

Abstract
Resistance and susceptibility to Leishmania major in mice are determined by multiple genes and correlate with the preferential development of Th1 and Th2 responses, respectively. Here, we found that CD11b+ dendritic cells (DCs) prime parasite-specific CD4+ T cells in both susceptible BALB/c (H2-d) and resistant B10.D2 (H2-d) mice. However, BALB/c and B10.D2 DCs from L. major–infected mice differ in their ability to polarize naive T cells into Th1 or Th2 effector cells. This difference is cell-intrinsic, is not restricted to H2-d mice, and is observed with both parasite-specific and allospecific CD4+ T cells. Thus, strain-specific differences within CD11b+ DCs influence the ability of inbred mice to mount polarized CD4+ T cell responses.
Keywords: Ag presentation, T cell differentiation, intracellular pathogen, cytokine secretion, Leishmania major
Introduction
Leishmania major is a protozoan parasite which invades macrophages (1). Most strains of mice, including C57BL/6, C3H, and B10.D2, develop of a protective Th1 response and self-healing lesions (1). In contrast, mice of the BALB genetic background develop a Th2-biased counterprotective immune response resulting in the development of progressive lesions and in the dissemination of the parasite to internal organs. Because BALB/c (H2-d) and B10.D2 (H2-d) mice differ in several polymorphic genes which control resistance to L. major, elucidating how these genes work should provide valuable information on the mechanisms leading to differentiation of naive T cells. Susceptibility to L. major is a multigenic phenotype which depends on several genetic loci (2–5). Candidate loci were identified on chromosomes 6, 7, 10, 11, 15, and 16 (2). The presence of all six loci is not necessary to confer resistance and no single locus is required. Rather, various combinations of these loci are capable of conferring resistance. Furthermore, both T cell and non-T cell compartment can independently determine resistance to L. major (6). Thus, several genes are impaired in BALB strains and the accumulation of these defects results in susceptibility to L. major.
Previous studies have shown that T cells from BALB/c mice are intrinsically different from those of other strains (7–10). For example, BALB/c T cells exhibit an intrinsic ability to secrete high amounts of IL-4 and to rapidly lose IL-12 responsiveness in vitro. However, it is not known whether this latter phenomenon accounts for the impaired ability of BALB/c mice to mount a Th1 protective response upon infection with L. major. During the course of L. major infection, Langerhans cells (LCs)* transport L. major promastigotes from the skin to the draining LN (11–13). Ingestion of L. major amastigotes by LC-like immature DCs derived from fetal skin results in the up-regulation of CD80, CD86, CD40, ICAM-1, and MHC class II molecules (14, 15). However, it is not known whether DCs from different mouse strains differ in their ability to promote the differentiation of naive CD4+ T cells into Th1 or Th2 effector cells. To address this issue, we have infected mice with L. major and purified DCs from their draining LN. By comparing the ability of these cells to induce the differentiation of naive TCR transgenic CD4+ T cells which are specific for the L. major LACK Ag, we have discovered an intrinsic difference in DCs from BALB/c versus B10.D2 mice.
Materials and Methods
Mice and Parasites.
BALB/c and B10.D2 mice were purchased from Harlan. 16.2β (16) and WT15 (17) transgenic mice have been previously described. Animals were housed under SPF conditions and used between 7 and 10 wk of age. L. major promastigotes (World Health Organization strain WHOM/IR/-/173) were grown in M199 medium containing 20% FCS. Mice were infected with 2 × 106 stationary phase promastigotes as described (18). In some experiments, BALB/c mice were injected i.p. with 200 ng of recombinant murine IL-1β 1 d before infection, at the time of infection, and 2 d later. Parasite loads were measured as described (19). All experiments were performed in compliance with the CNRS institutional guidelines and have been approved by the local committee for animal experimentation.
Reagents and Antibodies.
The following mAb were purchased from Becton Dickinson: GK1.5, anti-CD4; 53–6.7, anti-CD8α; KT4.1, anti-Vβ4; RA3–6B2, anti-B220; 2G9; anti-I-Ad/I-Ed; M1/70, anti-CD11b; HL3, anti-CD11c; 1G10, anti-CD80; GL1, anti-CD86; 3/23, anti-CD40; 145–2C11, anti-CD3. Recombinant murine IL-1β, IFN-γ, IL-2, IL-4, and IL-5 were purchased from R&D Systems. LACK peptide (aa 158 through 173: FSPSLEHPIVVSGSWD) was synthesized by Mimotopes France.
Cells.
LMR7.5 hybridomas express a high affinity TCR specific for the LACK peptide (aa 158 through 163) bound to I-Ad MHC class II molecules (16). To prepare DCs, LNs were digested with a cocktail of DNase I fraction IX (Sigma-Aldrich; 100 μg/ml) and 1.6 mg/ml of collagenase (400 Mandl U/ml) at 37°C for 45 min. DCs were positively selected by MACS using N418 (anti-CD11c) magnetic beads (Miltenyi Biotec) in the presence of 10% mouse serum and 5 mM EDTA according to the manufacturer instructions. Positively selected cells were pure to more than 95% as determined by flow cytometry analysis following staining with anti-CD11c mAb. Cells were either used immediately, or further sorted by flow cytometry into CD11b+ CD11c+ and CD11b− CD11c+ subsets after staining with anti-CD11b and anti-CD11c mAb. In some experiments, LN cells were depleted of CD3+ cells using Dynabeads (Dynal) prior purification of CD11c+ cells. Bone marrow–derived DCs were prepared as described (20). CD4+ LN cells were purified by negative depletion of CD8+, B220+, CD11b+, and I-Ad+ cells using sheep anti-rat Ig-coated Dynabeads (Dynal). Naive CD44low CD62Lhigh CD4+ T cells were further purified by flow cytometry.
T Cell Assays.
For stimulation of T cell hybridomas, CD11c+ cells or purified subsets of DCs were incubated with 105 LMR7.5 hybridomas with or without 1 μM of LACK peptide, in DMEM supplemented with l-glutamine (2 mM), heat-inactivated FCS (10%), 2-ME (5 × 10−5 M), penicillin (100 μg/ml), and streptomycin (100 U/ml) in U-bottomed 96-well plates. Supernatants were harvested 24 h later and IL-2 contents were measured by ELISA. For stimulation of TCR transgenic T cells, DCs were incubated with 5 × 105 CD44low CD62Lhigh CD4+ cells from F1 (BALB/c × B10.D2) WT15 or 16.2β transgenic mice with the indicated concentrations of LACK peptide in U-bottomed 96-well plates or flat-bottomed 48-well plates. Supernatants were harvested on day 3 or 6 and analyzed for IFN-γ, IL-4, and IL-5 contents by ELISA. When indicated, T cells were purified and restimulated with 1 μM of LACK peptide in the presence of 2 × 106 mitomycin C-treated splenocytes from F1 (BALB/c × B10.D2) mice. For stimulation of T cells from infected mice, 5 × 105 purified CD4+ T cells were incubated with 2 × 106 mitomycin C–treated syngeneic splenocytes with or without 30 μg/ml of soluble Leishmania antigens (SLA) in flat-bottomed 96-well plates. Supernatants were harvested 48 h later and analyzed for IFN-γ, IL-4, and IL-5 contents by ELISA. For intracellular staining of cytokines, cells were incubated for 4 h at 37°C with PMA (10 ng/ml), ionomycin (1.5 μg/ml), and brefeldin A (10 μg/ml). Cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) and stained with anti–IL-4 and anti–IFN-γ mAb.
Real-Time RT-PCR.
Total RNA from CD11c+ or CD11b+ CD11c+ cells was extracted and reverse transcribed as described (21). Complementary DNA was quantitatively analyzed for the expression of murine cytokines (IFN-γ, GM-CSF, IFN-α, IL-12 p35, IL-12 p40, IL-3, IL-4, IL-6, IL-1α, IL-1β, TNF-α, IL-10, TGF-β1, TGF-β2, and TGF-β3), 18S ribosomal RNA and ubiquitin mRNA, using the fluorogenic 5′-nuclease PCR assay (22) as described (21). Specific primers were obtained from DNAX. Gene-specific PCR products were continuously measured by means of an ABI PRISM 5700 Sequence Detection System (Applied Biosystems) during 40 cycles. Ubiquitin mRNA was used for normalization.
Flow Cytometry.
Cells were analyzed by flow cytometry using a FACSCalibur™ and the CELLQuest™ software (Becton Dickinson). Cell sorting was performed using a FACSVantage SE™ (Becton Dickinson). In some experiments, positively selected CD11c+ cells were incubated on ice for 30 min with anti-CD8α, anti-CD11b, anti-CD11c mAb, and either anti-CD80, or anti-CD86, anti-I-Ad, or anti-CD40 mAb. Staining was performed in PBS containing 0.5% BSA.
Results
MHC Class II Presentation of LACK In Vivo.
Infection of BALB/c and B10.D2 mice with L. major induces LACK-specific CD4+ T cells to up-regulate CD69 and CD44 and to progress through the cell cycle (16). Activated LACK-specific CD4+ T cells are first detected in the draining LN and their frequency peaks at day 3, suggesting that APCs that have captured and processed LACK are present in the LN on day 2. As DCs have been shown to be responsible for the priming of CD4+ T cells in some experimental models (23–25), we sought to investigate the role of DCs in the priming of parasite-specific CD4+ T cells. To this aim, CD11c+ and CD11c− cells were purified from the draining LN of BALB/c and B10.D2 mice which had been infected 2 d earlier with L. major. We next incubated these cells with LACK-specific LMR7.5 T cell hybridomas with or without LACK peptide. In the absence of peptide, CD11c+ cells, but not CD11c− cells, induced LMR7.5 hybridomas to secrete IL-2 in a dose-dependent manner (Fig. 1) . In contrast, both CD11c+ and CD11c− cells stimulated LMR7.5 hybridomas when incubated with LACK peptide. Thus, although both CD11c+ and CD11c− cells can present LACK to T cells in vitro, only CD11c+ cells do so in vivo.
Figure 1.

MHC class II presentation of LACK by CD11c+ cells. BALB/c (left panels) and B10.D2 (right panels) mice were infected with L. major promastigotes into the hind footpads. LN cells were prepared on day 2 and depleted of CD3+ cells. Cells were either used directly (▴) or sorted into CD11c+ (▪) and CD11c− (○) cells. The indicated numbers of cells were incubated with 105 LMR7.5 hybridomas without (top panels) or with (bottom panels) 1 μM of LACK peptide. Supernatants were harvested 24 h later and analyzed for IL-2 content. Data are representative of five experiments.
Polarization of CD4+ T Cells by BALB/c or B10.D2 DCs.
Previous studies have shown that CD4+ T cells from infected BALB/c and B10.D2 mice secrete different amounts of IFN-γ, IL-5, and IL-4 when restimulated with SLA. Likewise, in a typical experiment, LN CD4+ T cells from BALB/c mice secreted 10- to 20-fold less IFN-γ than those from B10.D2 mice (Fig. 2 A). In contrast, while T cells from BALB/c mice secreted relatively high amounts of IL-4 and IL-5, these cytokines were below the levels of detection in the wells containing B10.D2 T cells. To investigate whether DCs could induce the differentiation of naive LACK-specific CD4+ T cells in vitro, we used WT15 TCR transgenic mice in which CD4+ T cells express the α and β chains of a high avidity LACK-specific TCR (17). We incubated LN CD11c+ cells from infected BALB/c or B10.D2 mice with naive CD4+ T cells from F1 (BALB/c × B10.D2) WT15 transgenic mice with or without LACK peptide, and analyzed supernatants for cytokine contents on day 3 and 6, respectively. In the absence of peptide, B10.D2 DCs induced WT15 transgenic T cells to secrete twofold more IFN-γ and fourfold less IL-5 than BALB/c DCs (Fig. 2 B, left panel). While the levels of IL-5 and IFN-γ varied from one experiment to another, the IL-5/IFN-γ ratio was 5.8 ± 0.7-fold higher when T cells were primed with BALB/c DCs as compared with B10.D2 DCs (P < 0.05). We obtained qualitatively similar results when T cells were stimulated with LACK peptide (Fig. 2 B, right panel). IL-4 secretion was below the limit of detection in all samples. T cells did not secrete any detectable amounts of IL-5 and IFN-γ when incubated without DCs (unpublished data).
Figure 2.

DCs from infected BALB/c and B10.D2 mice differ in their ability to polarize naive CD4+ T cells. BALB/c (□) and B10.D2 (▪) mice were infected with L. major. (A) LN CD4+ T cells were purified 5 wk after infection and 5 × 105 cells were incubated with SLA (30 μg/ml) and 2 × 106 mitomycin C–treated syngeneic splenocytes. Supernatants were analyzed after 48 h for IFN-γ, IL-5, and IL-4 contents by ELISA. Data show the amounts of IFN-γ (ng/ml), IL-5 (U/ml), and IL-4 (ng/ml). (B and C) LN CD11c+ cells were purified on day 2. (B) CD44low CD62Lhigh CD4+ T cells were purified from F1 (BALB/c × B10.D2) WT15 transgenic mice and 5 × 105 cells were incubated with 1.2 × 105 CD11c+ cells without (left panel) or with (right panel) 1 μM of LACK peptide. Supernatants were harvested on day 6 (left panel) or on day 3 (right panel) and analyzed for IFN-γ and IL-5 contents. Data are representative of six experiments and show the amounts of IFN-γ (ng/ml) and IL-5 (U/ml). (C) CD44low CD62Lhigh CD4+ T cells were purified from F1 (BALB/c × B10.D2) 16.2β transgenic mice and 1.5 × 106 cells were incubated with 3 × 105 CD11c+ cells without LACK peptide. (left) Supernatants were harvested on day 6 and analyzed for IFN-γ and IL-5 contents. Live cells were purified and restimulated with mitomycin C–treated F1 splenocytes and 1 μM of LACK peptide. (right) Supernatants were harvested on day 3 and analyzed for IFN-γ, IL-4, and IL-5 contents. Data are representative of three experiments and show the amounts of IFN-γ (ng/ml), IL-5 (U/ml), and IL-4 (ng/ml).
To generalize this phenomenon to polyclonal populations of T cells, we used 16.2β single chain transgenic mice which exhibit both an increased frequency of LACK-specific T cells and a polyclonal T cell repertoire as the result of the transgenic expression of the β chain of a LACK-specific hybridoma (16). In the absence of LACK peptide, naive CD4+ T cells from F1 (BALB/c × B10.D2) 16.2β transgenic mice secreted fourfold more IFN-γ and twofold less IL-5 when incubated with B10.D2 DCs than when incubated with BALB/c DCs (Fig. 2 C, left). To confirm that the effector T cells which were generated in these experiments secreted different amounts of Th1 and Th2 cytokines, live cells were recovered after primary stimulation and further incubated for 2 d with LACK peptide and mitomycin C–treated splenocytes from F1 (BALB/c × B10.D2) mice. As observed in primary cultures, T cells which had been primed with BALB/c DCs secreted more IL-5 and less IFN-γ than those which had been primed with B10.D2 DCs. Furthermore, while IL-4 was below the level of detection in primary cultures, T cells which had been primed with BALB/c DCs secreted 8- to 10-fold more IL-4 than those which had been primed with B10.D2 DCs (Fig. 2 C, right panel).
LACK Is Presented by CD11b+ DCs In Vivo.
LN CD11c+ cells have been classified into CD11b+CD8α−, CD11b− CD8α+, and CD11b−CD8α− cells (26). To identify which DCs are responsible for the activation of LACK-specific T cells in vivo, we purified CD11b+ and CD11b− DCs from the LN of infected mice and incubated various numbers of these cells with LMR7.5 hybridomas. In the absence of LACK peptide, CD11b+ DCs were 1,000- to 10,000-fold more efficient than CD11b− DCs in stimulating LMR7.5 hybridomas to secrete IL-2 (Fig. 3 A). In contrast, both cell types were equally efficient in stimulating LMR7.5 hybridomas when incubated with LACK peptide. We obtained similar results using BALB/c and B10.D2 DCs. Thus, while both CD11b+ and CD11b− DCs could present LACK peptide to CD4+ T cells in vitro, CD11b+ DCs were solely responsible for the priming of LACK-specific T cells in vivo.
Figure 3.

MHC class II presentation of LACK by CD11b+ DCs. BALB/c (left panels) and B10.D2 (right panels) mice were injected with L. major promastigotes into the hind footpads. LN cells were prepared on day 2 and depleted of CD3+ cells. CD11c+ cells were purified by MACS and stained with anti-CD11b and anti-CD11c mAb. (A) CD11b+ (▪) and CD11b− (○) DCs were sorted by flow cytometry, and the indicated numbers of cells were incubated with 105 LMR7.5 hybridomas without (top panels) or with (bottom panels) 1 μM of LACK peptide. Supernatants were harvested 24 h later and analyzed for IL-2 content. Data are representative of five experiments. (B) CD11b+ and CD11b− DCs were sorted by flow cytometry, and mixtures of these cells (6 × 104 cells of the indicated subset), or 1.2 × 105 CD11b+ cells alone, were incubated with 5 × 105 CD44low CD62Lhigh CD4+ T cells from F1 (BALB/c × B10.D2) WT15 transgenic mice. Supernatants were harvested on day 6 and analyzed for IFN-γ (white bars) and IL-5 (black bars) contents. The IL-5/IFN-γ ratio (gray bars) was calculated for each sample. Data are representative of three experiments.
We next investigated whether strain-specific differences within CD11b+ DCs accounted for the ability of B10.D2 and BALB/c DCs to differentially polarize naive CD4+ T cells in vitro. CD11b+ and CD11b− DCs were purified from the LN of infected BALB/c and B10.D2 mice, and these cells or mixtures of these cells were incubated with naive F1 WT15 TCR transgenic T cells. While WT15 TCR transgenic T cells did not secrete any detectable amount of IFN-γ or IL-5 after incubation with CD11b− DCs (unpublished data), these T cells secreted twofold more IFN-γ and 10-fold less IL-5 when primed with CD11b+ DCs from B10.D2 mice as compared with CD11b+ DCs from BALB/c mice (Fig. 3 B). Further experiments using mixtures of CD11b+ and CD11b− DCs showed that CD11b− DCs had little effect on T cell polarization, further confirming that the amounts of IFN-γ and IL-5 which were secreted by T cells were mainly determined by CD11b+ DCs.
Several parameters influence the differentiation of naive CD4+ T cells, including the dose of Ag, the nature and the amounts of cytokines in the extracellular medium, and the levels of accessory molecules expressed by APCs (27). To determine whether BALB/c and B10.D2 DCs expressed different amounts of cytokine mRNA, we purified CD11b+ CD11c+ cells from infected mice on day 0 and day 2 after infection. We next measured cytokine mRNA levels using real time RT-PCR. CD11b+ DCs from infected B10.D2 mice expressed 10- to 15-fold more IL-1β mRNA than those from infected BALB/c animals (Fig. 4 A). CD11b+ DCs from naive B10.D2 mice also expressed more IL-1β mRNA than those from naive BALB/c mice. All other cytokine mRNA, including those coding for IFN-γ, GM-CSF, IFN-α, IL-12 p35, IL-12 p40, IL-6, IL-1α, TNF-α, TGF-β1, and TGF-β2 mRNA were expressed at similar levels by CD11b+ DCs in the two strains before and after infection. IL-3, IL-4, IL-10, and TGF-β3 mRNA were below the level of detection in all samples (unpublished data).
Figure 4.

Cytokine mRNA and costimulatory molecules expressed by BALB/c and B10.D2 DCs. Mice were infected or not with L. major promastigotes. (A) LNs were harvested on day 2 and CD11b+ CD11c+ cells were purified by flow cytometry. mRNA levels for the indicated cytokines were measured using Real Time RT-PCR. Values are expressed as the number of Ct for each cytokine mRNA after normalization to ubiquitin mRNA for infected BALB/c (white bars) and B10.D2 (black bars) mice, and for PBS-injected BALB/c (light gray bars) and B10.D2 (dark gray bars) mice. Data are representative of three experiments. (B and C) LN CD11c+ were purified at the indicated times after infection and cells were analyzed by flow cytometry after staining with anti-CD11b, anti-CD11c, and either anti-CD80, anti-CD86, anti-CD40, or anti-I-Ad mAb. (B) Typical flow cytometry profiles are shown after gating on CD11b+ DCs for BALB/c (thin lines) and B10.D2 (thick lines) mice. Data show the mean fluorescence intensity (MFI) expressed as geometric mean for BALB/c (plain) and B10.D2 (bold) mice. (C) Surface levels of CD80 (left panel) and CD86 (right panel) were measured at the indicated times after infection in BALB/c (○) and B10.D2 (•) mice. Data show MFI expressed as geometric mean.
T cell activation is induced by the simultaneous engagement of the costimulatory molecule CD28 expressed on T cells with its ligands CD80 and CD86 expressed at the surface of APCs (28). Some investigators have suggested that CD80 and CD86 provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTLs (29). However, others have reported significant biological differences between these molecules, including differences in their ability to induce maturation toward polarized Th1 or Th2 effector cells (30). To measure the levels of expression of CD80 and CD86 at the surface of CD11b+ DCs, we purified LN DCs from BALB/c and B10.D2 mice on day 0 and 2 after infection and analyzed these cells by flow cytometry after staining with anti-CD11b and anti-CD11c mAb, and mAb to CD80, CD86, CD40, and I-Ad MHC class II molecules. CD11b+ DCs from naive B10.D2 mice expressed 1.5- to 2-fold more CD80 than those from naive BALB/c mice (Fig. 4 B). Furthermore, while CD80 was only slightly and transiently up-regulated by L. major in BALB/c mice, its level of expression was increased by 2.5 ± 0.6-fold in B10.D2 animals. In both BALB/c and B10.D2 mice, L. major induced the down-regulation of CD86 and the up-regulation of CD40 and I-Ad. However, in contrast to CD80, CD11b+ DCs from BALB/c and B10.D2 mice expressed roughly similar levels of CD86, CD40, and I-Ad before as well as 1, 2, and 3 d after infection (Fig. 4, B and C, and unpublished data).
IL-1β Inhibits the Development of Th2 Cells In Vivo.
To determine whether IL-1β could have an effect on the polarization of parasite-specific T cells in the setting of L. major infection, BALB/c mice were treated or not with recombinant murine IL-1β and infected with L. major together with control B10.D2 mice. Mice were killed 5 wk later and parasite loads were measured by limiting dilution (Fig. 5 A). While untreated BALB/c mice exhibited 6 ± 2.7 × 109 parasites in their LN, parasite loads in B10.D2, and IL-1β-treated BALB/c mice were 4.6 ± 2.0 × 104 and 2.1 ± 1 × 108, respectively. In parallel experiments, LN cells were analyzed by flow cytometry after intracellular staining of IFN-γ and IL-4 (Fig. 5 B). While 5.1 ± 0.8% of CD4+ T cells secreted IFN-γ in B10.D2 mice, the frequency of IFN-γ–secreting CD4+ T cells was 2.0 ± 0.2% in both IL-1β–treated and untreated BALB/c mice. In contrast, the frequency of IL-4–secreting CD4+ T cells was 2.1 ± 0.3% in untreated BALB/c mice, as compared with 1.0 ± 0.2% and 0.4 ± 0.1% in IL-1β–treated mice and B10.D2 mice, respectively. Thus, treatment of BALB/c mice with IL-1β resulted in a significantly reduced Th2 response (P < 0.02) and in a 20-fold reduction in the number of parasites in the draining LN.
Figure 5.
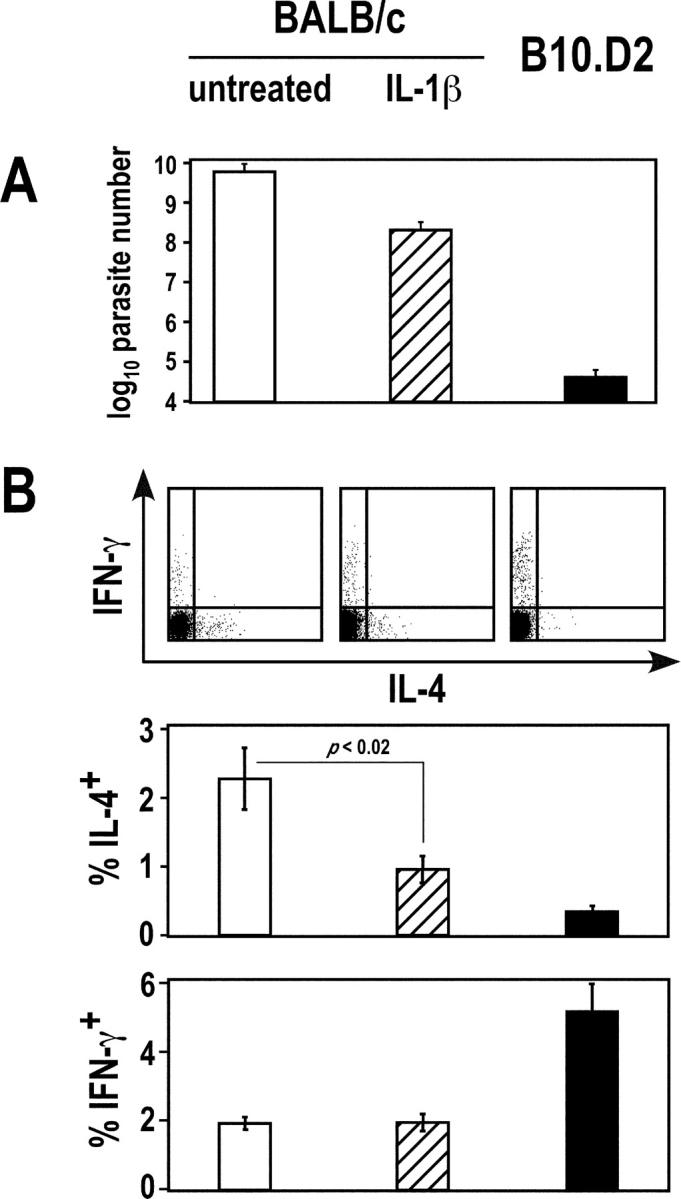
Effect of IL-1β on T cell polarization and parasite load. BALB/c mice (five mice per group) were treated or not with recombinant murine IL-1β and infected with L. major, together with control B10.D2 mice. Mice were killed 5 wk later. (A) Parasite loads were measured in draining LN. Data show mean ± SEM for untreated BALB/c mice (empty bars), IL-1β–treated BALB/c mice (dashed bars), and B10.D2 mice (filled bars). (B) LN cells were stimulated with PMA and ionomycin, and further analyzed by flow cytometry after surface staining with anti-CD4 mAb and intracellular staining with anti-IL-4 and anti-IFN-γ mAb. Data show representative FACS® profiles after gating on CD4+ T cells (top panels), and the frequency of IL-4– and IFN-γ–secreting cells (mean ± SEM) for untreated BALB/c mice (empty bars), IL-1β-treated BALB/c mice (dashed bars), and B10.D2 mice (filled bars; bottom panels). Statistical analysis was performed using Student's t test.
BALB/c and B10.D2 DCs Are Intrinsically Different.
To determine whether the differential ability of BALB/c and B10.D2 DCs to polarize CD4+ T cells was the result of L. major infection or was an intrinsic property of these DCs, we incubated LN DCs from naive mice with naive CD4+ T cells from F1 WT15 transgenic mice and different amounts of LACK peptide, and analyzed supernatants for IFN-γ and IL-5 contents on day 3. At low or intermediate peptide concentrations (≤200 nM), the IL-5/IFN-γ ratio was two- to threefold higher for BALB/c than for B10.D2 DCs (Fig. 6 A). While the levels of cytokines produced varied from one experiment to another, the IL-5/IFN-γ ratio at 50 nM of LACK peptide was 3.4 ± 0.6-fold higher when T cells were primed with BALB/c DCs as compared with B10.D2 DCs (P < 0.05). Similar results were obtained using bone marrow-derived DCs (Fig. 6 B). Thus, the differential ability of BALB/c and B10.D2 DCs to polarize CD4+ T cells in vitro was an intrinsic property of these cells rather than the result of L. major infection. To determine whether differences found in the amounts of cytokines produced resulted from differences in the frequencies of cytokine-secreting cells, we incubated LN DCs from naive BALB/c and B10.D2 mice with naive CD4+ T cells from F1 WT15 transgenic mice and an intermediate amount of LACK peptide (50 nM). Cells were then restimulated with an optimal amount of LACK peptide in the presence of syngeneic splenocytes, and cellular supernatants were analyzed for cytokine contents by ELISA. Alternatively, cells were restimulated with PMA and ionomycin and further analyzed by flow cytometry for intracellular staining of IFN-γ, IL-4, and IL-5. Upon restimulation, T cells primed with DCs from naive BALB/c mice secreted more IL-4 and IL-5 and less IFN-γ than those primed with DCs from naive B10.D2 mice (Fig. 6 C). While IL-4- and IL-5–secreting T cells were hardly detectable in all samples, the frequency of IFN-γ–secreting T cells was three- to fourfold higher upon priming with B10.D2 DCs as compared with BALB/c DCs (24.6 versus 8.4% in a typical experiment). Qualitatively similar results were obtained when cells were restimulated with anti-CD3 mAb (unpublished data).
Figure 6.

DCs from naive BALB/c and B10.D2 mice differ in their ability to polarize CD4+ T cells. CD11c+ cells were purified from the popliteal LNs (A and C) or generated from the bone marrow (B) of naive BALB/c (□) and B10.D2 (▪) mice. 2 × 105 cells were pulsed for 18 h with the indicated concentrations (A and B) or 50 nM (C) of LACK peptide, and further incubated with 5 × 105 CD44low CD62Lhigh CD4+ T cells from F1 (BALB/c × B10.D2) WT15 TCR transgenic mice. (A and B) Supernatants were harvested on day 3 and analyzed for IFN-γ and IL-5 contents. Data are representative of six and three experiments, respectively, and show the ratios between IL-5 (U/ml) and IFN-γ (ng/ml). (C) Cells were recovered on day 3 and restimulated with 2 μM of LACK peptide in the presence of syngeneic splenocytes. Supernatants were analyzed on day 3 for IFN-γ, IL-4, and IL-5 contents (left panels). Data show the amounts of IFN-γ (ng/ml), IL-5 (U/ml), and IL-4 (ng/ml). Cells were restimulated with PMA and ionomycin, stained with anti-CD4 mAb and analyzed by flow cytometry for intracellular staining of IFN-γ and IL-4 (right panels). Data are representative of three experiments and show FACS® profiles after gating on CD4+ T cells.
We next investigated whether BALB/c and B10.D2 DCs differed in their ability to polarize allospecific CD4+ T cells. To this aim, LN CD11c+ cells from naive BALB/c or B10.D2 mice were incubated with naive CD4+ T cells from either C3H (H2-k) or C57BL/6 (H2-b) mice. Whatever DC numbers were used, IL-5/IFN-γ ratios were two- to eightfold higher for BALB/c DCs than for B10.D2 DCs (Fig. 7 A). Similar results were obtained using naive T cells from C57BL/6 mice (Fig. 7 B). To investigate whether the differential ability of BALB/c and B10.D2 DCs to polarize CD4+ T cell responses could be observed with DCs expressing other MHC molecules, we incubated LN CD11c+ cells from BALB.B (H2-b) and C57BL/6 (H2-b) mice with naive CD4+ T cells from C3H (H2-k) mice. As observed with BALB/c and B10.D2 DCs, T cells incubated with BALB.B DCs secreted more IL-5 and less IFN-γ than those incubated with C57BL/6 DCs (Fig. 7 C).
Figure 7.
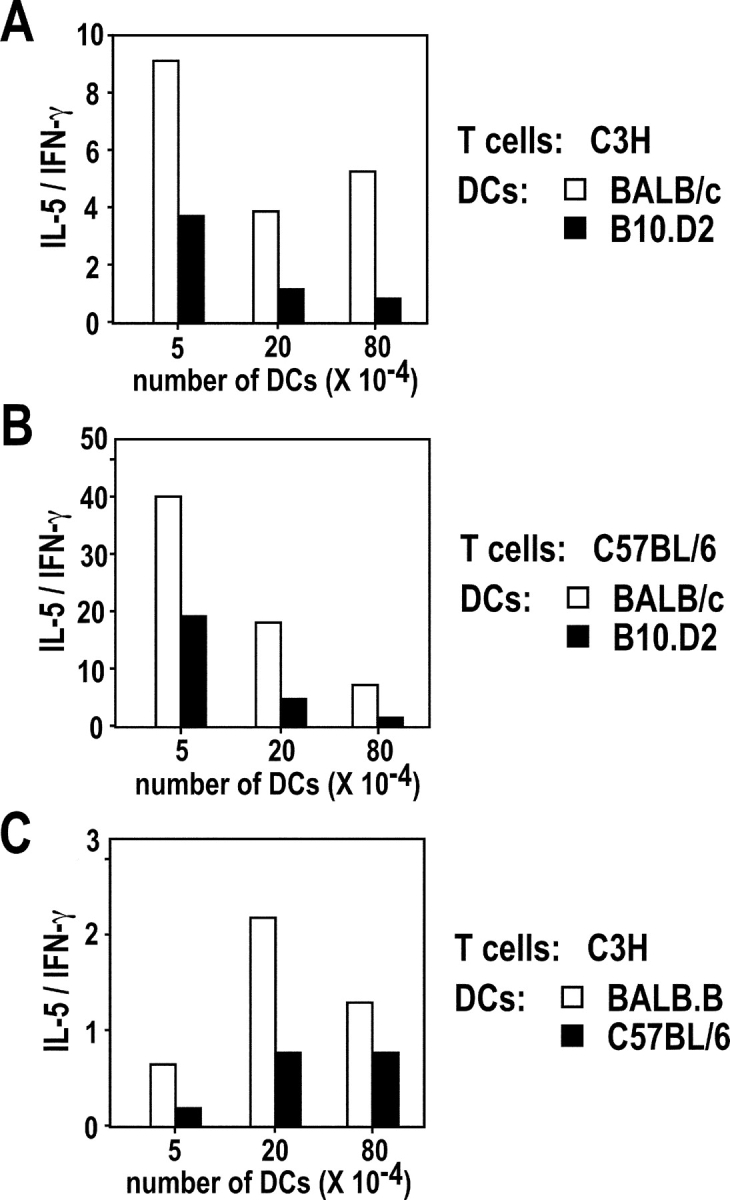
DCs from different strains of mice differ in their ability to polarize allospecific CD4+ T cells. (A and B) CD11c+ cells were purified from the popliteal LN of naive BALB/c (□) or B10.D2 (▪) mice and the indicated numbers of cells were incubated with 5 × 105 CD44low CD62Lhigh CD4+ T cells from C3H (A) or C57BL/6 (B) mice. Supernatants were harvested on day 3 and analyzed for IFN-γ and IL-5 contents. Data are representative of four experiments and show the ratios between IL-5 (U/ml) and IFN-γ (ng/ml). (C) CD11c+ cells were purified from the popliteal LN of naive BALB.B (□) or C57BL/6 (▪) mice and the indicated numbers of cells were incubated with 5 × 105 CD44low CD62Lhigh CD4+ T cells from C3H mice. Supernatants were harvested on day 3 and analyzed for IFN-γ and IL-5 contents. Data show the ratios between IL-5 (U/ml) and IFN-γ (ng/ml) in a representative out of two experiments.
Discussion
Previous studies have shown that L. major promastigotes are transported from the inflammatory site to the LN by LCs (12) and that LN DCs from infected mice are capable of stimulating parasite-specific T cells in vitro (13). However, it was not known whether parasite Ag are processed by other types of cells in vivo. Furthermore, despite the characterization of several L. major Ag, no study had been performed to identify the APCs which are responsible for the processing of defined parasite Ag and their presentation to CD4+ T cells. Lastly, although many subsets of DCs have been described (26), it was not known whether all DCs present Ag to T cells in vivo or whether this task is performed by a restricted subset of DCs. To address these issues, we used LMR7.5 T cell hybridomas which express a TCR exhibiting a high avidity for a LACK-derived peptide bound to I-Ad. In both infected BALB/c and B10.D2 mice, CD11c+, but not CD11c− cells stimulated LMR7.5 hybridomas. Furthermore, CD11b+ DCs were far more efficient than CD11b− DCs in stimulating LMR7.5 hybridomas. However, CD11b− DCs were as efficient as CD11b+ DCs in stimulating LMR7.5 hybridomas when incubated with LACK peptide. Thus, while both CD11b+ and CD11b− DCs can present LACK to CD4+ T cells in vitro, CD11b+ DCs are solely responsible for the priming of LACK-specific CD4+ T cells in vivo, at least 2 d after infection. It remains to be determined whether this remains true during the late stages of the disease, and whether CD11b+ DCs also stimulate CD4+ T cells reacting to other parasite Ag. It would also be interesting to determine whether CD11b+ DCs are responsible for the stimulation of CD4+ T cells when LACK is not delivered upon infection with L. major. In this respect, we have recently found that LN CD11b+ DCs are responsible for the tolerization of islet-specific CD4+ T cells in NOD mice in which β cell apoptosis has been induced (31). Likewise, Zhao and colleagues have found that only CD11b+ submucosal DCs present viral Ag to CD4+ T cells in mice which have been infected with Herpes Simplex Virus (HSV)-2 (32). Thus, while other APCs have the ability to present antigenic peptides in vitro, it seems that CD11b+ DCs play a critical role in the priming of CD4+ T cells in vivo, at least when these T cells react to cell-associated Ag.
Although CD11b+ DCs were responsible for the priming of CD4+ T cells in both B10.D2 and BALB/c mice, naive CD4+ T cells secreted more IFN-γ and less IL-5 when incubated with B10.D2 DCs as compared with BALB/c DCs. This was observed using both unseparated CD11c+ and purified CD11b+ DCs, suggesting that strain-specific differences within the CD11b+ DC subset are sufficient to influence the differentiation of CD4+ T cells. We obtained similar results with DCs from naive mice incubated with either LACK-specific TCR transgenic T cells and LACK peptide, or allospecific CD4+ T cells from C3H or C56BL/6 mice. Likewise, CD4+ T cells from C3H mice secreted more IL-5 and less IFN-γ when incubated with BALB.B DCs as compared with C57BL/6 DCs. Thus, the preferential ability of B10.D2 and BALB/c DCs to polarize CD4+ T cells into Th1 or Th2 effector cells, respectively, is an intrinsic property of these DCs, is not dependent on L. major infection, is not restricted to mice of the H2-d haplotype, and can be observed with both LACK-specific and allospecific CD4+ T cells.
It remains to be elucidated why DCs from different strains differ in their ability to polarize naive CD4+ T cells into cytokine-secreting cells. Previous studies have shown that the Ag dose influences the differentiation of naive CD4+ T cells (27). We found that LN DCs from infected BALB/c and B10.D2 mice expressed similar levels of I-Ad MHC class II molecules. Furthermore, B10.D2 and BALB/c DCs pulsed with relatively high amounts of LACK peptide retained their preferential ability to polarize naive T cells into Th1 and Th2 effector cells, respectively. Thus, the differential ability of BALB/c and B10.D2 DCs to polarize naive CD4+ T cells did not reflect possible differences in the amounts of peptide/MHC complexes expressed at the cell surface. Several pathogens induce DCs to secrete polarizing cytokines such as IL-12 (33). However, in contrast to other pathogens and to L. major amastigotes, L. major promastigotes evade IL-12 induction (34). In agreement with this latter study, L. major promastigotes did not induce DCs to up-regulate IL-12 p35 or p40 mRNA. Furthermore, many cytokine mRNA, including those coding for IFN-γ, GM-CSF, IFN-α, IL-6, IL-1α, TNF-α, TGF-β1, and TGF-β2, were expressed at similar levels in BALB/c and B10.D2 DCs both before and after infection. Thus, as previously suggested (34), infection with L. major promastigotes is rather silent at least in the draining LN on day 2. In contrast to other cytokine mRNA, CD11b+ DCs from B10.D2 mice expressed significantly more IL-1β mRNA than those from BALB/c animals both before and after infection. Although it remains to be determined whether there is a causal relationship between this latter phenomenon and the differential ability of B10.D2 and BALB/c DCs to polarize T cell responses, it is noteworthy that mice deficient for the IL-1 type I receptor (IL-1RI) exhibit reduced Th1 and increased Th2 responses upon infection with L. major (35). Furthermore, we have shown here that BALB/c mice treated with recombinant IL-1β at the time of infection developed a decreased Th2 response and exhibited less parasites than untreated BALB/c mice. Thus, the fact that CD11b+ DCs from B10.D2 mice expressed more IL-1β mRNA than those from BALB/c mice may be one of the parameters accounting for the increased ability of B10.D2 DCs to promote Th1 responses. We have also found that CD11b+ DCs from B10.D2 mice expressed more CD80 than those from BALB/c mice. As interactions between CD80 and CD28 have been shown to promote the development of Th1 cells and to down-regulate Th2 responses (30), it is also possible that the differential expression of CD80 by BALB/c and B10.D2 DCs may contribute to their differential ability to polarize T cells.
Despite a large body of evidence showing that BALB/c (H2-d) and BALB.B (H2-b) mice are more capable than most other strains to mount vigorous Th2 responses upon immunization or infection, the molecular basis for this difference are not known. Although it is difficult to compare the amounts of cytokines produced by TCR transgenic T cells primed in vitro with those produced by polyclonal populations of T cells primed in vivo, we found that the levels of IFN-γ and IL-5 produced in these two experimental settings were quite similar (Fig. 2, A and C, right panel). However, TCR transgenic T cells primed in vitro with BALB/c DCs produced less IL-4 than polyclonal populations of BALB/c T cells primed in vivo. This could be due to the fact that CD4+ T cells from BALB/c mice have an intrinsic ability to produce IL-4 upon stimulation (36). Indeed, previous studies have shown that CD4+ T cells from BALB/c and B10.D2 mice are different (8–10, 37, 38). However, it has not yet been possible to determine whether these differences account for the unusual ability of BALB/c mice to mount a Th2 response in vivo. Whatever the response to this question, we focused on the non-T cell compartment and showed that DCs from BALB/c and BALB.B mice differ from those from B10.D2 and C57BL/6 mice in their ability to polarize CD4+ T cells in vitro. Our results suggest that the intrinsic ability of different individuals to mount Th1- or Th2-dominated responses may be governed by polymorphic genes which are expressed by DCs. Identifying the molecular mechanisms which are at work and the genes which are involved may be valuable for understanding why some individuals are more susceptible than others to specific infectious or autoimmune diseases.
Acknowledgments
We thank researchers at the DNAX Research Institute for Real Time RT PCR reagents.
This work was supported by grants from the Ministère de l'Education Nationale, de la Recherche et de l'Enseignement Supérieur, the EU (contract QLGI-CT-1999-00050), and the Association pour la Recherche contre le Cancer (ARC). SU and SH were supported by fellowships from the Ligue Nationale Contre le Cancer (LNCC).
Footnotes
Abbreviations used in this paper: LC, Langerhans cell; SLA, soluble Leishmania antigens.
References
- 1.Reiner, S.L., and R.M. Locksley. 1995. The regulation of immunity to Leishmania major. Annu. Rev. Immunol. 13:151–177. [DOI] [PubMed] [Google Scholar]
- 2.Beebe, A.M., S. Mauze, N.J. Schork, and R.L. Coffman. 1997. Serial backcross mapping of multiple loci associated with resistance to Leishmania major in mice. Immunity. 6:551–557. [DOI] [PubMed] [Google Scholar]
- 3.Roberts, L.J., T.M. Baldwin, J.M. Curtis, E. Handman, and S.J. Foote. 1997. Resistance to Leishmania major is linked to the H2 region on chromosome 17 and to chromosome 9. J. Exp. Med. 185:1705–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coffman, R.L., and A.M. Beebe. 1998. Genetic control of the T cell response to Leishmania major infection. Adv. Exp. Med. Biol. 452:61–66. [DOI] [PubMed] [Google Scholar]
- 5.Roberts, L.J., T.M. Baldwin, T.P. Speed, E. Handman, and S.J. Foote. 1999. Chromosomes X, 9, and the H2 locus interact epistatically to control Leishmania major infection. Eur. J. Immunol. 29:3047–3050. [DOI] [PubMed] [Google Scholar]
- 6.Shankar, A.H., and R.G. Titus. 1995. T cell and non-T cell compartments can independently determine resistance to Leishmania major. J. Exp. Med. 181:845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bix, M., and R.M. Locksley. 1998. Independent and epigenetic regulation of the IL-4 alleles in CD4+ T cells. Science. 281:1352–1354. [DOI] [PubMed] [Google Scholar]
- 8.Güler, M.L., N.G. Jacobson, U. Gubler, and K.M. Murphy. 1997. T cell genetic background determines maintenance of IL-12 signaling: effects on BALB/c and B10.D2 T helper cell type 1 phenotype development. J. Immunol. 159:1767–1774. [PubMed] [Google Scholar]
- 9.Gorham, J.D., M.L. Guler, and K.M. Murphy. 1997. Genetic control of IL-12 responsiveness: implications for disease pathogenesis. J. Mol. Med. 75:502–511. [DOI] [PubMed] [Google Scholar]
- 10.Güler, M.L., J.D. Gorham, W.F. Dietrich, T.L. Murphy, R.G. Steen, C.A. Parvin, D. Fenoglio, A. Grupe, G. Peltz, and K.M. Murphy. 1999. Tpm1, a locus controlling IL-12 responsiveness, acts by a cell-autonomous mechanism. J. Immunol. 162:1339–1347. [PubMed] [Google Scholar]
- 11.Will, A., C. Blank, M. Rollinghoff, and H. Moll. 1992. Murine epidermal Langerhans cells are potent stimulators of an antigen-specific T cell response to Leishmania major, the cause of cutaneous leishmaniasis. Eur. J. Immunol. 22:1341–1347. [DOI] [PubMed] [Google Scholar]
- 12.Moll, H., H. Fuchs, C. Blank, and M. Rollinghoff. 1993. Langerhans cells transport Leishmania major from the infected skin to the draining lymph node for presentation to antigen-specific T cells. Eur. J. Immunol. 23:1595–1601. [DOI] [PubMed] [Google Scholar]
- 13.Blank, C., H. Fuchs, K. Rappersberger, M. Rollinghoff, and H. Moll. 1993. Parasitism of epidermal Langerhans cells in experimental cutaneous leishmaniasis with Leishmania major. J. Infect. Dis. 167:418–425. [DOI] [PubMed] [Google Scholar]
- 14.von Stebut, E., Y. Belkaid, T. Jakob, D.L. Sacks, and M.C. Udey. 1998. Uptake of Leishmania major amastigotes results in activation and IL-12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J. Exp. Med. 188:1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Stebut, E., Y. Belkaid, B.V. Nguyen, M. Cushing, D.L. Sacks, and M.C. Udey. 2000. Leishmania major-infected murine Langerhans cell-like dendritic cells from susceptible mice release IL-12 after infection and vaccinate against experimental cutaneous leishmaniasis. Eur. J. Immunol. 30:3498–3506. [DOI] [PubMed] [Google Scholar]
- 16.Malherbe, L., C. Filippi, V. Julia, G. Foucras, M. Moro, H. Appel, K. Wucherpfennig, J.C. Guery, and N. Glaichenhaus. 2000. Selective activation and expansion of high-affinity CD4+ T cells in resistant mice upon infection with Leishmania major. Immunity. 13:771–782. [DOI] [PubMed] [Google Scholar]
- 17.Wang, Q., L. Malherbe, D. Zhang, K. Zingler, N. Glaichenhaus, and N. Killeen. 2001. CD4 promotes breadth in the TCR repertoire. J. Immunol. 167:4311–4320. [DOI] [PubMed] [Google Scholar]
- 18.Mougneau, E., F. Altare, A.E. Wakil, S. Zheng, T. Copolla, Z.-E. Wang, R. Waldmann, R. Locksley, and N. Glaichenhaus. 1995. Expression cloning of a Leishmania major protective T cell antigen. Science. 268:563–566. [DOI] [PubMed] [Google Scholar]
- 19.Julia, V., M. Rassoulzadegan, and N. Glaichenhaus. 1996. Resistance to Leishmania major induced by tolerance to a single antigen. Science. 274:421–423. [DOI] [PubMed] [Google Scholar]
- 20.Thery, C., A. Regnault, J. Garin, J. Wolfers, L. Zitvogel, P. Ricciardi-Castagnoli, G. Raposo, and S. Amigorena. 1999. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 147:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holland, P.M., R.D. Abramson, R. Watson, and D.H. Gelfand. 1991. Detection of specific polymerase chain reaction product by utilizing the 5′-3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA. 88:7276–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muller, A., B. Homey, H. Soto, N. Ge, D. Catron, M.E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S.N. Wagner, et al. 2001. Involvement of chemokine receptors in breast cancer metastasis. Nature. 410:50–56. [DOI] [PubMed] [Google Scholar]
- 23.Guéry, J.-C., F. Ria, and L. Adorini. 1996. Dendritic cells but not B cells present antigenic complexes to class II-restricted T cells after administration of protein in adjuvant. J. Exp. Med. 183:751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingulli, E., A. Mondino, A. Khoruts, and M.K. Jenkins. 1997. In vivo detection of dendritic cell antigen presentation to CD4+ T cells. J. Exp. Med. 185:2133–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porgador, A., K.R. Irvine, A. Iwasaki, B.H. Barber, N.P. Restifo, and R.N. Germain. 1998. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J. Exp. Med. 188:1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henri, S., D. Vremec, A. Kamath, J. Waithman, S. Williams, C. Benoist, K. Burnham, S. Saeland, E. Handman, and K. Shortman. 2001. The dendritic cell populations of mouse lymph nodes. J. Immunol. 167:741–748. [DOI] [PubMed] [Google Scholar]
- 27.Constant, S.L., and K. Bottomly. 1997. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu. Rev. Immunol. 15:297–322. [DOI] [PubMed] [Google Scholar]
- 28.Sharpe, A.H., and G.J. Freeman. 2002. The B7-CD28 superfamily. Nat. Rev. Immunol. 2:116–126. [DOI] [PubMed] [Google Scholar]
- 29.Lanier, L.L., S. O'Fallon, C. Somoza, J.H. Phillips, P.S. Linsley, K. Okumura, D. Ito, and M. Azuma. 1995. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J. Immunol. 154:97–105. [PubMed] [Google Scholar]
- 30.Kuchroo, V.K., M.P. Das, J.A. Brown, A.M. Ranger, S.S. Zamvil, R.A. Sobel, H.L. Weiner, N. Nabavi, and L.H. Glimcher. 1995. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 80:707–718. [DOI] [PubMed] [Google Scholar]
- 31.Hugues, S., E. Mougneau, W. Ferlin, D. Jeske, P. Hofman, D. Homann, L. Beaudoin, C. Schrike, M. Von Herrath, A. Lehuen, and N. Glaichenhaus. 2002. Tolerance to islet antigens and prevention from diabetes induced by limited apoptosis of pancreatic Beta cells. Immunity. 16:169–181. [DOI] [PubMed] [Google Scholar]
- 32.Zhao, X., E. Deak, K. Soderberg, M. Linehan, D. Spezzano, J. Zhu, D.M. Knipe, and A. Iwasaki. 2003. Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to Herpes Simplex Virus-2. J. Exp. Med. 197:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reis e Sousa, C. 2001. Dendritic cells as sensors of infection. Immunity. 14:495–498. [DOI] [PubMed] [Google Scholar]
- 34.Reiner, S.L., S. Zheng, Z.E. Wang, L. Stowring, and R.M. Locksley. 1994. Leishmania promastigotes evade interleukin 12 (IL-12) induction by macrophages and stimulate a broad range of cytokines from CD4+ T cells during initiation of infection. J. Exp. Med. 179:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satoskar, A.R., M. Okano, S. Connaughton, A. Raisanen-Sokolwski, J.R. David, and M. Labow. 1998. Enhanced Th2-like responses in IL-1 type 1 receptor-deficient mice. Eur. J. Immunol. 28:2066–2074. [DOI] [PubMed] [Google Scholar]
- 36.Bix, M., Z.E. Wang, B. Thiel, N.J. Schork, and R.M. Locksley. 1998. Genetic regulation of commitment to IL-4 production by a CD4+ T cell-intrinsic mechanism. J. Exp. Med. 188:2289–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Güler, M.L., J.D. Gorham, C.-S. Hsieh, A.J. Mackey, R.G. Steen, W.F. Dietrich, and K.M. Murphy. 1996. Genetic suceptibility to Leishmania: IL-12 responsiveness in Th1 development. Science. 271:984–987. [DOI] [PubMed] [Google Scholar]
- 38.Guler, M.L., J.D. Gorham, W.F. Dietrich, R.G. Steen, C. Parvin, D. Fenoglio, A. Grupe, G. Peltz, and K.M. Murphy. 1999. Loci influencing development of Th responses. Identification from in vitro analysis. Microbes Infect. 1:79–88. [DOI] [PubMed] [Google Scholar]