

Abstract
A variety of data suggest that in vivo production of interferon (IFN)-γ is necessary, but not sufficient, for expression of secondary protective immunity against intracellular pathogens. To discover specific IFN-γ–independent T cell mediated mechanisms, we took advantage of an in vitro culture system that models in vivo immune responses to the intracellular bacterium Francisella tularensis live vaccine strain (LVS). LVS-immune lymphocytes specifically controlled 99% of the growth of LVS in wild-type murine bone marrow–derived macrophages. Surprisingly, LVS-immune lymphocytes also inhibited LVS intracellular growth by as much as 95% in macrophages derived from IFN-γ receptor knockout (IFNγR KO) mice. CD8+ T cells, and to a lesser degree CD4+ T cells, controlled LVS intracellular growth in both wild-type and IFNγR KO macrophages. Further, a unique population of Thy1+αβ+CD4−CD8− cells that was previously suggested to operate during secondary immunity to LVS in vivo strongly controlled LVS intracellular growth in vitro. A large proportion of the inhibition of LVS intracellular growth in IFNγR KO macrophages by all three T cell subsets could be attributed to tumor necrosis factor (TNF) α. Thus, T cell mechanisms exist that control LVS intracellular growth without acting through the IFN-γ receptor; such control is due in large part to TNF-α, and is partially mediated by a unique double negative T cell subpopulation.
Keywords: protective immunity, Francisella tularensis LVS, interferon γ, TNF-α, T cells
Introduction
Francisella tularensis is a small Gram-negative facultative intracellular bacterium, and the causative agent of an acute febrile illness known as tularemia (1). This highly virulent pathogen, which can be transmitted via aerosol, has been classified as a Category A bioterrorism agent (2). The attenuated live vaccine strain (LVS)* of F. tularensis was derived by repeated passage of a virulent strain on agar (3). However, the efficacy and safety of this vaccine remains poorly defined, and it is not licensed for use in humans in the United States today. Thus, there is an interest in the development of a new F. tularensis vaccine, as well as the identification of correlates of protection for tularemia infection that can be used during testing of vaccine candidates.
Murine infection with LVS is a well-established and convenient model for characterizing the mechanisms of protection against tularemia infection, as well as for other intracellular bacteria in general (4, 5). Although F. tularensis LVS is avirulent for humans, it is highly virulent for laboratory mice, and when administered i.p. causes a fulminent infection in these animals that is histopathologically similar to human tularemia (1, 4, 6). In the mouse model, the outcome of a primary LVS infection is dependent on the route of inoculation: the 50% lethal dose (LD50) when infection is initiated via the intradermal (i.d.) route is ∼106 bacteria in BALB/cByJ mice, whereas infection via the i.v. or i.p. routes has an LD50 that approaches a single bacterium (6, 7). Mice given a sublethal LVS infection via the i.d. route are solidly immune to a secondary i.p. challenge of as much as 100,000 LD50s. In both mice and humans, survival and resolution of a primary Francisella infection confers a long-lasting specific protective immunity to reinfection (1, 4, 6).
Mice genetically deficient for IFN-γ, as well as mice treated with neutralizing anti–IFN-γ antibodies, quickly succumb to a primary LVS infection with doses that are sublethal for normal mice (8). However, studies investigating the role of IFN-γ during a secondary LVS infection suggest that IFN-γ may be less critical for survival during a secondary LVS challenge. Treatment of LVS-infected mice with neutralizing antibodies to IFN-γ had no detectable effect on the outcome of a secondary i.p. challenge (8) or on a secondary i.d. challenge (9) except at high i.d. challenge doses. Thus, there appear to be immune mechanisms that function during a late primary and a secondary immune response to LVS that do not require IFN-γ.
Here we investigate mechanisms that control LVS intracellular growth in the absence of signaling through the macrophage IFN-γ receptor. To this end, we measured the ability of different LVS-immune splenic T cell subsets to control LVS intracellular growth in either wild-type (10) or IFN-γ receptor deficient (IFNγR KO) bone marrow macrophages (BMMØs). Surprisingly, LVS-immune spleen cells inhibited LVS intracellular growth by as much as 95% in the IFNγR KO BMMØ monolayer. CD4+ cells, and to a greater extent CD8+ T cells, controlled LVS intracellular growth in both wild-type and IFNγR KO BMMØs. Further, a unique population of Thy1+CD4− CD8− cells that was previously suggested to be operative during secondary immunity to LVS in vivo (11, 12) strongly controlled LVS intracellular growth in vitro. In the absence of IFN-γ stimulation of macrophages, these T cell subsets relied to varying extents upon TNF-α production to inhibit LVS intracellular growth.
Materials and Methods
Animals.
6–8-wk-old male specific-pathogen-free 129S1/SvlmJ or IFNγR-deficient mice on a 129S1/SvlmJ background were purchased from The Jackson Laboratory. Animals were housed in sterile microisolator cages in barrier environment at the Center for Biologics Evaluation and Research. Mice were fed autoclaved food and water ad libitum. All experiments were performed under Institutional Animal Care and Use Committee guidelines.
Culture and Infection of BMMØ with Bacteria.
BMMØ were used as the target cells for the in vitro system. BMMØ were cultured as described previously (10). Bone marrow was flushed from femurs of healthy IFNγR-deficient or wild-type mice with DMEM (Life Technologies) supplemented with 10% heat-inactivated FCS (HyClone), 10% L-929 conditioned medium, 0.2 mM l-glutamine (Life Technologies), 1 mM HEPES buffer (Life Technologies), and 0.1 mM nonessential amino acids (Life Technologies; complete DMEM [cDMEM]). Cells were washed, a single cell suspension prepared, and cells plated at 2 × 106 in 24-well plates (Costar) or at 106 in 48-well plates, in cDMEM supplemented with 50 μg/ml gentamycin (Life Technologies) and incubated at 37°C in 5% CO2. After 1 d of incubation, the medium was replaced with antibiotic-free cDMEM, and the cells were incubated for an additional 6 d at 37°C in 5% CO2. The medium was replaced with fresh, gentamycin-free cDMEM every 2 d during the 7-d incubation.
After the 7-d culture period, the BMMØ formed a confluent monolayer, and the concentration of BMMØ was estimated to be 107 cells/well in the 24-well plates and 5 × 106 cells/well in the 48-well plates (10). BMMØ were then infected with F. tularensis LVS according to the following protocol: bacteria were diluted from frozen stocks in cDMEM and added at a multiplicity of infection (MOI) of 1:20–1:40 (bacterium-to-BMMØ ratio). A low MOI was chosen to permit controlled infection of the macrophage monolayer over 3 to 5 d, permitting time for lymphocyte–macrophage interactions and modeling in vivo relationships. LVS was coincubated with BMMØ at 37°C in 5% CO2 for 2 h and then washed three times with PBS (Life Technologies). The monolayers were then incubated for 45 min–1 h in cDMEM supplemented with 50 μg/ml gentamicin to eliminate extracellular bacteria. The BMMØ were then washed a further three times with PBS. After the last wash, PBS was replaced with 1 ml/well cDMEM and the cells were incubated at 37°C in 5% CO2 for the remainder of the experiment. To determine bacterial uptake, some BMMØs were lysed with sterile distilled water for 3 min immediately after washing with PBS. Culture lysates were serially diluted in PBS and plated on Mueller Hinton agar plates. The plates were incubated for 2–3 d at 37°C in 5% CO2, and LVS colonies were counted. Growth of LVS in BMMØ was monitored by lysing cultures at the indicated time points, plating lysates, and counting LVS colonies as described above.
Infection of Mice with LVS and Listeria monocytogenes EGD.
Wild-type mice were infected i.d. with 104 LVS or L. monocytogenes strain EGD (ATCC 15313). These concentrations of bacteria were chosen to establish a sublethal infection that is cleared from organs by 2 to 3 wk and that engenders long-term protective immunity to a lethal secondary infection; such mice were therefore used as a source of LVS-primed or Listeria-primed splenocytes.
Harvesting and Enrichment of Splenocytes, and Assay for Control of Intracellular LVS Growth.
Spleens were aseptically removed from selected mice and disrupted with a 3-ml syringe plunger. A single-cell suspension was prepared, and erythrocytes were lysed with ammonium chloride. Cells were washed, viability was assessed by exclusion of trypan blue, and cells were resuspended in Dulbecco PBS-2% FCS to the appropriate concentration. In most experiments, wild-type macrophages in the spleen were depleted by adherence; spleen cells in cDMEM were added to 75 cm2 tissue culture flasks for 2 h at 37°C and nonadherent cells recovered by gentle pipetting. The recovered nonadherent splenocytes were added to BMMØs cultures at various concentrations as indicated. Unless otherwise stated, 5 × 106 splenocytes were added to each well in the 24-well plate cultures, and 2.5 × 106 splenocytes were added to the 48 well cultures (∼1 splenocyte to 2 BMMØs).
Depletion of various lymphocyte subsets before their addition to the in vitro system was achieved by performing in vivo depletion of the desired cell type before harvesting of the splenocytes (11). On days −3 and −1 before harvest, mice were given either 500 μg of anti-CD4 mAb (clone GK1.5), 300 μg anti-CD8 mAb (clone 2.43), or 500 μg of anti-Thy1.2 mAb (clone 30-H12) via the i.p. route. Cell depletions routinely reduced the depleted cell type to less than 1% of the total cell population. In all cases, starting and depleted splenocyte populations were analyzed by flow cytometry as described below.
To obtain separated and purified cell populations for the in vitro assay, CD4+ T cells, CD8+ T cells, and Thy1+CD4−CD8− populations were enriched using CD4+ cell, CD8+ cell, and Thy1.2+ cell enrichment columns (MACS magnetic cell sorting system; Miltenyi Biotec) according to the manufacturer's instructions. To obtain the Thy1+CD4−CD8− splenocyte population, mice were depleted in vivo of CD4+ and CD8+ T cells before harvesting of the spleens, and then these spleens were depleted of B220+ B cells using a B220+ MACS cell depletion column. The remaining Thy1+ cell population was obtained using a Thy1.2+ MACS cell enrichment column; these cells were ∼2% of total splenocytes. Cell populations were routinely greater than 90% of the desired population using this method of enrichment. In all cases, starting and enriched splenocyte populations were analyzed by flow cytometry using a fluorescence-activated cell sorter scan as described below.
In some experiments, TNF-α was neutralized in the culture by addition of a pair of azide-free, low-endotoxin mouse anti–TNF-α neutralizing antibodies (MP6-XT3 and G281–2626; BD Biosciences) at a concentration of 20 μg/ml. In other experiments, 1 mM N-monomethyl-l-arginine (NMMA; Sigma-Aldrich) was added to inhibit synthesis of nitric oxide in the cultures. Antibodies and NMMA were added to the cocultures with splenocytes at the initiation of the culture period.
Quantitation of Cytokines and NO in BMMØ Culture Supernatants.
Culture supernatants were assayed for IFN-γ, IL-12p40, and TNF-α by standard sandwich ELISAs. All antibody pairs and standards were purchased from BD Biosciences, and all ELISAs performed in accordance with the manufacturer's instructions. Samples were read at 405 nm on a VersaMax tunable microplate reader with a reference wavelength of 630nm (Molecular Devices). Cytokines were quantified by comparison to recombinant standards (all purchased from BD Biosciences) using four-parameter fit regression in the SoftMax Pro ELISA analysis software (Molecular Devices).
Nitric oxide (NO) was detected in culture supernatants by the Griess reaction (13). Briefly, 100 μl aliquots of culture supernatants were incubated with an equal volume of commercial Griess reagent (Sigma-Aldrich) for 5 min at room temperature, and the absorbance of each sample at 490 nm was measured. NO2 was quantified by comparison to serially diluted NaNO2 as a standard using four-parameter fit regression in the SoftMax Pro ELISA analysis software (Molecular Devices).
Analysis of Splenocyte Populations Using Flow Cytometry.
Cells were prepared as described above and stained for B220+, CD4+, CD8+, Thy1.2+, CD11b+, and DX5+ cell surface markers as described previously (10, 11). Briefly, single cell suspensions were mixed with anti-CD16 (Fc Block; BD Biosciences) for 10 min on ice. FITC-conjugated rat IgG2a, PE-conjugated rat IgG2b, and allophycocyanin (APC)-conjugated rat IgG2a were used as isotype controls. FITC-conjugated anti-CD45/B220 (RA3–6B2), PE-conjugated anti-CD4 (RM4–4), PE-conjugated anti-CD8a (53–6.7), PE-conjugated anti-CD11b (M1/70), PE-conjugated anti-DX5 (DX5), or APC-conjugated anti-CD90/Thy1.2 (53–2.1) monoclonal antibody was added, and cells were incubated for an additional 30 min on ice. All antibodies were obtained from BD Biosciences, and optimal concentrations for staining were determined in separate experiments. Cells were washed three times in PBS-2% FCS, fixed in 0.5% buffered paraformaldehyde, and analyzed using a BD Biosciences LSR flow cytometer with gates set to viable lymphocytes and monocytes according to forward and side-scatter profiles. Among the total population of splenocytes, 129S1/SvlmJ mice had ∼25–30% CD4+ cells, 11–15% CD8+ cells, 40% B220+ cells, 15% CD11b+ cells, and 2% DX5+ cells.
Results
Growth Inhibition of LVS in Wild-Type and IFNγR-deficient BMMØ.
We tested the ability of LVS-immune splenocytes from wild-type mice to control the intracellular growth of LVS in BMMØs derived from both wild-type and IFNγR KO mice. Splenocytes were obtained from LVS-immune wild type mice that were infected (primed) 4 wk earlier with a sublethal in vivo LVS infection. To minimize the influence of wild-type macrophages in the splenocyte population, adherent cells in the spleen were depleted by plastic adherence before addition to the BMMØ cultures. Nonadherent immune splenocytes were cocultured with LVS-infected wild-type and IFNγR KO BMMØ monolayers. Because previous experiments demonstrated that the maximal control of LVS growth in BMMØ by immune splenocytes occurred after 72 h of coculture (10), here we used the 72 h time point to compare the ability of splenocytes to control LVS growth in the two different BMMØ monolayers. As seen in Fig. 1 , addition of LVS-immune, but not naive, splenocytes to both types of infected BMMØ monolayers resulted in readily measurable and significant (P < 0.01) inhibition of LVS growth compared with LVS growth in cultures containing either naive splenocytes or no splenocytes. LVS replicated to a similar extent in both the wild-type and IFNγR KO BMMØs, and growth was not significantly affected by the addition of naive splenocytes to the cultures. Coculture of LVS-immune splenocytes with wild-type BMMØs resulted in a 3.5 log10 reduction in growth of the bacteria, while coculture with the IFNγR KO BMMØs resulted in a 1.6 log10 reduction of LVS growth. Thus, as expected, a substantial fraction of the control of LVS growth was mediated by IFN-γ stimulation of infected macrophages. Nonetheless, in multiple replicate experiments, control of LVS growth in the IFNγR KO BMMØs by wild-type immune splenocytes ranged from a 1 to 1.7 log10 reduction. In absolute numbers, this represents inhibition of more than 95% of LVS growth in the IFNγR KO BMMØs by LVS-immune wild-type splenocytes.
Figure 1.

Control of LVS growth by wild-type LVS-immune splenocytes in (A) wild-type BMMØ and (B) BMMØ that lack the IFNγR. BMMØ from wild-type and IFNγR KO mice were infected with LVS at an MOI of 1:40 (bacterium-to-macrophage ratio). Infected BMMØs were cocultured with splenocytes from either uninfected mice (naive spleen), mice infected intradermally with LVS 4 wk previously (primed spleen), or no spleen cells (macrophages + LVS). Immediately after LVS infection of the BMMØs, splenocytes were added to the indicated wells at a ratio of 1:2 (splenocyte:BMMØ). 72 h after infection, the BMMØ were washed, lysed, and plated to determine the levels of intracellular bacteria. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Asterisks (*) indicate P values <0.01 as compared with cocultures containing naive spleen cells. These data are representative of five experiments of similar design.
To determine whether the observed control of LVS intracellular growth in the IFNγR KO BMMØs is a specific event, we tested the ability of splenocytes harvested from mice infected with another intracellular pathogen, Listeria monocytogenes EGD, to control LVS intracellular growth. As shown in Fig. 2, A and B , splenocytes from LVS-immune mice controlled LVS growth in both sets of BMMØ monolayers. However, neither Listeria-immune nor naive splenocytes had a significant (P < 0.01) effect on control of LVS growth in the wild-type and IFNγR KO BMMØ monolayers. These results indicate that the control of LVS growth in the IFNγR KO BMMØs is specific.
Figure 2.

Specificity of control of LVS growth in (A) wild-type BMMØ and (B) IFNγR KO BMMØs. Infected BMMØs were cocultured with wild-type splenocytes from either uninfected mice (naive spleen), mice infected intradermally 4 wk previously with LVS (LVS primed spleen), or with L. monocytogenes (Listeria primed spleen). Immediately following LVS infection of the BMMØs, splenocytes were added to the indicated wells at a ratio of 1:2 (splenocyte:BMMØ). 72 h after infection, the BMMØ were assessed for levels of intracellular bacteria as described in the legend to Fig. 1. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Asterisks (*) indicate P values <0.01 as compared with cocultures containing naive spleen cells. These results are representative of three experiments of similar design.
TNF-α and NO Contribute to LVS Growth Inhibition in the IFNγR KO BMMØs.
We next examined the mechanisms used by the LVS-immune splenocytes to control LVS growth in the IFNγR KO BMMØs. As TNF-α appears to be important for survival of both a primary and secondary LVS infection (8, 9), we tested the impact of abrogation of TNF-α activity on the control of LVS growth in the culture system. As seen in Fig. 3, A and B , inhibition of TNF-α activity by addition of neutralizing anti–TNF-α Abs to the cultures significantly reversed the ability of the splenocytes to control LVS growth in both the IFNγR KO and wild-type BMMØ monolayers (P < 0.01 compared with cultures with control IgG or no antibodies). The IFNγR KO BMMØ cultures that were treated with anti–TNF-α Ab exhibited no significant control of intracellular LVS growth compared with that seen with naive cells (P > 0.01), while the wild-type cultures treated with anti–TNF-α Ab retained significant control compared with that seen with naive cells (P < 0.01).
Figure 3.

Effects of TNF-α neutralization and iNOS inhibition on control of LVS growth in (A) wild-type BMMØ and (B) IFNγR KO BMMØs. Infected BMMØs were cocultured with splenocytes from either uninfected mice (naive spleen), or wild-type mice infected intradermally with LVS 4 wk previously (primed spleen). Immediately following LVS infection of the BMMØs, splenocytes were added to the indicated wells at a ratio of 1:2 (splenocyte:BMMØ). In the indicated cultures, either anti–TNF-α Abs or control IgG Ab (20 μg/ml), or 1 mM NMMA, were added to the cultures at the time of addition of the splenocytes. 72 h after infection, the BMMØ were assessed for levels of intracellular bacteria as described in the legend to Fig. 1. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Asterisks (*) indicate P values <0.01 as compared with cocultures containing naive spleen cells. These results are representative of three experiments of similar design.
It has previously been shown that LVS-infected murine macrophages treated in vitro with recombinant IFN-γ use NO as a primary mechanism for controlling LVS intracellular growth, and that this NO production is partially dependent on the autocrine action of macrophage TNF-α (14, 15). Thus, we next investigated the possibility that the role of TNF-α in our IFNγR KO BMMØ cocultures was to induce macrophage production of NO independently of IFN-γ stimulation of macrophages. As seen in Fig. 3, A and B, addition of the nitric oxide synthase inhibitor NMMA significantly reversed splenocyte control of LVS growth in both the IFNγR KO and wild-type BMMØ monolayers compared with untreated control cultures (P < 0.01). This reversal was comparable in magnitude to that seen with anti–TNF-α Ab treatment.
Depletion and Enrichment of Spleen Cell Subsets Reveals Roles for CD8+ T Cells as well as for Unique Thy1+CD4− CD8− Cells in Control of LVS Intracellular Growth in the Absence of IFN-γ.
We next sought to determine the T cell subpopulations required to control LVS intracellular growth in both wild-type and IFNγR KO BMMØ monolayers. As our previous experiments revealed that growth inhibition of LVS in the wild-type and IFNγR KO BMMØ monolayers was specific, and as survival of an LVS infection is dependent upon specific αβ+ T cells (10–12, 16, 17), we depleted mice in vivo of various T cell subsets before addition of their splenocytes to the in vitro culture system. Mice were depleted of total Thy1+ cells, CD4+ cells, CD8+ cells, or both CD4+ and CD8+ cells simultaneously. The splenocytes that remained following depletion were then added to the BMMØ cultures, and LVS growth was determined after 72 h of coculture. As shown in Fig. 4, A and B , splenocytes obtained from LVS-immune mice depleted of CD4+ cells, CD8+ cells, or CD4+ and CD8+ cells simultaneously, all controlled LVS intracellular growth in both wild-type and IFNγR KO BMMØs in a manner similar to whole primed splenocytes. In contrast, splenocytes obtained from LVS-immune mice depleted of Thy1+ cells exhibited a dramatic and significant (P < 0.01) reversal of control of growth when cocultured with both types of BMMØs (Fig. 4, A and B). In some experiments, depletion of Thy1+ cells resulted in a population that did not significantly control intracellular LVS growth as compared with naive cells. These results indicate that a Thy1+CD4−CD8− cell population is involved in control of LVS intracellular growth.
Figure 4.
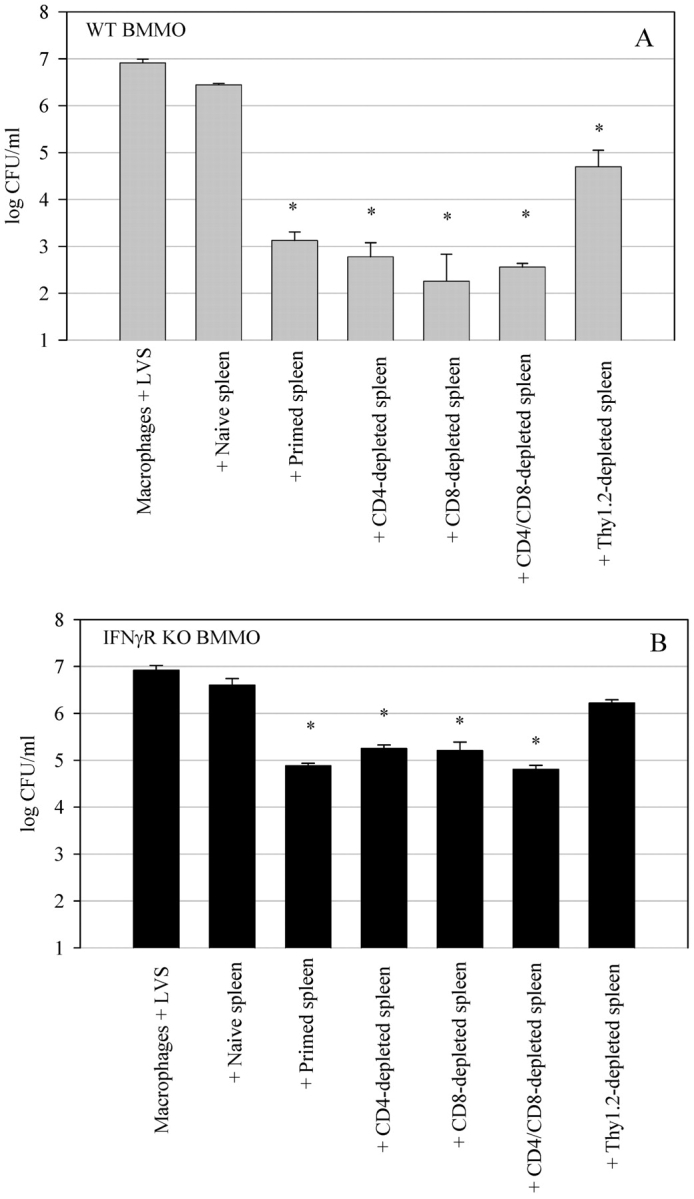
Effects of T cell depletion on control of LVS growth in (A) wild-type BMMØ and (B) IFNγR KO BMMØs. Infected BMMØs were cocultured with splenocytes obtained from either uninfected mice (naive spleen), wild-type mice infected intradermally with LVS 4 wk previously (primed spleen), or LVS-primed wild-type mice that had been in vivo depleted of the indicated T cell subsets by intraperitoneal injection with Abs specific for CD4, CD8, CD4 and CD8 simultaneously, or Thy1.2 cell surface markers. Immediately after LVS infection of the BMMØs, splenocytes were added to the indicated wells at a ratio of 1:2 (splenocyte:BMMØ). 72 h after infection, the BMMØ were assessed for levels of intracellular bacteria as described in the legend to Fig. 1. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Asterisks (*) indicate P values <0.01 as compared with cocultures containing naive spleen cells. These results are representative of three experiments of similar design.
To further define the T cell subsets responsible for control of LVS intracellular growth, we positively selected CD4+ T cells, CD8+ T cells, and Thy1+CD4−CD8− cells from LVS-immune mouse splenocytes (see Materials and Methods). The purified T cell subpopulations were cocultured with LVS-infected wild-type and IFNγR KO BMMØs. All cell populations were added in equivalent numbers to the BMMØ monolayer (1:2 ratio of splenocyte: BMMØ). As shown in Fig. 5 A, purified CD4+ T cells, CD8+ T cells, and Thy1+CD4−CD8− T cells were all capable of controlling LVS growth in wild-type BMMØs, and to an even greater extent than whole primed splenocytes. All of these LVS-immune T cell subpopulations reduced LVS numbers in wild-type macrophages significantly, by over 99% as compared with naive splenocytes (P < 0.01 compared with naive cells).
Figure 5.

Ability of various T cell subsets to control LVS growth in (A) wild-type BMMØ and (B) IFNγR KO BMMØs. Infected BMMØs were cocultured with splenocytes obtained from either uninfected mice (naive spleen), whole primed splenocytes from immune wild-type mice (primed spleen), or various T cell subsets enriched from immune wild-type mice (CD4+ cells, CD8+ cells, and Thy1+CD4−CD8− cells). All splenocyte populations were added at a 1:2 ratio (splenocyte to BMMØ). 72 h after infection, the BMMØ were assessed for levels of intracellular bacteria as described in the legend to Fig. 1. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Asterisks (*) indicate P values <0.01 as compared with cocultures containing naive spleen cells. These results are representative of three experiments of similar design.
All three T cell subsets also controlled LVS growth in IFNγR KO BMMØs as compared with naive splenocytes (P < 0.001; Fig. 5 B). However, the control of LVS growth in IFNγR KO BMMØs clearly varied in magnitude with the different T cell subsets. Repeated experiments consistently found that CD4+ T cells were the least effective T cell subset at mediating growth inhibition of LVS in macrophages that cannot respond to IFN-γ, while CD8+ T cells and Thy1+CD4−CD8− T cells were consistently more effective and comparable to each other (see Fig. 7).
Figure 7.
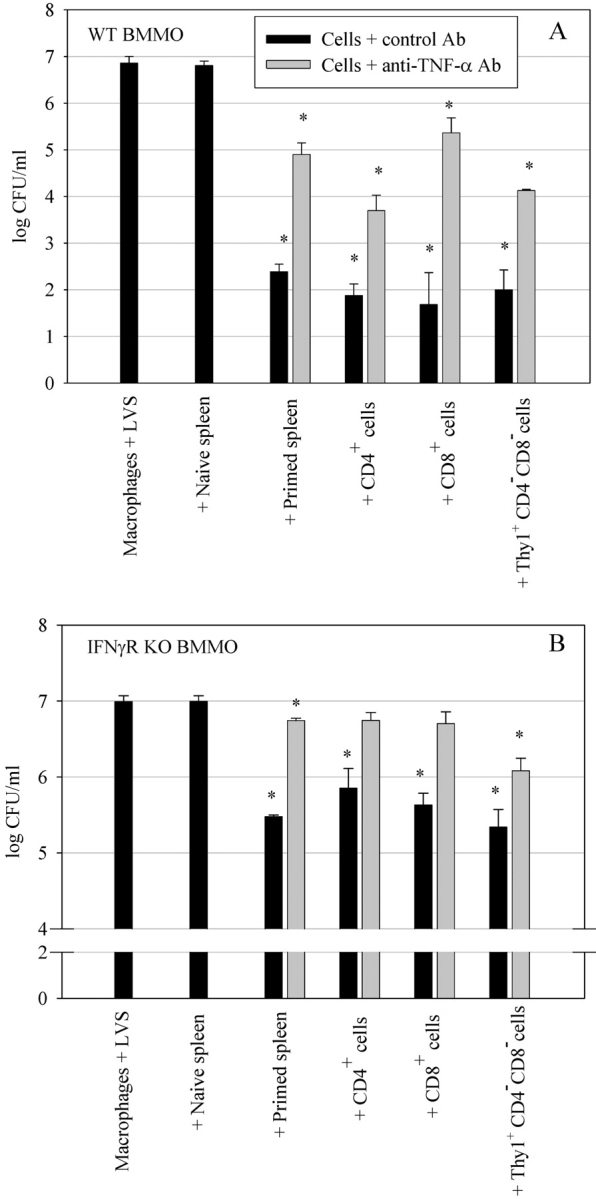
Effects of TNF-α neutralization on the ability of different T cell subsets to control LVS growth in (A) wild-type BMMØ and (B) IFNγR KO BMMØs. Infected BMMØs were cocultured with splenocytes obtained from either uninfected mice (naive spleen), whole primed splenocytes from immune wild-type mice (primed spleen), or various T cell subsets enriched from immune wild-type mice (CD4+ cells, CD8+ cells, and CD4−CD8−Thy1.2+ cells). All splenocyte populations were added at a 1:2 ratio (splenocyte to BMMØ). Anti–TNF-α Abs were added to the wells at the time of addition of the splenocytes to the infected BMMØs. 72 h after infection, the BMMØ were assessed for levels of intracellular bacteria as described in the legend to Fig. 1. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Asterisks (*) indicate P values <0.01 as compared with cocultures containing naive spleen cells. These results are representative of three experiments of similar design.
The phenotype of purified Thy1+CD4−CD8− was examined in detail by flow cytometry for the expression of other cell surface markers. These cells were largely positive for αβ T cell receptors (>98%) and CD3 (>85%), but mostly negative for B220 (<3%), DX5 (<3%), CD11b (7.5%), and γδ T cell receptors (<3%). Determination of NK1.1 staining was not possible, as mice of the 129 background lack the NK1.1 marker.
To study the mechanisms by which these three T cell subsets contribute to control of LVS growth in the wild-type and IFNγR KO BMMØ cultures, secretion of cytokines and NO into the coculture supernatants was examined. As shown in Fig. 6 A, supernatants from wild-type BMMØ cultures containing infected macrophages alone or naive splenocytes had undetectable levels of IFN-γ. In contrast, cultures containing whole primed splenocytes, purified CD4+ cells, CD8+ cells, or Thy1+CD4−CD8− cells, all produced IFN-γ (Fig. 6 A). However, while cultures containing whole primed spleen cells and purified CD4+ cells produced high quantities of IFN-γ, cultures containing wild-type BMMØs and purified CD8+ cells or Thy1+CD4−CD8− cells produced relatively low levels of IFN-γ. The levels of IFN-γ were consistently found to be several-fold higher in the cultures containing IFNγR KO BMMØs, likely as a result of the inability of the macrophages in those cultures to utilize and remove the IFN-γ from the cultures. This is particularly notable for the CD8+ T cells. Interestingly, however, it appears that the Thy1+CD4− CD8− cells produce less net IFN-γ than the CD4+ and CD8+ cells (Fig. 6 A).
Figure 6.

Secretion of cytokines and NO into culture supernatants after coculture of LVS infected BMMØs and immune splenocytes. Wild-type (black bars) and IFNγR KO (gray bars) BMMØ were infected with LVS and cocultured with the indicated splenocyte populations. Splenocytes from either unprimed mice or mice infected 4 wk previously with an intradermal LVS infection were cocultured with LVS-infected macrophages at a 1:2 ratio (splenocytes to BMMØ). Similarly, CD4+ and CD8+ and Thy1+CD4−CD8− splenocyte subpopulations were added to infected BMMØ cultures at a 1:2 ratio. Culture supernatants were collected 72 h later and tested for IFN-γ (A), TNF-α (B), IL-12 (C), and NO (D). Values shown are the mean ng/ml ± the SEM of the indicated cytokine (A–C) or mean μM/ml nitrite (D) (triplicate samples). These results are representative of three experiments of similar design.
Similarly, TNF-α production was found to be very low in both the wild-type and IFNγR KO BMMØ cultures containing either LVS-infected macrophages alone or plus naive splenocytes, and was significantly increased upon addition of LVS-immune whole splenocytes or each of the three T cell subsets (Fig. 6 B). The IFNγR KO BMMØ cultures exhibited lower levels of TNF-α as compared with the wild-type BMMØ cultures. In contrast, IL-12 production was similar in both the wild-types and IFNγR KO BMMØ cultures (Fig. 6 C); small amounts of IL-12 were evident in cultures containing naive splenocytes, and these amounts increased substantially upon addition of either whole primed splenocytes, CD4+ cells, CD8+ cells, or Thy1+CD4−CD8− cells.
The secretion of NO by macrophages in the cultures was examined and found to correlate most obviously with TNF-α production (Fig. 6 D). NO was undetectable in both the wild-type and IFNγR KO BMMØ cultures containing naive splenocytes, and significantly increased upon addition of LVS-immune whole splenocytes, as well as each of the three T cell subsets. The IFNγR KO BMMØ cultures exhibited lower levels of NO as compared with the wild-type BMMØ cultures. Overall, NO production most closely paralleled TNF-α production.
CD4+, CD8+, and Thy1+CD4−CD8− Cells All Utilize TNF-α to Mediate Inhibition of LVS Intracellular Growth in Both Wild-Type and IFNγR KO BMMØs.
As TNF-α was found to be a key cytokine that contributes to inhibition of LVS growth in both wild-type and IFNγR KO BMMØs by whole primed splenocytes (Fig. 3), we next investigated the effect of TNF-α neutralization on the ability of the three different T cell subsets to control LVS growth. As seen in Fig. 7, A and B , inhibition of TNF-α activity by addition of neutralizing anti–TNF-α Abs to the cultures significantly (P < 0.01) reversed the ability of all three T cell subsets to control LVS growth in both the IFNγR KO and wild-type BMMØ monolayers as compared with control cocultures. In repeated experiments, CD4+ and CD8+ T cells were consistently affected more strongly by neutralization of TNF-α in the IFNγR KO BMMØ cultures than were Thy1+CD4−CD8− T cells. Thus, LVS growth inhibition mediated by either CD4+ T cells or CD8+ T cells in the IFNγR KO BMMØ cocultures was completely reversed by treatment with anti–TNF-α Ab, and was not significantly different from that of naive splenocytes (P > 0.01). In contrast, IFNγR KO BMMO cocultures with Thy1+CD4−CD8− cells that were treated with anti–TNF-α Ab still exhibited a significant 88% reduction of LVS numbers as compared with naive spleen cell cultures (P < 0.01). Therefore, growth inhibition by Thy1+CD4−CD8− T cells is mediated by IFN-γ, TNF-α, and other mechanisms that remain to be identified.
Discussion
Development of vaccines for intracellular pathogens such as Mycobacterium tuberculosis, a global public health problem, and Francisella tularensis, a potential bioterrorism pathogen, is limited by difficulties in conducting field trials, lack of correlates of protection, and a generally incomplete understanding of mechanisms of protective immunity. Although IFN-γ is often proposed as a correlate, the available evidence suggests that production of IFN-γ is likely to be necessary but not sufficient for successful protection against intracellular bacteria (18). Thus, correlates of protection can best be determined by expanding our understanding of the components of the immune system that are involved in limiting intracellular growth of these pathogens. To this end, we have previously developed an in vitro model system to evaluate the cell types, cytokines, and receptors involved in inhibition of F. tularensis growth inside its primary host cell, the macrophage (10). Here we have applied this in vitro system to examine the T cell subsets responsible for F. tularensis LVS growth inhibition in the absence of IFN-γ stimulation of the macrophage.
Using IFNγR KO BMMØs, we showed that as much as 95% of LVS intracellular growth can specifically be inhibited in macrophages that cannot respond to IFN-γ (Figs. 1 B and 2). Further, we demonstrated that both CD4+ T cells and especially CD8+ T cells are capable of inhibiting LVS intracellular growth in the absence of IFN γ stimulation of the macrophage (Figs. 4 and 5). Consistent with the well-known importance of CD4+ T cells as sources of IFN-γ, CD4+ T cells were notably less efficient than CD8+ T cells at inhibiting LVS growth in IFNγR KO BMMØs. Surprisingly, we also identified an unusual T cell subset that strongly controlled LVS intracellular growth in both wild-type and IFNγR KO BMMØs: the double negative (CD4−/CD8−) Thy1+ T cell (Fig. 5). The ability of all three T cell subsets to inhibit LVS intracellular growth in the IFNγR KO BMMØs depended upon TNF-α production to varying extents (Figs. 3 and 7 B). While much of the intracellular growth control in vitro by CD4+ and CD8+ T cells could be attributed to the combination of IFN-γ and TNF-α, it appears that yet another (to be identified) mechanism(s) is operative with the double negative Thy1.2+ T cells.
In many intracellular infections, IFN-γ is considered a primary candidate as a correlate of protection. This is largely due to its pivotal role in activating macrophages, as well as the extreme sensitivity of IFN-γ–deficient mice and humans to many intracellular pathogens. Indeed, studies using the F. tularensis LVS murine infection model have also underscored the importance of IFN-γ for the successful development and survival of primary infections with this intracellular bacterium. Mice deficient in T cells survive more than a month after a primary i.d. LVS infection, but then succumb (11, 16, 17). The early T cell–independent phase of immunity is dependent upon both IFN-γ and TNF-α production, but the final resolution and clearance of LVS infection is clearly dependent upon αβ+ T cells (8, 16). The large amounts of IFN γ produced during the T cell–independent phase of infection are obviously insufficient to resolve the infection, and thus another T cell function must be necessary to mediate clearance.
The role of IFN-γ in secondary intracellular infections, which is relevant to the case of vaccination and secondary challenge, has not been thoroughly studied, but there are a few pertinent examples. IFN-γ–deficient (GKO) mice primed with an attenuated L. monocytogenes mutant strain were fully protected against secondary lethal challenge with virulent L. monocytogenes (19). Protection was mediated largely by CD8+ T cells, and included production of TNF-α and perforin (20, 21). We have recently demonstrated that IFN-γ–independent immunity to secondary challenge, mediated primarily by CD4+ T cells, also exists in the Mycobacterium tuberculosis murine infection model (unpublished data). In the studies presented here, much of the immunity to LVS observed in vitro using IFNγR KO BMMØs also relied largely upon TNF-α, but was mediated by multiple T cell subsets. The few other studies that have examined this topic in Francisella infection further suggest that IFN-γ may have relatively less importance during late primary and secondary LVS infections than in early primary infection. For example, Leiby et al. (8) found that mice treated with neutralizing anti–IFN-γ mAbs did not survive primary sublethal LVS infection if the mAbs were administered within the first 2–3 d of infection, but survived if anti–IFN-γ was administered at later times. Further, treatment with neutralizing anti–IFN-γ mAbs during a secondary lethal i.p. challenge had no detectable effect on survival (8). Similarly, Sjöstedt et al. (9) found that treatment with neutralizing anti–IFN-γ mAbs during secondary lethal i.d. challenge only reduced survival of the animals when the challenge inoculum was extremely high. Collectively, these data indicate that very significant IFN-γ–independent mechanisms of immunity exist in multiple infection models, and may be mediated by a variety of different T cell subsets.
In contrast to depletion of IFN-γ, in vivo depletion of TNF-α during secondary i.d. LVS challenge resulted in loss of control of infection and death at all challenge doses (9). Similarly, here blockade of TNF-α activity by addition of neutralizing anti–TNF-α antibodies reduced the control of intracellular LVS growth in wild-type macrophages, and largely ablated it in IFNγR KO macrophages (Fig. 3 B). The exact cellular source and form of this TNF-α, as well as the receptors used, remain to be determined in future experiments, but the data to date suggest that it is likely to be T cell derived rather than macrophage derived. The form may be suggested by studies that take advantage of transgenic mice that express membrane TNF-α but do not secrete soluble TNF-α (22). Intriguingly, Mycobacteria infection of such memTNF transgenic mice progresses similarly to that in wild-type mice (23), even though mice lacking all TNF-α are acutely susceptible to Mycobacteria infections (24, 25). Preliminary results similarly suggest that transgenic memTNF-α mice, unlike TNF-α–deficient mice, also survive primary i.d. LVS infection (unpublished data). Thus, we propose that membrane TNF-α expressed on LVS-specific T cells is (at least in part) responsible for IFN-γ–independent control of LVS infection, and are actively pursuing studies to address this hypothesis.
Surprisingly, depletion experiments as well as positive selection of T cell subsets led us to an unexpected T cell subset that has a powerful capacity to control LVS growth in macrophages. These cells were CD4−CD8−B220−, and αβ+Thy1.2+CD3+, and used IFN-γ, TNF-α, and another unidentified mechanism to mediate LVS growth inhibition (Fig. 7, A and B). To the best of our knowledge, these data are the first direct demonstration of growth inhibition of an intracellular pathogen by an αβTCR+ double negative T cell. The relationship of these cells to other double negative T cells that have been previously described remains to be clarified, but the available data suggest that these may be novel subset. Cells with a similar phenotype, although rare in murine splenocytes, have been previous recognized in autoimmune mice, but are usually NK1.1+, B220+, and have a restricted T cell receptor repertoire (26, 27). Because the 129 mouse strain used here (due to the availability of IFNγR KO mice on this background) does not express the NK1.1 allele, we cannot directly examine this marker. However, the overwhelming majority of the cells we describe here do not express B220 or another natural killer cell marker, DX5. Other double negative T cells that have been described include CD1-restricted T cells that recognize lipid antigens. For example, Stenger et al. have previously shown that human double negative T cell clones can lyse Mycobacterium tuberculosis–infected macrophages in a FasL-dependent manner; however, this activity had no direct effect on the viability or growth of the mycobacteria (28, 29). Our experiments to date indicate that both perforin and Fas–FasL interactions have minimal contributions to the course of LVS infection or to intracellular growth control (unpublished data). A handful of other studies have shown that various populations of double negative T cells can either expand (30, 31), or express Th1-type cytokines (31, 32), in response to an in vivo challenge with intracellular bacteria or viruses.
Importantly, the existence of double negative αβ+T cells that have an in vivo role in secondary immunity to LVS infection has already been implicated in previous studies. Simultaneous depletion of CD4+ and CD8+ T cells during a secondary LVS challenge had little effect on the outcome of infection; the mice survived and cleared the infection, albeit at a slower rate than wild-type mice (11, 12). Conlan et al. found that although the CD4+ and CD8+ T cells in these mice were successfully depleted, a substantial Thy1+CD4−CD8− population of cells remained (12). Similarly, studies in the murine Listeria model also indicate a major role for such cells in secondary protection (33). The precise origin, phenotype, role, and mechanism of action of these cells in Francisella infection are therefore the subjects of ongoing study in our lab. Such studies should help to further define correlates of protective immunity for future vaccine candidates.
Acknowledgments
We are grateful to our CBER colleagues Dr. Ronald Rabin and Dr. Suzanne Epstein, and to Dr. Dragana Jankovic (NIAID/NIH), for their thorough and thoughtful reviews of the manuscript, and to Ms. Elizabeth Hamilton for excellent technical assistance.
Footnotes
Abbreviations used in this paper: BMMØ, bone marrow derived macrophage(s); cDMEM, complete DMEM; i.d., intradermal; LVS, live vaccine strain.
References
- 1.Tarnvik, A. 1989. Nature of protective immunity to Francisella tularensis. Rev. Infect. Dis. 11:440–451. [PubMed] [Google Scholar]
- 2.Dennis, D.T., T.V. Inglesby, D.A. Henderson, J.G. Bartlett, M.S. Ascher, E. Eitzen, A.D. Fine, A.M. Friedlander, J. Hauer, M. Layton, et al. 2001. Tularemia as a biological weapon: medical and public health management. JAMA. 285:2763–2773. [DOI] [PubMed] [Google Scholar]
- 3.Eigelsbach, H.T., and C.M. Downs. 1961. Prophylactic effectiveness of live and killed tularemia vaccines. J. Immunol. 87:415–425. [PubMed] [Google Scholar]
- 4.Elkins, K.L., S.C. Cowley, and C.M. Bosio. 2003. Innate and adaptive immune responses to an intracellular bacterium, Francisella tularensis live vaccine strain. Microbes Infect. 5:132–142. [DOI] [PubMed] [Google Scholar]
- 5.Tarnvik, A., M. Eriksson, G. Sandstrom, and A. Sjostedt. 1992. Francisella tularensis - a model for studies of the immune response to intracellular bacteria in man. Immunology. 76:349–354. [PMC free article] [PubMed] [Google Scholar]
- 6.Fortier, A.H., M.V. Slayter, R. Ziemba, M.S. Meltzer, and C.A. Nacy. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 59:2922–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elkins, K.L., R.K. Winegar, C.A. Nacy, and A.H. Fortier. 1992. Introduction of Francisella tularensis at skin sites induces resistance to infection and generation of protective immunity. Microb. Pathog. 13:417–421. [DOI] [PubMed] [Google Scholar]
- 8.Leiby, D.A., A.H. Fortier, R.M. Crawford, R.D. Schreiber, and C.A. Nacy. 1992. In vivo modulation of the murine immune response to Francisella tularensis LVS by administration of anticytokine antibodies. Infect. Immun. 60:84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sjostedt, A., R.J. North, and J.W. Conlan. 1996. The requirement of tumour necrosis factor-alpha and interferon-gamma for the expression of protective immunity to secondary murine tularaemia depends on the size of the challenge inoculum. Microbiology. 142:1369–1374. [DOI] [PubMed] [Google Scholar]
- 10.Bosio, C.M., and K.L. Elkins. 2001. Susceptibility to secondary Francisella tularensis LVS infection in B cell deficient mice is associated with neutrophilia but not with defects in specific T cell mediated immunity. Infect. Immun. 69:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yee, D., T.R. Rhinehart-Jones, and K.L. Elkins. 1996. Loss of either CD4+ or CD8+ T cells does not affect the magnitude of protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 157:5042–5048. [PubMed] [Google Scholar]
- 12.Conlan, J.W., A. Sjostedt, and R.J. North. 1994. CD4+ and CD8+ T-cell-dependent and -independent host defense mechanisms can operate to control and resolve primary and secondary Francisella tularensis LVS infection in mice. Infect. Immun. 62:5603–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green, L.C., A. Wagner, J. Glogowski, P.L. Skipper, J.S. Wishnok, and S.R. Tannenbaum. 1982. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 126:131–138. [DOI] [PubMed] [Google Scholar]
- 14.Fortier, A.H., T. Polsinelli, S.J. Green, and C.A. Nacy. 1992. Activation of macrophages for destruction of Francisella tularensis: identification of cytokines, effector cells, and effector molecules. Infect. Immun. 60:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green, S.J., C.A. Nacy, R.D. Schreiber, D.L. Granger, R.M. Crawford, M.S. Meltzer, and A.H. Fortier. 1993. Neutralization of gamma interferon and tumor necrosis factor alpha blocks in vivo synthesis of nitrogen oxides from L-arginine and protection against Francisella tularensis infection in Mycobacterium bovis BCG-treated mice. Infect. Immun. 61:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elkins, K.L., T. Rhinehart-Jones, C.A. Nacy, R.K. Winegar, and A.H. Fortier. 1993. T-cell-independent resistance to infection and generation of immunity to Francisella tularensis. Infect. Immun. 61:823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elkins, K.L., T.R. Rhinehart-Jones, S.J. Culkin, D. Yee, and R.K. Winegar. 1996. Minimal requirements for murine resistance to infection with Francisella tularensis LVS. Infect. Immun. 64:3288–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaible, U.E., H.L. Collins, and S.H. Kaufmann. 1999. Confrontation between intracellular bacteria and the immune system. Adv. Immunol. 71:267–377. [DOI] [PubMed] [Google Scholar]
- 19.Harty, J.T., and M.J. Bevan. 1995. Specific immunity to Listeria monocytogenes in the absence of IFNγ. Immunity. 3:109–117. [DOI] [PubMed] [Google Scholar]
- 20.White, D.W., V.P. Badovinac, G. Kollias, and J.T. Harty. 2000. Cutting edge: antilisterial activity of CD8+ T cells derived from TNF-deficient and TNF/perforin double-deficient mice. J. Immunol. 165:5–9. [DOI] [PubMed] [Google Scholar]
- 21.Badovinac, V.P., and J.T. Harty. 2000. Adaptive immunity and enhanced CD8+ T cell response to Listeria monocytogenes in the absence of perforin and IFN-gamma. J. Immunol. 164:6444–6452. [DOI] [PubMed] [Google Scholar]
- 22.Ruuls, S.R., R.M. Hoek, V.N. Ngo, T. McNeil, L.A. Lucian, M.J. Janatpour, H. Korner, H. Scheerens, E.M. Hessel, J.G. Cyster, et al. 2001. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity. 15:533–543. [DOI] [PubMed] [Google Scholar]
- 23.Olleros, M.L., R. Guler, N. Corazza, D. Vesin, H.-P. Eugster, G. Marchal, P. Chavarot, C. Mueller, and I. Garcia. 2002. Transmembrane TNF induces an efficient cell-mediated immunity and resistance to Mycobacterium bovis bacillus Calmette-Guerin infection in the absence of secreted TNF and lymphotoxin-alpha. J. Immunol. 168:3394–3401. [DOI] [PubMed] [Google Scholar]
- 24.Roach, D.R., A.G. Bean, C. Demangel, M.P. France, H. Briscoe, and W.J. Britton. 2002. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol. 168:4620–4627. [DOI] [PubMed] [Google Scholar]
- 25.Roach, D.R., H. Briscoe, B. Saunders, M.P. France, S. Riminton, and W.J. Britton. 2001. Secreted lymphotoxin-alpha is essential for the control of an intracellular bacterial infection. J. Exp. Med. 193:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 27.MacDonald, H.R. 1995. NK1.1+ T cell receptor-alpha/beta+ cells: new clues to their origin, specificity, and function. J. Exp. Med. 182:633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stenger, S., D.A. Hanson, R. Teitelbaum, P. Dewan, K.R. Niazi, C.J. Froelich, T. Ganz, S. Thoma-Uszynski, A. Melian, C. Bogdan, et al. 1998. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. 282:121–125. [DOI] [PubMed] [Google Scholar]
- 29.Stenger, S., R.J. Mazzaccaro, K. Uyemura, S. Cho, P.F. Barnes, J.-P. Rosat, A. Sette, M.B. Brenner, S.A. Procelli, B.R. Bloom, and R.L. Modlin. 1997. Differential effects of cytolytic T cell subsets on intracellular infection. Science. 276:1684–1687. [DOI] [PubMed] [Google Scholar]
- 30.Phyu, S., S. Sornes, T. Mustafa, A. Tadesse, R. Jonsson, and G. Bjune. 1999. Changes in T-lymphocyte subsets in lungs and spleens of mice with slowly progressive primary Mycobacterium tuberculosis infection: involvement of unconventional T-cell subsets. Scand. J. Immunol. 50:137–144. [DOI] [PubMed] [Google Scholar]
- 31.Hossain, M.S., H. Takimoto, T. Ninomiya, H. Yoshida, K. Kishihara, G. Matsuzaki, G. Kimura, and K. Nomoto. 2000. Characterization of CD4-CD8-CD3+ T-cell receptor-alphabeta+ T cells in murine cytomegalovirus infection. Immunology. 101:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadena, T., G. Matsuzaki, S. Fujise, K. Kishihara, H. Takimoto, M. Sasaki, M. Beppu, S. Nakamura, and K. Nomoto. 1997. TCR alpha beta+ CD4− CD8− T cells differentiate extrathymically in an lck-independent manner and participate in early response against Listeria monocytogenes infection through interferon-gamma production. Immunology. 91:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunn, P.L., and R.J. North. 1991. Resolution of primary murine listeriosis and acquired resistance to lethal secondary infection can be mediated predominatly by Thy-1+ CD4− CD8− cells. J. Infect. Dis. 164:869–877. [DOI] [PubMed] [Google Scholar]