

Abstract
Flt3 ligand (Flt3L) is a growth factor for hemopoietic progenitors and can promote the expansion of both conventional dendritic cells (DCs) and plasmacytoid predendritic cells (p-preDCs). The cells responding to Flt3L treatment and the precursors for the DCs and p-preDCs had not been fully characterized. We examined different mouse bone marrow (BM) hemopoietic precursor populations for the surface expression of Flt3 and tested them for early DC and p-preDC precursor activity. Most DC precursor activity, other than that given by multipotent hemopoietic stem cells, was within the downstream precursors expressing Flt3. The majority of mouse BM common lymphoid precursors expressed high levels of Flt3 and these were the most efficient precursors of both DCs and p-preDCs. In contrast, only a small proportion of the common myeloid precursors (CMPs) expressed Flt3, but the precursor activity for both DCs and p-preDCs was within this minor Flt3+ CMP fraction. The granulocyte and macrophage precursors and pro-B cells did not express Flt3 and had no DC or p-preDC precursor activity. These findings demonstrate that the early precursors for all DC subtypes are within the BM Flt3+ precursor populations, regardless of their lymphoid or myeloid lineage orientation.
Keywords: dendritic cells, plasmacytoid predendritic cells, Flt3, bone marrow, hemopoietic progenitors
Introduction
The ligand for the receptor kinase fms-like tyrosine kinase 3 (Flt3L)*is a growth factor for hemopoietic progenitors (1–3). Early studies have shown that the receptor for this growth factor, namely Flt3, is expressed by a restricted subset of early hemopoietic progenitors (4–7). Recent studies have demonstrated that up-regulation of Flt3 expression within the mouse BM Lin− Sca-1+ c-kit+ stem cell compartment is associated with the loss of self-renewal capacity and sustained lymphoid-restricted potential (8). Moreover, these Flt3+ Lin− Sca-1+ c-kit+ cells were shown to serve as the precursors of BM common lymphoid precursors (CLPs). Based on these findings, it was suggested that Flt3 expression could be used as a marker to distinguish the short-term reconstituting hemopoietic progenitors from the long-term multipotent hemopoietic stem cells (HSCs; 9).
Recently, the importance of Flt3L in the development of DCs and plasmacytoid predendritic cells (p-preDCs) has been recognized. Mice treated with Flt3L showed dramatic increase in numbers of DCs and p-preDCs in blood and lymphoid tissues (10–16). In addition, mice lacking Flt3L showed deficiency not only in hemopoietic progenitor cells, but also in DCs and natural killer cells (17). Flt3L has been shown to promote the development of DCs in culture, especially from lymphoid precursors (18, 19). Recent studies have also shown that the addition of Flt3L can promote the growth of large number of p-preDCs from mouse BM cultures (20, 21). All above evidence supports an important role of FLt3L in the development of DCs and p-preDCs. It is not clear, however, at which stage of DC and p-preDC development and on what precursor cells Flt3L exerts its function.
Whether conventional DCs and the p-preDCs have a related origin is not clear. Mouse p-preDCs have only been identified recently (16, 22–26). Like their human counterpart, mouse p-preDCs produce large amount of type I IFNs upon stimulation by viruses or microbial stimuli, such as CpG (16, 22–24, 26). Also, like their human counterpart, mouse p-preDCs can produce DCs in culture, and these DCs are of the CD8+ type. However, despite this, p-preDCs are not the normal precursors of CD8+ conventional DCs and apparently only produce DC progeny in vivo on microbial infection (26). Nevertheless, it is generally assumed that DCs and p-preDCs have common progenitors within the BM.
In this study we identified early precursors for mouse DCs and p-preDCs in BM and thymus. Regardless of the lymphoid or myeloid lineage orientation, the precursor activity for DCs and p-preDCs is primarily associated with those BM precursors showing surface expression of Flt3. However, within the thymus there is evidence for a separation of the stream leading to the thymic CD8+ DC and thymic p-preDC because in contrast to BM CLPs, the early T cell precursors can produce DCs but are very poor precursors of p-preDCs.
Materials and Methods
Mice.
C57BL/Ka-Thy-1.1 (Ly 5.2) mice at 6–7 wk of age were used as donors. C57BL/6 Pep3b Ly 5.1 mice at 8–10 wk of age were used as recipients. All mice were bred in the animal facility of The Walter and Eliza Hall Institute under specific pathogen-free conditions.
Antibodies.
The following monoclonal antibodies were used as supernatants for immunomagnetic bead depletion of lineage marker–positive BM cells: anti-CD3 (KT3-1.1), anti-CD8 (53-6.7), anti-CD2 (RM2-1), anti-B220 (RA3-6B2), anti–Mac-1 (M1/70), anti–Gr-1 (RA6-8C5), and anti-erythrocyte antigen (TER-119). The monoclonal antibody supernatants used for depletion in splenic DC and splenic plasmacytoid cell preparations were: anti-CD3 (KT3-1.1), anti-CD19 (ID3), anti-CD90 (T24/31.7), anti–Gr-1 (RA6-8C5), and anti-erythrocyte antigen (TER-119). For thymic DC and thymic plasmacytoid cell preparation, anti-CD11b (M1/70) and anti-macrophage antigen F4/80 (F4/80) were also added to the antibodies used for spleen preparations. Note that the use of anti–Gr-1 did not cause any depletion of plasmacytoid cells in our mice (26).
The following purified antibodies were used as fluorescent conjugates for cell staining and sorting: anti–Ly 5.2 (AL1-4A2) used as an FITC conjugate; anti–Thy-1.1 (19XE5) used as an FITC conjugate; anti–FcγRII/III (anti–CD16/32[2.4G2]) used as an FITC conjugate; anti-CD19 (ID3) used as an FITC conjugate; anti-Flt3 (A2F10.1) used as a PE conjugate; anti–c-kit (2B8) used as an allophycocyanin (APC) conjugate; anti–Sca-1 (E13–161–7) and anti–IL-7Rα (A7R34) used as Alexa 594 conjugates; and anti–IL-7Rα (A7R34) and anti-CD34 (RAM34) used as biotin conjugates. Anti–rat immunoglobulin–Texas Red and PE–avidin were used for second stage staining. The following fluorescent conjugated antibodies were used for staining the DCs: anti–MHC class II–FITC (M5/114), anti–CD11c–Alexa 594 (N418), anti–CD4-PE (GK1.5), anti–CD8-Cy5 (YTS 169.4), and anti–DEC-205–biotin (NLDC-145) with PE-avidin used as the second stage.
Isolation of Precursor Populations.
The early intrathymic lymphoid precursor population (CD3− CD8− CD4low c-kit+ CD25− Thy-1low) was purified by means of a previously described procedure (27). The BM precursor populations were isolated by procedures described elsewhere (28). In brief, the CLP population from mouse BM was purified by immunomagnetic bead depletion of lineage marker–positive cells, followed by enrichment sorting for Sca-1low/+ cells. These enrichment-sorted cells were stained with anti–Thy-1.1–FITC, anti–c-kit–APC, anti–Sca-1–Alexa 594, and anti–IL-7Rα–biotin, followed by PE-avidin as the second stage. CLPs were then sorted as IL-7Rα+ Sca-1int c-kitint Thy-1.1− cells on a FACstarPLUS™ instrument (Becton Dickinson). The myeloid-committed precursor populations from BM were isolated by first depleting lineage marker–positive cells by means of immunomagnetic beads. The remaining cells were then stained with goat anti–rat immunoglobulin Texas Red and anti–c-kit–APC and then enrichment sorted for lin− c-kit+ cells. The enrichment-sorted cells were then stained with anti-FcRγII/III (CD16/32)–FITC, anti–c-kit–APC, anti–IL-7Rα–Alexa 594, anti–Sca-1–Alexa 594, and anti–CD34-biotin, followed by PE-avidin. The common myeloid precursor (CMP) population was sorted as CD16/32low CD34+ c-kit+ Sca-1− IL-7Rα− cells. The granulocyte and macrophage precursor (GMP) population was sorted as CD16/32+ CD34+ c-kit+ Sca-1− IL-7Rα− cells. Further division of the CMP population by Flt3 expression was performed by staining the enrichment-sorted lin− c-kit+ cells with anti–CD16/32-FITC, anti–Flt3-PE, anti–c-kit–APC, anti–IL-7Rα–Alexa 594, and anti–Sca-1–Alexa 594, and then sorting for Flt-3+ CD16/32low c-kit+ Sca-1− IL-7Rα− and Flt3− CD16/32low c-kit+ Sca-1− IL-7Rα− fractions was performed. The BM pro-B cells were purified by immunomagnetic bead depletion of lineage marker–positive (except B220+) cells, and then stained with anti–CD19-FITC, anti–B220-PE, and anti–c-kit–APC, and sorted for CD19+ B220+ c-kit+ cells. The purity of sorted cells was determined by reanalyzing a small sample of the collected cells and was usually >97%.
In Vivo Assays for p-preDC and DC Production by Different Precursor Populations.
To examine the capacity for p-preDC and DC production, purified precursor cells (1–4 × 104) from C57BL Ly5.2 mice were i.v. injected into lethally irradiated (5.5 Gy twice with a 3-h interval) C57BL Ly5.1 recipients, along with 5 × 104 recipient type Ly 5.1 unfractionated BM cells to ensure mouse survival. At various times after precursor transfer, the thymus and spleens of recipients were collected, and DCs and p-preDCs were enriched from these tissues by light density separation and then immunomagnetic bead depletion using the procedure described elsewhere (26). These DC and p-preDC–enriched preparations were then stained in four fluorescent colors using conjugated antibodies to Ly 5.2 to reveal donor-derived cells, together with conjugated antibodies to other markers expressed by DCs and p-preDCs. These included pan-DC markers CD11c and MHC class II, p-preDC marker CD45RA, and DC subset–specific markers CD4, CD8α, DEC-205, and CD11b. Donor-derived DCs and p-preDCs were revealed by electronic gating for Ly5.2+ CD45RA− CD11c+ and Ly5.2+ CD45RA+ CD11cint cells, respectively, during flow cytometric analysis. The expression of other DC markers by these donor-derived cells was further analyzed and the proportion of the individual donor-derived DC subpopulations was then determined.
Results
Flt3 Expression on BM Cells and on Different Hemopoietic Progenitor Populations.
Based on the fact that both in vivo and in vitro treatment with Flt3L drastically increase the number of DCs and p-preDCs in mouse and human (10–15), we proposed that the precursors with the potential to generate DCs and p-preDCs express the receptor for Flt3L. Therefore, we examined the surface Flt3 expression on BM cells and on different defined hemopoietic precursor populations using multicolor flow cytometric analysis. We used PE-conjugated anti-Flt3 antibody together with a range of fluorochrome-conjugated antibodies to other cell surface markers for distinguishing different hemopoietic cell lineages and different precursor populations. The different hemopoietic lineage cells were distinguished as B cells (B220+ CD19+), myeloid cells, (Mac-1+ Gr-1+), and T cells (CD3+). For distinguishing the BM precursor populations, we used the procedures established in the Stanford laboratories (29, 30) and in this laboratory (28), which defined the precursor populations as: multipotent HSCs, Lin− Sca-1+ c-kithi; the CLPs, Lin− IL-7Rα+ Sca-1lo c-kitlo; the CMPs, Lin− IL-7Rα− Sca-1− c-kithi CD34+ CD16/32lo; and GMPs, Lin− IL-7Rα− Sca-1− c-kithi CD34+CD16/32hi. The surface expression of Flt3 on the more committed precursors for T and B lymphoid cells was also examined. These precursors were distinguished as: intrathymic lymphoid precursors (CD4lo precursors), Lin− CD3− 8− 25− 4lo Thy-1lo c-kit+; and BM pro-B cells, Lin− CD19+ B220+ c-kit+. The results are shown in Fig. 1 .
Figure 1.

Surface expression of Flt3 by mouse BM cells and by different hemopoietic precursor populations. Total BM cells were stained with PE-conjugated anti-Flt3 antibody together with other fluorescence-conjugated antibodies against different surface molecules expressed by BM cells including CD19, B220, CD3, Mac-1, Gr-1, MHC class II, and c-kit. The expressions of these molecules on the gated Flt3+ BM cells are shown in A. The solid lines represent the staining of different surface molecules and the dashed lines represent the isotype controls. The precursor populations from mouse BM or thymus were first enriched for lineage marker–negative cells and then distinguished by four color fluorescence staining as (B) CLP (c-kit+ CD127+), (C) CMP (CD34+ CD16/32lo) and GMP (CD34+ CD16/32hi), (D) intrathymic CD4lo lymphoid precursors (c-kit+ Thy-1lo), and (E) BM pro-B precursors (c-kit+ CD19+). The surface expression of Flt3 by each precursor population was then examined. The contour plots display the gating for each precursor population (cells within the boxes). The histograms show the Flt3 expression on each gated precursor population. The solid lines represent the Flt3 staining and the dashed lines represent the isotype controls. Data presented in this figure is representative of at least three similar experiments on each precursor population.
First, the expression of Flt3 by different hemopoietic lineage cells in the BM was examined. Only a small proportion of total BM cells (2.0–2.5%) was found expressing Flt3, and these Flt3+ cells were mainly within the lineage marker–negative (CD19− Mac-1− Gr-1− CD3−, Lin−) and MHC class II−cell fraction (Fig. 1 A). In addition, we found that the majority of the Flt3+ BM cells (65–70%) was within the Lin− c-kit+ fraction. Interestingly, a small fraction of these BM Flt3+ cells (25–30%) expressed B220 and relatively lower levels of Flt3 (Fig. 1 A). These Flt3+ B220+ cells did not express CD19, MHC class II, Mac-1, Gr-1, and c-kit (unpublished data), indicating that they were neither B lineage cells nor early precursors. Overall, these results demonstrated that the BM Flt3+ cells were mainly amongst the Lin− c-kit+ precursor populations of the BM.
Because most of the early hemopoietic precursor populations are within the BM Lin− c-kit+ fraction, for further characterizing these Lin− Flt3+ c-kit+ BM cells, we also examined the expression of Flt3 on different early hemopoietic precursor populations. As we expected, the majority (66–75%) of BM CLPs, the most efficient precursors for both lymphoid cells and DCs (28, 31), expressed high levels of Flt3 (Fig. 1 B). Interestingly and in contrast, only a small fraction (22–25%) of the BM CMPs expressed Flt3, the majority being Flt3− (Fig. 1 C). The more committed GMPs, the least efficient precursors for DCs (28), did not express detectable levels of Flt3 on the surface (Fig. 1 C). When the more committed precursors for T and B lymphoid lineages were examined, only 10–13% of the intrathymic CD4lo precursors expressed Flt3, and this was a low level of expression (Fig. 1 D). Only a few pro-B cells (<3%) expressed low levels of Flt3 (Fig. 1 E). These results demonstrate that Flt3 was mainly expressed by the early hemopoietic progenitors that were not yet restricted to the development of a single hemopoietic lineage.
The Flt3+ BM Cells Contain Precursors for DCs and p-preDCs.
Because Flt3 was expressed by a minor population of BM cells as described above, it would be interesting to test whether these Flt3+ BM cells contain precursors for DCs and p-preDCs. Therefore, we tested Flt3+ and Flt3− fractions of BM cells for their DC and p-preDC precursor activity. The Flt3+ and Flt3− populations were purified from the BM of Ly5.2 mice and transferred (2–5 × 104) i.v. together with the host-type BM cells (5 × 104) into lethally irradiated Ly5.1 recipient mice. The Ly5.2+ donor-derived DCs and p-preDCs were then examined at different times after cell transfer. As shown in Fig. 2 and Table I , both Flt3+ and Flt3− BM cells could generate DCs and p-preDCs in the recipient spleen 2 wk after cell transfer. However, on a per cell basis, the Flt3+ BM cells were much more efficient (20–30-fold) than the Flt3− BM cell in generating DCs and p-preDCs (Table I). These results indicated that the DC and p-preDC precursor activity was enriched in the Flt3+ BM cell fraction. The lower DC and p-preDC precursor activity of the Flt3− BM cell fraction most likely originated from the few self-renewable HSCs contained within the Flt3− fraction (8). Although the Flt3+ BM cells were enriched for DC precursor activity, they could also generate T cells, B cells, and myeloid cells (unpublished data) and therefore were not DC-restricted precursors.
Figure 2.

Generation of DCs by BM Flt3+ and Flt3− cell populations. BM cells from Ly5.2 mice were stained with PE-conjugated Flt3 antibody. The Flt3+ and Flt3− cell fractions were purified by cytometric sorting. The purified cells were then i.v. transferred into lethally irradiated Ly5.1 recipient mice. The generation of DCs in the spleen of recipient mice was examined 2 wk after cell transfer. The donor-derived DCs were distinguished as Ly5.2+ CD11c+ cells from the DC-enriched spleen cell preparation. The data presented is typical of two such experiments.
Table I.
Numbers of DCs Generated by Flt3+ and Flt3− BM Cell Populations
| Precursor populations (104) |
||||
|---|---|---|---|---|
| Flt3−BM | Flt3+BM | Flt3+B220+ | Flt3+B220− | |
| Splenic DCs (×103) |
6.3–12.4 | 227.2–262.6 | 21.0–27.9 | 278.7–321.3 |
| Splenic p-preDCs (×103) |
5.0–7.5 | 103.1–148.5 | 19.4–25.2 | 124.0–179.3 |
The precursor populations were purified by FACS® sorting and transferred i.v. into the irradiated Ly 5.1 recipient mice. 2 wk after precursor transfer, the recipient spleens were analyzed for donor-derived DCs and p-preDCs. The data presented are the ranges of donor-derived DC and p-preDC numbers generated by 104 transferred BM cell populations from two experiments. Each experimental group contained two to three recipient mice.
Because a small fraction of the Flt3+ BM cells expressed B220 (Fig. 1 A), we also tested the Flt3+ B220+ and the Flt3+ B220− populations, respectively, for their capacity to produce DCs. As shown in Table I, both B220+ and B220− fractions of the Flt3+ BM cells contained precursors of DCs. However, the Flt3+ B220− cells were more efficient than the Flt3+ B220+ cells in generating DCs (Table I). In addition to DCs, the Flt3+ B220− cells were also able to produce other hemopoietic cell lineages including B cells, T cells, and myeloid cells, whereas the Flt3+ B220+ cells only produced a few B cells and myeloid cells (unpublished data).
The results shown above, together with the finding that the majority of the Flt3+ B220− cells was also Lin− c-kit+ (Fig. 1 A), suggest that the DC precursors were mainly contained within the Lin− Flt3+ B220− c-kit+ BM precursor population. Because most of the early hemopoietic precursor populations (including CLP, CMP, and GMP) are within the Lin− c-kit+ fraction of the BM and some of these precursors expressed surface Flt3 (Fig. 1, B and C), we examined the possible correlation of Flt3 expression with the capacity to generate DCs by these precursor populations.
DC Precursor Activities Are Enriched in the Flt3+ Fraction of CMPs.
As described above, the BM CMP population appeared to be heterogeneous and separable into two fractions based on the surface expression of Flt3. Because previous studies (28, 31, 32) have shown that CMPs are able to generate DCs in vivo, it therefore became interesting to examine which of these CMP fractions contained the DC precursor activity. To test this, the Flt3+ and Flt3− fractions of CMPs were first purified and sorted from the BM of Ly5.2 mice. A purity of 97–98% was obtained. The purified precursor populations (1–3 × 104) together with host-type BM cells (5 × 104) were injected i.v. into lethally irradiated Ly5.1 recipient mice. The Ly 5.2+ DC generated from the Flt3+ and Flt3− CMP fractions was examined at different times after injection. As shown in Fig. 3 A and Table II , the minor Flt3+ fraction of CMPs was highly efficient in DC production. In contrast, the major Flt3− CMPs produced only a small number of DCs, although it retained ability to produce granulocytes and macrophages (Fig. 3 B and Table II). Similar results were obtained from all the time points analyzed (unpublished data). These observations indicated that the DC precursor activity of CMPs was highly enriched in the Flt3+ fraction, whereas the Flt3− CMPs had very poor DC precursor activity.
Figure 3.
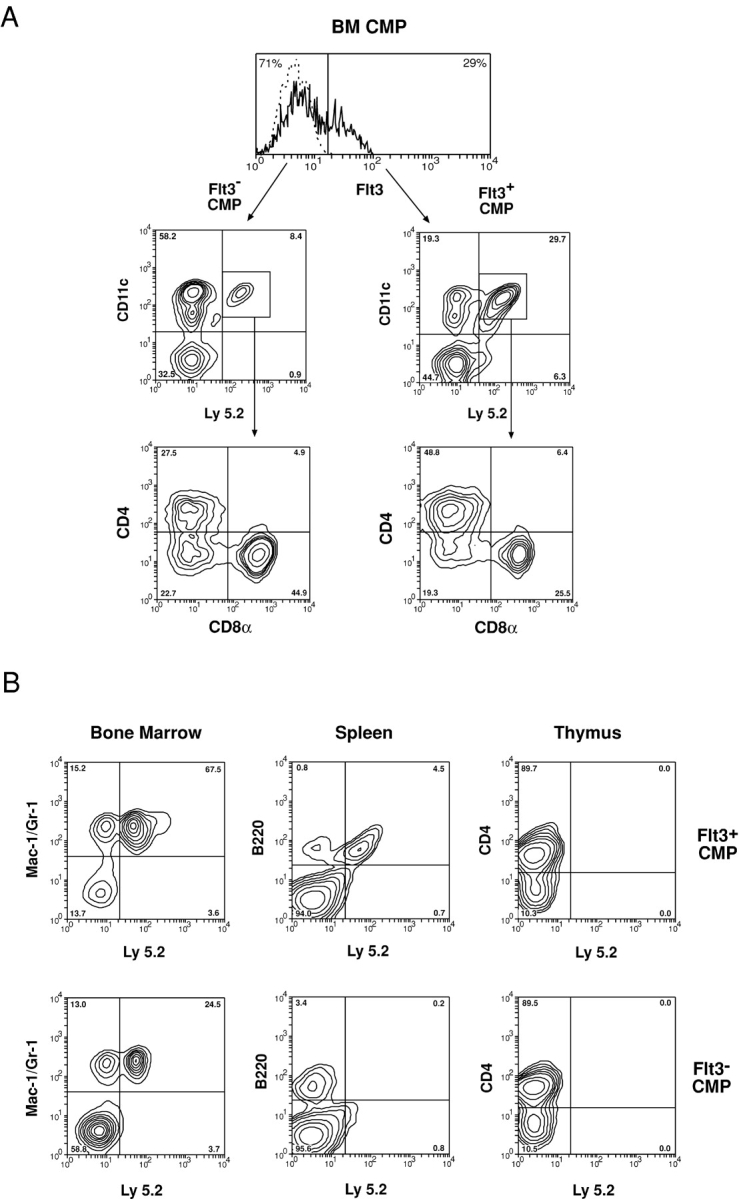
Generation of DCs and other hemopoietic lineage cells by Flt3+ and Flt3− fractions of CMPs. The Flt3+ and Flt3− fractions of CMPs were purified from the BM of Ly5.2 mice and i.v. transferred to lethally irradiated Ly5.1 recipient mice. The donor-derived cells were distinguished as Ly5.2+ cells. For DC production, recipient spleens were analyzed 2 wk after precursor transfer. Donor-derived DCs were identified as Ly5.2+ CD11c+ cells from DC-enriched spleen cell preparations (A). The subsets of donor-derived DCs were further segregated by their expression of CD4 and CD8 (A). For myeloid cell production, the recipient BM cells were analyzed 1 wk after precursor transfer for donor-derived Ly5.2+ Mac-1+ Gr-1+ cells (B). For B cell and T cell production, the recipient spleen and thymus were analyzed 2 wk after precursor transfer for donor-derived Ly5.2+ B220+ or Ly5.2+ CD4+ cells (B). The data presented is typical of three such experiments. Each experimental group contained two to three recipient mice.
Table II.
Numbers of DCs, Myeloid Cells, and Lymphoid Cells Generated by Flt3+ and Flt3− Fractions of CMPs
| Splenic DCs(×106) | BMmyeloid cells(×106) | Spleen B cells(×106) |
Thymic T cells(×106) |
|
|---|---|---|---|---|
| Flt3+
CMPs (104) |
0.27 ± 0.04 | 0.26 ± 0.09 | 3.70 ± 0.50 | <0.1 |
| Flt3−
CMPs (104) |
0.04 ± 0.01 | 0.12 ± 0.02 | <0.1 | <0.1 |
The Flt3+ and Flt3− fractions of CMPs were purified and i.v. transferred to lethally irradiated Ly5.1 recipient mice. For DC production by the precursors, splenic DCs were analyzed 2 wk after precursor transfer. For myeloid cell production, the BM cells were prepared 1 wk after precursor transfer from the femoral and tibial bones of the recipient mice and analyzed for donor-derived Mac-1+ and Gr-1+ cells. For B cell and T cell production, the recipient spleen and thymus were analyzed 2 wk after precursor transfer. The cell numbers presented are the mean ± standard error from three experiments. Each experimental group contained two to three recipient mice.
To determine whether the differences in DC precursor activity between the Flt3+ and Flt3− fractions of CMPs were simply quantitative, or whether there was segregation of the precursors for different DC subpopulations, we further analyzed the types of DCs generated. As shown in Fig. 3 A, 2 wk after injection, all three DC populations identified in mouse spleen, namely the CD4+ 8−, CD4− 8−, and CD4− 8+ DCs, were found within the large number of DCs generated from Flt3+ CMPs as well as within the very small number of DCs generated from Flt3− CMPs.
The Flt3+ CMP Contains Precursors for Both B Lineage Cells and Myeloid Cells.
It has been reported that although most of the cells within the CMP fraction are myeloid-restricted precursors, some residual B cell precursor activity can still be detected (30). We obtained similar findings. In an attempt to distinguish the cells with B cell precursor activity from those fully restricted to myeloid development, we tested the Flt3+ and Flt3− fractions of CMPs for precursors of hemopoietic lineages other than myeloid and DC. The purified Flt3+ and Flt3− CMPs were injected i.v. into lethally irradiated Ly 5 disparate recipient mice. The BM of the recipients was analyzed 1 wk after precursor transfer for donor-derived myeloid cells, and the thymus and spleen of the recipient mice were analyzed 2 wk after precursor transfer for donor-derived T and B lineage cells. These were determined to be the peak times for the responses analyzed. The results are presented in Fig. 3 B and Table II.
Interestingly, the Flt3+ fraction of CMPs displayed an enriched precursor activity for B lineage cells, but no precursor activity for T cells was detected (Fig. 3 B and Table II). In contrast, the Flt3− fraction of CMPs showed no precursor activities for either B cells or T cells (Fig. 3 B and Table II). However, both the Flt3+ and Flt3− fractions of CMPs showed precursor activity for granulocytes and macrophages, so there was no segregation of the capacity to form myeloid cells (Fig. 3 B and Table II). Thus, the originally described BM CMP was a heterogeneous population, the Flt3+ subgroup of CMPs containing precursors of DCs and B cells as well as of myeloid cells, whereas only the Flt3− fraction contained precursors fully committed to myeloid development.
The Correlation of DC Precursor Activity with the Level of Flt3 Expression on Different Precursor Populations.
Because different hemopoietic precursor populations expressed different levels of surface Flt3, it was interesting to determine if this correlated with their capacity to produce DCs. To examine this, purified BM CLPs, CMPs (unseparated), Flt3+ CMPs, Flt3− CMPs, GMPs, thymic CD4lo precursors, and BM pro-B cells were transferred i.v. into lethally irradiated recipients. 2 wk after precursor transfer, the thymus and spleen of the recipient mice were analyzed for donor-derived DCs. As shown in Fig.4 , the CLP population, in which the majority of cells expressed high levels of Flt3, was the most efficient DC precursor population. Almost all of the DC precursor activity was within the Flt3+ CLP fraction, whereas the minor Flt3− CLP fraction produced very few DCs (<5 × 103 for 104 transferred Flt3− CLP, in contrast to 350 × 103 for the Flt3+ CLP) as well as some B cells, but not myeloid cells (unpublished data), suggesting that the Flt3− CLP may represent a small fraction of cells at a relatively later developmental stage within the CLP population. Flt3+ CMPs generated DCs with an efficiency that was lower than that of CLPs, but much higher than the unseparated CMPs and Flt3− CMPs. GMPs did not express detectable levels of Flt3 and showed very poor DC precursor activity. A proportion of the thymic CD4lo precursors expressed low levels of Flt3 and these lymphoid precursors displayed a lower DC precursor activity than either CLPs or Flt3+ CMPs. Only a few pro-B cells expressed low levels of Flt3 and these pro-B cells did not show any precursor activity for DCs. These findings demonstrate a general association of Flt3 expression with DC precursor potential.
Figure 4.

Comparison of the generation of DCs by different hemopoietic precursor populations. Different precursor populations were purified from C57BL/6 Ly5.2+ mice by FACS® sorting and transferred i.v. into irradiated Ly5.1 recipient mice. 2 wk after precursor transfer, the recipient thymus and spleen were analyzed for donor-derived DCs. Each bar represents the average DC number produced by 104 precursor cells from three experiments. Each experimental group contained two to three recipient mice.
BM CLPs and the Flt3+ Fraction of CMPs Are Potent Precursors of p-preDCs.
The mouse p-preDCs were identified recently (16, 22–26). However, the early precursors for these p-preDCs have not yet been identified and characterized. Flt3L treatment of mice increased the numbers of DCs and p-preDCs in blood and lymphoid tissues (10–16), and the addition of Flt3L alone efficiently promoted p-preDC development in mouse BM cultures (20, 21). These findings suggested that the precursors of p-preDCs should express the receptor for Flt3L. To test this, purified BM precursor populations were transferred i.v. into irradiated recipients and the recipient thymus and spleen were then analyzed for the donor-derived p-preDCs (Ly5.2+ CD11cint CD45RA+) at different times after precursor transfer. The peak of p-preDC production was at 2 wk after transfer. Results at this time point are presented in Figs. 5 and 6 and Table III , but the same relative p-preDC precursor activity was obtained at 1, 2, and 3 wk after transfer.
Figure 5.

Generation of p-preDCs by different hemopoietic precursor populations. Different BM precursor populations were purified from C57BL/6 Ly5.2 mice and i.v. transferred to lethally irradiated Ly5.1 recipient mice. 2 wk after precursor transfer, recipient spleens were analyzed for donor-derived p-preDCs. The DC-enriched cell preparations were stained with anti–Ly5.2-FITC, anti–CD45RA-PE, and anti-CD11c-Alexa 594. The donor-derived cells were distinguished as Ly5.2+ cells (top). The donor-derived p-preDCs were identified as CD45RA+ CD11cint cells within the gated Ly5.2+ cells (bottom, cells within the boxes). The data presented is representative of three similar experiments on each precursor population. Each experimental group contained two to three recipient mice.
Figure 6.

Comparison of the generation of p-preDCs by different hemopoietic precursor populations. Different precursor populations were purified from C57BL/6 Ly5.2+ mice by FACS® sorting and transferred i.v. into irradiated Ly5.1 recipient mice. 2 wk after precursor transfer, the recipient thymus and spleen were analyzed for donor-derived p-preDCs. Each bar represents the average number of p-preDCs produced by 104 precursor cells from three experiments. Each experimental group contained two to three recipient mice.
Table III.
Generation of p-preDCs by Different Precursor Populations
| Precursor population (104) |
||||||
|---|---|---|---|---|---|---|
| CLP | Flt3+ CMP | Flt3− CMP | GMP | Thymic CD4lo | Pro-B | |
| Thymic p-preDCs (×103) |
1.60 ± 0.15 | 0.55 ± 0.08 | 0.20 ± 0.01 | 0.03 ± 0.01 | 0.22 ± 0.05 | <0.1 |
| Splenic p-preDCs (×103) |
75.7 ± 1.75 | 49.0 ± 8.20 | 2.81 ± 0.45 | 0.80 ± 0.08 | 2.21 ± 0.35 | <0.1 |
The precursor populations were purified by FACS® sorting and transferred i.v. into the irradiated Ly 5.1 recipient mice. 2 wk after precursor transfer, the recipient thymus and spleen were analyzed for donor-derived p-preDCs. The numbers of p-preDCs presented are the mean ± standard error from three experiments. Each experimental group contained two to three recipient mice.
BM CLPs, which expressed high levels of Flt3 and were the most potent precursors for DCs, were also found to be the most efficient precursors for p-preDCs (Figs. 4 and 5 and Table III). The Flt3+ CMPs could also efficiently produce p-preDCs in both the thymus and spleen of the recipients, although with relatively lower efficiency than the CLPs, whereas the Flt3− CMPs showed very poor if any p-preDC precursor activity (Figs. 5 and 6 and Table III). The more committed myeloid precursor GMP showed almost no precursor activity for p-preDCs (Figs. 5 and 6 and Table III). Nor did BM pro-B cells display any p-preDC precursor activity. Thus, these BM precursor populations showed a similar hierarchy of precursor activities for DCs and p-preDCs, and both were within the Flt3+ populations.
The Thymic CD4lo Precursor Population Displays DC but Not p-preDC Precursor Activity.
The results described above for BM precursor populations suggest that DC and p-preDC precursor activity resides in the same precursor populations, both being correlated with populations expressing surface Flt3. As shown in this and our previous studies (27, 28), a small proportion of the intrathymic CD4lo precursors express low levels of Flt3 and this population is capable of producing DCs, although with a lower efficiency than the BM CLPs and the Flt3+ CMPs (Fig. 4). Therefore, we tested whether thymic p-preDCs could also be generated within thymus by the thymic CD4lo precursors. To our surprise, the thymic CD4lo precursors showed relatively little precursor activity for either thymic or splenic p-preDCs (Fig. 6 and Table III), in marked contrast to their ability to produce DCs (Fig. 4). This marked differential in DCs compared with p-preDC production was sustained at all the time points from 1–3 wk after precursor transfer (unpublished data).
Discussion
This study was prompted by the finding that the cytokine tyrosine kinase receptor Flt3 is expressed mainly on early hemopoietic progenitors and that Flt3L promotes the development of both DCs and p-preDCs. Accordingly, we examined the expression of Flt3 on the surface of total BM cells and on different mouse hemopoietic precursor cells, and then correlated this with their capacity to form DCs and p-preDCs. The majority of BM Flt3+ cells were within the Lin− c-kit+ fraction of BM that contains the established CLP and CMP populations. Our study showed that the DC precursor activity was enriched in this Flt3+ BM cell fraction. Our further studies revealed high levels of Flt3 expression on BM CLPs but a bimodal expression of Flt3 on CMPs, so the CMPs were not a homogeneous population. Other downstream precursors had much lower levels of Flt3 expression.
Our central finding is that these differences in Flt3 expression show a general correlation with the capacity to produce DCs and p-preDCs. The point of particular interest is the CMPs, because of the bimodal Flt3 expression and reports that they could produce both DCs and some B cells (28, 30–32). In fact, both the B cell and the DC and p-preDC generation capacity were concentrated in the minor Flt3+ fraction. Thus, the CMP fraction actually had two distinct components, the major Flt3− fraction was clearly myeloid restricted and a very poor DC progenitor, and a minor Flt3+ fraction that was oligopotent. It was of interest that this Flt3+ fraction, although unable to form T cells, was capable of B cell production as well as DC and p-preDC production, but the efficiency in B cell production was >20-fold lower than that of CLPs. Elsewhere we have shown that thymic DCs and p-preDCs, but not splenic conventional DCs, do have a lymphoid link in that they have D-J rearrangements at the IgH gene locus (33). A developmental linkage of B lineage cells and DCs has also been suggested (34). However, the DC precursor activity appears to branch off before the pro-B cells stage. Consistent with this, we found that the Flt3− pro-B cells were completely devoid of a capacity to form either DCs or p-preDCs, suggesting that the p-preDC precursor activity also branches off between the stages of CLPs to pro-B cells.
It was interesting that the ability of BM precursors to form both DCs and p-preDCs cut across the myeloid versus lymphoid precursor segregation. In fact, DC precursor potential was best defined by the levels of expression of Flt3 rather than the established myeloid or lymphoid precursor makers. At this level the same precursor in BM generates all DC subtypes and p-preDCs, with similar kinetics. However, a separation of the pathways leading to thymic DCs and thymic p-preDCs has clearly occurred at some point downstream because the earliest T precursor within the thymus, the “lymphoid-restricted” CD4lo precursor, retains a capacity to form DCs but has very little capacity to form p-preDCs. It is not clear whether this separation of pathways occurred before or after thymus seeding.
Interestingly, in this study we found that a small proportion of the Flt3+ BM cells also expressed B220 and low levels of CD11c (unpublished data), but was negative for CD19, MHC class II, and c-kit, a phenotype similar to that of recently described “common DC precursors” in mouse blood (35). However, the DC precursor activity of this Flt3+ B220+ population was much lower than that of other Flt3+ BM precursor populations. These cells could also generate some B cells and a few myeloid cells upon i.v. transfer (unpublished data) and therefore were not DC-restricted precursors. Whether this cell population contains separate precursors for DCs, B cells, and myeloid cells is yet to be clarified. We could not exclude the possibility that this cell population also includes some already formed B220+ CD11clo p-preDCs. Currently, the relationship of this BM Lin− Flt3+ B220+ precursor population to the blood common DC precursors remains unclear.
Overall, our study suggests a distinct developmental pathway for DCs and p-preDCs, with the precursors in BM defined by the surface expression of Flt3 (Fig. 7) . However, the developmental pathways leading to both DCs and p-preDCs downstream of these early Flt3+ BM precursors remain to be mapped. In particular, the relationship of these early Flt3+ DC and p-preDC precursors to the recently described “common DC precursors” (35) or to “preimmunocytes” (22) is yet to be clarified.
Figure 7.

The proposed developmental pathways for DCs and p-preDCs. Both conventional DCs and p-preDCs can develop from BM Flt3+ precursors regardless of whether this involves the “myeloid pathway” or the “lymphoid pathway.” The relationship of the Flt3+ lymphoid or myeloid precursors to the recently described common DC precursors currently remains unknown. HSC, hemopoietic stem cells; HPC, multipotent hemopoietic progenitor cells; CLP, common lymphoid precursors; CMP, common myeloid precursors; GMP, granulocyte and macrophage precursors; pro-T, intrathymic CD4lo precursors; pro-B, B220+ CD19+ c-kit+ B cell precursors. The most efficient precursors of DCs and p-preDCs are the Flt3-expressing BM precursors, shown as cells within the boxes. The thickness of the lines is relative to the efficiency of the precursors in producing DCs or p-preDCs. The dashed lines represent proposed developmental pathways yet to be confirmed by direct evidence.
Acknowledgments
The authors wish to thank Drs. F. Battye, D. Kaminaris, V. Lapatis, C. Tarlinton, C. Clark, and A. Holloway for their assistance with flow cytometry analysis and sorting. We thank Prof. K. Shortman for critical reading of this manuscript.
This work is supported by an Australian National Health and Medical Research Council Program grant. L. Wu is a special scholar of the Cheung Kong Scholars Program of The Ministry of Education, People's Republic of China.
Footnotes
Abbreviations used in this paper: APC, allophycocyanin; CLP, common lymphoid precursor; CMP, common myeloid precursor; Flt3L, Flt3 ligand; GMP, granulocyte and macrophage precursor; HSC, hemopoietic stem cell; p-preDC, plasmacytoid predendritic cell.
References
- 1.Lyman, S.D., L. James, T. Vanden Bos, P. de Vries, K. Brasel, B. Gliniak, L.T. Hollingsworth, K.S. Picha, H.J. McKenna, R.R. Splett, et al. 1993. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell. 75:1157–1167. [DOI] [PubMed] [Google Scholar]
- 2.Hannum, C., J. Culpepper, D. Campbell, T. McClanahan, S. Zurawski, J.F. Bazan, R. Kastelein, S. Hudak, J. Wagner, J. Mattson, et al. 1994. Ligand for FLT3/FLK2 receptor tyrosine kinase regulates growth of haematopoietic stem cells and is encoded by variant RNAs. Nature. 368:643–648. [DOI] [PubMed] [Google Scholar]
- 3.Gilliland, D.G., and J.D. Griffin. 2002. The roles of FLT3 in hematopoiesis and leukemia. Blood. 100:1532–1542. [DOI] [PubMed] [Google Scholar]
- 4.Rasko, J.E., D. Metcalf, M.T. Rossner, C.G. Begley, and N.A. Nicola. 1995. The flt3/flk-2 ligand: receptor distribution and action on murine haemopoietic cell survival and proliferation. Leukemia. 9:2058–2066. [PubMed] [Google Scholar]
- 5.Brasel, K., S. Escobar, R. Anderberg, P. de Vries, H.J. Gruss, and S.D. Lyman. 1995. Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia. 9:1212–1218. [PubMed] [Google Scholar]
- 6.Turner, A.M., N.L. Lin, S. Issarachai, S.D. Lyman, and V.C. Broudy. 1996. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood. 88:3383–3390. [PubMed] [Google Scholar]
- 7.Rosnet, O., H.J. Buhring, S. Marchetto, I. Rappold, C. Lavagna, D. Sainty, C. Arnoulet, C. Chabannon, L. Kanz, C. Hannum, et al. 1996. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia. 10:238–248. [PubMed] [Google Scholar]
- 8.Adolfsson, J., O.J. Borge, D. Bryder, K. Theilgaard-Monch, I. Astrand-Grundstrom, E. Sitnicka, Y. Sasaki, and S.E. Jacobsen. 2001. Upregulation of Flt3 expression within the bone marrow Lin(−)Sca1(+)c-kit(+) stem cell compartment is accompanied by loss of self-renewal capacity. Immunity. 15:659–669. [DOI] [PubMed] [Google Scholar]
- 9.Christensen, J.L., and I.L. Weissman. 2001. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc. Natl. Acad. Sci. USA. 98:14541–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maraskovsky, E., K. Brasel, M. Teepe, E.R. Roux, S.D. Lyman, K. Shortman, and H.J. McKenna. 1996. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand–treated mice: multiple dendritic cell subpopulations identified. J. Exp. Med. 184:1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drakes, M.L., L. Lu, V.M. Subbotin, and A.W. Thomson. 1997. In vivo administration of flt3 ligand markedly stimulates generation of dendritic cell progenitors from mouse liver. J. Immunol. 159:4268–4278. [PubMed] [Google Scholar]
- 12.Lynch, D.H. 1998. Induction of dendritic cells (DC) by Flt3 Ligand (FL) promotes the generation of tumor-specific immune responses in vivo. Crit. Rev. Immunol. 18:99–107. [DOI] [PubMed] [Google Scholar]
- 13.Maraskovsky, E., E. Daro, E. Roux, M. Teepe, C.R. Maliszewski, J. Hoek, D. Caron, M.E. Lebsack, and H.J. McKenna. 2000. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood. 96:878–884. [PubMed] [Google Scholar]
- 14.McKenna, H.J. 2001. Role of hematopoietic growth factors/flt3 ligand in expansion and regulation of dendritic cells. Curr. Opin. Hematol. 8:149–154. [DOI] [PubMed] [Google Scholar]
- 15.O'Keeffe, M., H. Hochrein, D. Vremec, J. Pooley, R. Evans, S. Woulfe, and K. Shortman. 2002. Effects of administration of progenipoietin 1, Flt-3 ligand, granulocyte colony-stimulating factor, and pegylated granulocyte-macrophage colony-stimulating factor on dendritic cell subsets in mice. Blood. 99:2122–2130. [DOI] [PubMed] [Google Scholar]
- 16.Bjorck, P. 2001. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood. 98:3520–3526. [DOI] [PubMed] [Google Scholar]
- 17.McKenna, H.J., K.L. Stocking, R.E. Miller, K. Brasel, T. De Smedt, E. Maraskovsky, C.R. Maliszewski, D.H. Lynch, J. Smith, B. Pulendran, et al. 2000. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 95:3489–3497. [PubMed] [Google Scholar]
- 18.Saunders, D., K. Lucas, J. Ismaili, L. Wu, E. Maraskovsky, A. Dunn, D. Metcalf, and K. Shortman. 1996. Dendritic cell development in culture from thymic precursor cells in the absence of granulocyte-macrophage colony-stimulating factor. J. Exp. Med. 184:2185–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brasel, K., T. De Smedt, J.L. Smith, and C.R. Maliszewski. 2000. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 96:3029–3039. [PubMed] [Google Scholar]
- 20.Gilliet, M., A. Boonstra, C. Paturel, S. Antonenko, X.L. Xu, G. Trinchieri, A. O'Garra, and Y.J. Liu. 2002. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3 ligand and granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 195:953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brawand, P., D.R. Fitzpatrick, B.W. Greenfield, K. Brasel, C.R. Maliszewski, and T. De Smedt. 2002. Murine plasmacytoid pre-dendritic cells generated from flt3 ligand-supplemented bone marrow cultures are immature APCs. J. Immunol. 169:6711–6719. [DOI] [PubMed] [Google Scholar]
- 22.Bruno, L., T. Seidl, and A. Lanzavecchia. 2001. Mouse pre-immunocytes as non-proliferating multipotent precursors of macrophages, interferon-producing cells, CD8alpha(+) and CD8alpha(−) dendritic cells. Eur. J. Immunol. 31:3403–3412. [DOI] [PubMed] [Google Scholar]
- 23.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, et al. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 24.Nakano, H., M. Yanagita, and M.D. Gunn. 2001. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 194:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Keeffe, M., H. Hochrein, D. Vremec, B. Scott, P. Hertzog, L. Tatarczuch, and K. Shortman. 2002. Dendritic cell precursor populations of mouse blood: identification of the murine homologues of human blood plasmacytoid pre-DC2 and CD11c+ DC1 precursors. Blood. 101:1453–1459. [DOI] [PubMed] [Google Scholar]
- 26.O'Keeffe, M., H. Hochrein, D. Vremec, I. Caminschi, J.L. Miller, E.M. Anders, L. Wu, M.H. Lahoud, S. Henri, B. Scott, et al. 2002. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8+ dendritic cells only after microbial stimulus. J. Exp. Med. 196:1307–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu, L., C.-L. Li, and K. Shortman. 1996. Thymic dendritic cell precursors: relationship to the T lymphocyte lineage and phenotype of the dendritic cell progeny. J. Exp. Med. 184:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu, L., A. D'Amico, H. Hochrein, M. O'Keeffe, K. Shortman, and K. Lucas. 2001. Development of thymic and splenic dendritic cell populations from different hemopoietic precursors. Blood. 98:3376–3382. [DOI] [PubMed] [Google Scholar]
- 29.Kondo, M., I.L. Weissman, and K. Akashi. 1997. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 91:661–672. [DOI] [PubMed] [Google Scholar]
- 30.Akashi, K., D. Traver, T. Miyamoto, and I. Weissman. 2000. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 404:193–197. [DOI] [PubMed] [Google Scholar]
- 31.Manz, M.G., D. Traver, T. Miyamoto, I.L. Weissman, and K. Akashi. 2001. Dendritic cell potentials of early lymphoid and myeloid progenitors. Blood. 97:3333–3341. [DOI] [PubMed] [Google Scholar]
- 32.Traver, D., K. Akashi, M. Manz, M. Merad, T. Miyamoto, E.G. Engleman, and I.L. Weissman. 2000. Development of CD8alpha-positive dendritic cells from a common myeloid progenitor. Science. 290:2152–2154. [DOI] [PubMed] [Google Scholar]
- 33.Corcoran, L., I. Ferrero, D. Vremec, M. O'Keeffe, L. Wu, A. Wilson, and K. Shortman. 2003. The lymphoid past of mouse plasmacytoid cells and thymic dendritic cells. J. Immunol. 170:4926–4932. [DOI] [PubMed] [Google Scholar]
- 34.Izon, D., K. Rudd, W. DeMuth, W.S. Pear, C. Clendenin, R.C. Lindsley, and D. Allman. 2001. A common pathway for dendritic cell and early B cell development. J. Immunol. 167:1387–1392. [DOI] [PubMed] [Google Scholar]
- 35.del Hoyo, G.M., P. Martin, H.H. Vargas, S. Ruiz, C.F. Arias, and C. Ardavin. 2002. Characterization of a common precursor population for dendritic cells. Nature. 415:1043–1047. [DOI] [PubMed] [Google Scholar]