). We were therefore interested in the ability of the KJ+ CD25+ cells used in our study to respond to their local antigenic environment. Intriguingly, adoptive transfers into RIP-mOVA recipients suggested that CD25− and CD25+ cells might be inversely regulated after encounter with tissue-derived antigen. The antigen-specific CD25− cells underwent a reduction in absolute cell numbers in the pancreatic LN, likely reflecting the local induction of apoptosis as has been demonstrated for both CD8 and CD4 T cells after encounter with tissue-derived antigen (14, 23, 24). Conversely, the antigen-specific CD25+ population showed a net increase in cell number in the pancreatic LNs of antigen-bearing recipients compared with transgene-negative littermates. This may reflect the more modest proliferative response that fails to prime the cells for apoptosis. There is some precedent for a decreased susceptibility of Tregs to apoptosis in vivo (25), although these observations relate to Fas-dependent activation-induced cell death rather than the BIM-dependent pathway that is believed to mediate death in response to tissue-derived antigen (26). An alternative mechanism that may contribute to the increased number of CD25+ antigen-specific T cells at sites draining peripheral antigen expression is increased recruitment from the circulation. Intriguingly, the Tregs in this study exhibited relatively poor accumulation in lymphoid tissues in the absence of antigen compared with their CD25− counterparts (Fig. 4 B, unimmunized controls and Fig. 7, transgene-negative littermates), in line with previous observations by others (9). Exposure to antigen could provide Tregs with crucial cues for recruitment to or retention in lymphoid organs or peripheral tissues. The accumulation of CD25+ cells at the site of long-term pathogen retention in a recent study suggests that this paradigm could hold true for foreign as well as self-antigens (27). Regardless of the underlying mechanisms, the net effect of encounter with tissue-derived antigen in our study is to reciprocally regulate CD25+ and CD25− T cells numbers. Thus, encounter with self-antigen may positively regulate Treg homeostasis while negatively regulating the homeostasis of potentially pathogenic self-reactive T cells.
We were initially surprised that the expression of a transgene in a peripheral tissue appeared to drive Treg development in the thymus. However, mounting evidence suggests that so-called “tissue-specific” promoters, including that of the insulin gene, are in fact active in the thymus (12, 28). Indeed, insulin protein can be detected in medullary thymic epithelial cells (28) and its expression appears to be dependent on the activity of the transcription factor AIRE (29). Thus, T cells maturing in the thymus have the potential to interact with proteins whose expression is otherwise restricted to specific organs. Such interactions could mediate Treg development in addition to facilitating negative selection of immature T cells. Consistent with negative selection and Treg differentiation occurring in parallel, the absolute number of DO11 cells was decreased in the thymus of DO11 × RIP-mOVA mice compared with single transgenic DO11 mice. This mirrors the simultaneous negative selection and Treg development observed in double transgenic mice bearing the DO11 TCR and OVA expressed systemically in nuclei (30). Mechanistically, however, negative selection and Treg development can be uncoupled because transgenic T cells that bind antigen with sufficient affinity to mediate deletion do not necessarily exhibit Treg differentiation (31).
It has previously been shown that although antigen expression on thymic epithelium favors the development of CD25+ Tregs (31), expression of antigen by hematopoietic cells in the periphery can induce Tregs that are CD25− (32). The finding that the KJ+ CD25+ cells, but not the KJ+ CD25− cells, were suppressive in DO11 × RIP-mOVA mice is consistent with a model in which OVA expression in the thymus favors CD25+ Treg development, whereas the relative paucity of antigen in the periphery fails to induce CD25− Tregs. In contrast, in models where peripheral antigen is more widely expressed, for example where hemagglutinin is expressed under the control of the immunoglobulin κ promoter, the CD25− antigen-specific T cells are also anergic and suppressive (32). It will be of future interest to determine whether the same rules govern the antigen responsiveness and peripheral homeostasis of both CD25− and CD25+ Treg subsets. In addition, there is evidence that a distinct, yet phenotypically similar, population of CD25+ Tregs can be induced in the periphery after administration of intravenous or oral antigen (33). How closely the antigen responsiveness of these cells in vivo resembles that of thymic-derived CD25+ Tregs remains to be tested.
In conclusion, we have used Tregs bearing a transgenic TCR to enable us to examine their response to antigen in vivo. Tregs are known to be costimulation dependent (34), but the role of antigen in their peripheral homeostasis has not previously been addressed. The surprising ability of Tregs to proliferate in response to antigen in vivo offers a new perspective on the behavior of these cells within a physiological setting. Rather than constituting an anergic population, our results favor a model in which Tregs respond dynamically to their local antigenic environment. The ability to proliferate and accumulate after TCR engagement implies that local homeostatic mechanisms may serve to shape the repertoire of available Tregs at any given site.
Acknowledgments
We thank S. Jiang and C. MacArthur for cell sorting, M. Krummel and J. Cyster for helpful comments on the manuscript, and W. Heath for valuable advice.
L.S.K. Walker is funded by The Wellcome Trust. This work was supported by National Institutes of Health grants PO1-AI35297 and R37-AI 25022 to A.K. Abbas.
Footnotes
Abbreviations used in this paper: CD40L, CD40 ligand; CFSE, carboxy-fluorescein diacetate succinimidyl ester; CTLA-4, cytotoxic T lymphocyte–associated antigen 4; RIP, rat insulin promoter; Treg, regulatory T cell.
References
- 1.Takahashi, T., Y. Kuniyasu, M. Toda, N. Sakaguchi, M. Itoh, M. Iwata, J. Shimizu, and S. Sakaguchi. 1998. Immunologic self-tolerance maintained by CD25+ CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 12:1969–1980. [DOI] [PubMed] [Google Scholar]
- 2.Thornton, A.M., and E.M. Shevach. 1998. CD4+ CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Read, S., S. Mauze, C. Asseman, A. Bean, R. Coffman, and F. Powrie. 1998. CD38+ CD45RB(low) CD4+ T cells: a population of T cells with immune regulatory activities in vitro. Eur. J. Immunol. 28:3435–3447. [DOI] [PubMed] [Google Scholar]
- 4.Suri-Payer, E., A.Z. Amar, A.M. Thornton, and E.M. Shevach. 1998. CD4+ CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J. Immunol. 160:1212–1218. [PubMed] [Google Scholar]
- 5.Read, S., V. Malmstrom, and F. Powrie. 2000. Cytotoxic T lymphocyte–associated antigen 4 plays an essential role in the function of CD25+ CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pacholczyk, R., P. Kraj, and L. Ignatowicz. 2002. Peptide specificity of thymic selection of CD4+ CD25+ T cells. J. Immunol. 168:613–620. [DOI] [PubMed] [Google Scholar]
- 7.Seddon, B., and D. Mason. 1999. Peripheral autoantigen induces regulatory T cells that prevent autoimmunity. J. Exp. Med. 189:877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuniyasu, Y., T. Takahashi, M. Itoh, J. Shimizu, G. Toda, and S. Sakaguchi. 2000. Naturally anergic and suppressive CD25(+)CD4(+) T cells as a functionally and phenotypically distinct immunoregulatory T cell subpopulation. Int. Immunol. 12:1145–1155. [DOI] [PubMed] [Google Scholar]
- 9.Gavin, M.A., S.R. Clarke, E. Negrou, A. Gallegos, and A. Rudensky. 2002. Homeostasis and anergy of CD4(+) CD25(+) suppressor T cells in vivo. Nat. Immunol. 3:33–41. [DOI] [PubMed] [Google Scholar]
- 10.Annacker, O., R. Pimenta-Araujo, O. Burlen-Defranoux, T.C. Barbosa, A. Cumano, and A. Bandeira. 2001. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J. Immunol. 166:3008–3018. [DOI] [PubMed] [Google Scholar]
- 11.McHugh, R.S., M.J. Whitters, C.A. Piccirillo, D.A. Young, E.M. Shevach, M. Collins, and M.C. Byrne. 2002. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 16:311–323. [DOI] [PubMed] [Google Scholar]
- 12.Jolicoeur, C., D. Hanahan, and K.M. Smith. 1994. T-cell tolerance toward a transgenic beta-cell antigen and transcription of endogenous pancreatic genes in thymus. Proc. Natl. Acad. Sci. USA. 91:6707–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurts, C., W.R. Heath, F.R. Carbone, J. Allison, J.F. Miller, and H. Kosaka. 1996. Constitutive class I–restricted exogenous presentation of self-antigens in vivo. J. Exp. Med. 184:923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurts, C., H. Kosaka, F.R. Carbone, J.F. Miller, and W.R. Heath. 1997. Class I–restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 186:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itoh, M., T. Takahashi, N. Sakaguchi, Y. Kuniyasu, J. Shimizu, F. Otsuka, and S. Sakaguchi. 1999. Thymus and autoimmunity: production of CD25+ CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 162:5317–5326. [PubMed] [Google Scholar]
- 16.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 17.Khattri, R., T. Cox, S.-A. Yasayko, and F. Ramsdell. 2003. An essential role for scurfin in CD4+ CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 18.Schubert, L.A., E. Jeffery, Y. Zhang, F. Ramsdell, and S.F. Ziegler. 2001. Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. J. Biol. Chem. 276:37672–37679. [DOI] [PubMed] [Google Scholar]
- 19.Hoglund, P., J. Mintern, C. Waltzinger, W. Heath, C. Benoist, and D. Mathis. 1999. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J. Exp. Med. 189:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li, M., G.M. Davey, R.M. Sutherland, C. Kurts, A.M. Lew, C. Hirst, F.R. Carbone, and W.R. Heath. 2001. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J. Immunol. 166:6099–6103. [DOI] [PubMed] [Google Scholar]
- 21.Walker, L.S., L.J. Ausubel, A. Chodos, N. Bekarian, and A.K. Abbas. 2002. CTLA-4 differentially regulates T cell responses to endogenous tissue protein versus exogenous immunogen. J. Immunol. 169:6202–6209. [DOI] [PubMed] [Google Scholar]
- 22.Green, E.A., Y. Choi, and R.A. Flavell. 2002. Pancreatic lymph node-derived CD4+ CD25+ Treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity. 16:183–191. [DOI] [PubMed] [Google Scholar]
- 23.Forster, I., and I. Lieberam. 1996. Peripheral tolerance of CD4 T cells following local activation in adolescent mice. Eur. J. Immunol. 26:3194–3202. [DOI] [PubMed] [Google Scholar]
- 24.Hernandez, J., S. Aung, W.L. Redmond, and L.A. Sherman. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194:707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banz, A., C. Pontoux, and M. Papiernik. 2002. Modulation of Fas-dependent apoptosis: a dynamic process controlling both the persistence and death of CD4 regulatory T cells and effector T cells. J. Immunol. 169:750–757. [DOI] [PubMed] [Google Scholar]
- 26.Davey, G.M., C. Kurts, J.F.A.P. Miller, P. Bouillet, A. Strasser, A.G. Brooks, F.R. Carbone, and W.R. Heath. 2002. Peripheral deletion of autoreactive CD8 T cells by cross presentation of self-antigen occurs by a Bcl-2–inhibitable pathway mediated by Bim. J. Exp. Med. 196:947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belkaid, Y., C.A. Piccirillo, S. Mendez, E.M. Shevach, and D.L. Sacks. 2002. CD4+ CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 420:502–507. [DOI] [PubMed] [Google Scholar]
- 28.Derbinski, J., A. Schulte, B. Kyewski, and L. Klein. 2001. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2:1032–1039. [DOI] [PubMed] [Google Scholar]
- 29.Anderson, M.S., E.S. Venanzi, L. Klein, Z. Chen, S.P. Berzins, S.J. Turley, H. von Boehmer, R. Bronson, A. Dierich, C. Benoist, et al. 2002. Projection of an immunological self shadow within the thymus by the aire protein. Science. 298:1395–1401. [DOI] [PubMed] [Google Scholar]
- 30.Kawahata, K., Y. Misaki, M. Yamauchi, S. Tsunekawa, K. Setoguchi, J. Miyazaki, and K. Yamamoto. 2002. Generation of CD4(+)CD25(+) regulatory T cells from autoreactive T cells simultaneously with their negative selection in the thymus and from nonautoreactive T cells by endogenous TCR expression. J. Immunol. 168:4399–4405. [DOI] [PubMed] [Google Scholar]
- 31.Jordan, M.S., A. Boesteanu, A.J. Reed, A.L. Petrone, A.E. Holenbeck, M.A. Lerman, A. Naji, and A.J. Caton. 2001. Thymic selection of CD4+ CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2:301–306. [DOI] [PubMed] [Google Scholar]
- 32.Apostolou, I., A. Sarukhan, L. Klein, and H. von Boehmer. 2002. Origin of regulatory T cells with known specificity for antigen. Nat. Immunol. 3:756–763. [DOI] [PubMed] [Google Scholar]
- 33.Thorstenson, K.M., and A. Khoruts. 2001. Generation of anergic and potentially immunoregulatory CD25+ CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J. Immunol. 167:188–195. [DOI] [PubMed] [Google Scholar]
- 34.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+ CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]