

Abstract
Plasmacytoid dendritic cells (pDCs) have been identified as a potent secretor of the type I interferons (IFNs) in response to CpG as well as several viruses. In this study, we examined the molecular mechanism of virus recognition by pDCs. First, we demonstrated that the CD11c+Gr-1intB220+ pDCs from mouse bone marrow secreted high levels of IFN-α in response to either live or UV-inactivated Herpes simplex virus-2 (HSV-2). Next, we identified that IFN-α secretion by pDCs required the expression of the adaptor molecule MyD88, suggesting the involvement of a Toll-like receptor (TLR) in HSV-2 recognition. To test whether a TLR mediates HSV-2–induced IFN-α secretion from pDCs, various knockout mice were examined. These experiments revealed a clear requirement for TLR9 in this process. Further, we demonstrated that purified HSV-2 DNA can trigger IFN-α secretion from pDCs and that inhibitory CpG oligonucleotide treatment diminished HSV-induced IFN-α secretion by pDCs in a dose-dependent manner. The recognition of HSV-2 by TLR9 was mediated through an endocytic pathway that was inhibited by chloroquine or bafilomycin A1. The strict requirement for TLR9 in IFN-α secretion was further confirmed by the inoculation of HSV-2 in vivo. Therefore, these results demonstrate a novel mechanism whereby the genomic DNA of a virus can engage TLR9 and result in the secretion of IFN-α by pDCs.
Keywords: type I interferons, CpG motif, innate immunity, virus infection, DNA virus
Introduction
Herpes simplex virus-2 (HSV-2) is one of the most common sexually transmitted diseases with a high prevalence of 45 million in the USA (1). HSV-2 is primarily transmitted via exposure at the genital mucosal surfaces, which leads to the establishment of latency in the sacral ganglia. Although pharmacological interventions are used for the treatment of HSV-2–related symptoms, there are no preventative vaccines or curative measures available for this disease. Toward developing vaccines to prevent HSV-2 transmission, a clear understanding of the mechanism of how immunity is generated within the relevant mucosal sites is necessary.
DCs are the most potent APCs found throughout the body, particularly at the sites of potential encounter with pathogens such as the mucosal surfaces. In a recent study, using an in vivo mouse model for genital herpes, we demonstrated an essential role of DCs in Th1 induction following HSV-2 infection in the vaginal mucosa (2). Specifically, CD11b+ DCs were found to present virus antigens to CD4+ T cells in the draining lymph nodes, and this interaction was crucial in the generation of the potent Th1 response (2) needed to provide protective immunity. Consistent with the ability of these cells to induce T cell activation, the DCs in the draining lymph nodes were found to increase their levels of costimulatory molecules as early as 24 h after intravaginal infection with HSV-2 (2). One of the major pathways of DC activation involves the recognition of conserved molecules found on microbes via the Toll-like receptors (TLRs). Although the TLRs have been shown to play a crucial role in the innate recognition of bacterial and fungal pathogens (3), the role of TLRs in antiviral immunity in vivo is unclear.
Of the number of DC subtypes identified in recent years, the plasmacytoid DC (pDC) is characterized for its potent ability to secrete type I interferons in response to viruses (4–7). The phenotype of these type I IFN producing pDCs has been found to differ by species, as they are CD4+ CD11c− in humans (8) but in mice have been identified as CD11c+CD11b−Gr-1+B220+ cells (4, 9). Whereas DCs as a class are thought to be the professional antigen presenting cells specializing in antigen uptake, processing, and presentation to T cells, the predominant role of the pDCs appears to be the secretion of type I IFNs. In particular, pDCs can be induced to secrete large amounts of IFN-α when stimulated with oligodeoxynucleotides containing certain CpG motifs (5–7, 10) or viruses including sendai virus (7), influenza (4), and HSV-1 (5, 6). However, this situation may not be universal, as one study found that pDCs are the major producers of IFN-α and IFN-β after murine cytomegalovirus (MCMV), but not lymphocytic choriomeningitis virus (LCMV), infections in vivo (11), thereby highlighting a differing cell requirement for type I IFN responses dependent on the type of viral stimulus. Type I IFNs have an essential role in antiviral defense (12), mediated by pathways involving PKR and 2′-5′ oligoadenylate synthetase (13). Thus, pDCs serve an important role in providing innate antiviral immunity early during virus infection.
Although the ability of pDCs to secrete type I IFNs in response to viruses has been well characterized, the signaling events subsequent to viral infection that lead to type I IFN secretion have not yet been elucidated. In humans, pDCs, as compared with other subsets of DCs, express high levels of TLR7 and TLR9 (7, 14). In mice, pDCs, as well as other DC subsets, are found to express TLR9 (5, 15). The ligand for TLR9 has been identified as unmethylated CpG motifs found within microbial DNA sequences (16). Furthermore, it has been found that different CpG sequences have distinct stimulatory capacities and that certain DNA sequences can inhibit the stimulatory effects of CpG (17). Therefore, we hypothesized that the TLR9 expressed on pDCs could bind to unmethylated CpG motifs present on HSV-2 genomic DNA, thereby serving as the receptor responsible for secretion of type I IFNs by pDCs. Unlike small viruses, the HSV genome contains a high frequency of CpG motifs (18) which have been shown to have significant functional activity in vivo (19).
In this study, we describe a molecular mechanism of HSV-2 recognition by pDCs. We provide evidence that the ability of the pDCs to recognize and secrete IFN-α in response to HSV-2 required TLR9 expression by pDCs. The recognition of the virus does not require replication since live and UV-inactivated HSV-2 induced identical IFN-α secretion from pDCs. Further, we demonstrated that the ligand recognized by TLR9 is likely the genomic DNA of HSV-2. In vivo, the ability of HSV-2 to stimulate IFN-α secretion depended on the functional expression of TLR9. These results represent the first evidence for the requirement of TLR9 signaling in the generation of antiviral immunity, and suggest the importance of TLR9 mediated recognition of other DNA viruses in vivo.
Materials and Methods
Viruses and Viral DNA Isolation.
HSV-2 strain (186TKΔKpn) (20) and HSV-1 strain (KOS) were provided by Dr. David Knipe (Harvard University Medical School, Boston, MA) and were propagated and assayed on Vero cells. Briefly, confluent Vero cells were infected with HSV strains at a multiplicity of infection (MOI) of 0.005 and incubated with virus for 1 h at 37°C. Supernatant containing the virus was removed and cells were incubated at 37°C until complete infection had occurred (2–3 d) in the presence of DMEM with 1% FBS. Cells were centrifuged at 1,500 rpm for 5 min and supernatant was collected and saved. Cells were resuspended in a 1:1 mixture of sterile autoclaved milk and supernatant and frozen overnight at −80°C. Virus was thawed and sonicated, and the sonicated material was clarified by centrifugation at 1,600 rpm for 10 min at 4°C. Pure viral stocks were prepared by a similar method. Briefly, confluent Vero cells were infected as above and after a 2–3 d incubation, cells were centrifuged as described and the cell pellet was discarded. The supernatant was ultracentrifuged at 20,000 g at 15°C for 45 min. The pellet was resuspended in sterile PBS and stored at −80°C. All virus stocks were titered on the Vero cell line before use. In some cases, the purified virus was UV-inactivated by exposure to 1 J/cm2 UV light with a Stratalinker® UV Cross-linker (Stratagene) as described previously (21). To obtain HSV-2 genomic DNA, QIAamp DNA Mini Kit (QIAGEN) was used to isolate viral DNA from purified HSV-2 according to the manufacturer's instruction for Blood and Body Fluid Spin Protocol. The concentration of the resultant purified genomic DNA was determined by spectrophotometric reading of the optical density at 260 nm and the quality of DNA was assessed by agarose gel electrophoresis.
Animals.
6- to 8-wk-old male and female B6 129F2 mice were obtained from The Jackson Laboratory. MyD88−/− (22), TLR4−/− (23), and TLR9−/− (16) mice (all on the B6 129F2 background) were previously described. Mice received i.v. tail vein injections of UV-inactivated HSV-2 (107 PFU/mouse) in 200 μl volumes while restrained. All procedures used in this study complied with federal guidelines and institutional policies by the Yale Animal Care and Use Committee.
Antibodies.
The following antibodies were used for the identification of pDCs and other cell types: anti-CD11c (HL3), anti-CD11b (M1/70), anti-Ly-6G (Gr-1) (RB6–8C5), and anti-B220 (RA3–6B2), all purchased from BD Biosciences.
Bone Marrow Cell Preparation and Purification.
Bone marrow cells were prepared from the femurs and tibiae of mice. A single cell suspension was prepared and red blood cells were lysed. Total bone marrow populations were stimulated in the presence of the indicated reagents in 96-well, round-bottomed tissue culture plates at 106 cells in 200 μl of medium per well for 18 h. CD11c-depleted, CD11b-depleted, or B220-depleted bone marrow cells were prepared by negative selection using the LD-depletion columns (Miltenyi Biotech) according to the manufacturer's instructions. For FACS® sorting of pDCs and nonpDCs, bone marrow cells were stained with FITC-conjugated anti-CD11c, PE-conjugated anti-Gr-1, and CyChrome-conjugated anti-B220. Cells were washed extensively and sorted using a FACSVantage™. Sorted cells were incubated with various stimuli at 2 × 105 cells per well in 200 μl media. 18 h later, IFN-α levels were measured using ELISA.
In Vitro pDC Stimulation.
The following reagents were used to stimulate cells in vitro: Poly I:C (Amersham Biosciences) at 25 μg/ml, R-848 (InvivoGen) at 1 μg/ml, stimulatory CpG 2084 (TCCTGACGTTGAAGT), and inhibitory CpG 2088 (TCCTGGCGGGGAAGT; synthesized by Invitrogen) at various concentrations as indicated. Both of these oligonucleotides had the phosphodiester backbone. Purified HSV (live or UV-inactivated) was added to pDCs at a final concentration of 5 × 106 PFU/ml. DNase treatment of genomic HSV-2 DNA was performed at 37°C for 1 h using 312.5 Kunitz units/μl RNase-free DNase (QIAGEN). To prevent endosome acidification, chloroquine or bafilomycin A1 (Sigma-Aldrich) was used before in vitro stimulation. Briefly, bone marrow cells were preincubated with 0.01–0.001 mM chloroquine for 2 h or 10–500 nM bafilomycin A1 for 30 min at 37°C before the addition of various stimulants. Cells were then incubated for an additional 18 h at 37°C and cell viability was confirmed by trypan blue exclusion assays. Supernatants were analyzed for cytokine production by ELISA.
ELISA Measurement of Murine IFN-α and IL-12p40.
The levels of mouse IFN-α were determined by ELISA as described previously (11). Briefly, 96-well plates were coated with rat anti–mouse IFN-α (mAb F-18; HyCult Biotechnology) in carbonate buffer overnight. Nonspecific binding was blocked and plates were incubated with different concentrations of samples. Some wells were incubated with known concentrations of recombinant mouse IFN-α standard in duplicate (HyCult Biotechnology). After an overnight incubation, plates were sequentially incubated with polyclonal rabbit anti–mouse IFN-α (PBL Biomedical Laboratories) for 1 h and with horseradish peroxidase-conjugated donkey anti–rabbit F(ab′)2 (Jackson ImmunoResearch Laboratories) for 1 h. Plates were developed using substrate solution TMB (eBioscience) and the reaction was stopped with 2N H2SO4. Optical density at 450 nm was measured and the concentration of IFN-α was calculated from the standard curve. The concentrations of IL-12p40 from bone marrow cells stimulated with R848 were measured using the OptEIA kit (BD Biosciences) according to manufacturer's instructions.
Calculations of the Concentrations of the CpG Sequences.
These calculations were based on the assumption that all CpG sequences found in the HSV genome were immunologically active. Since many of the CpG sequences in the HSV genome are highly stimulatory, and others are non-stimulatory due to methylation (19), this assumption approximately reflects the average of the total functional stimulatory CpG content of the HSV genome. The amount of total DNA present within a virion is calculated as follows: 1 PFU = 1 virion which contains a genome of 154 kbp. The average molecular weight of a nucleotide is 323 g/mol 5 × 106 PFU/ml contains 154 kbp × 5 × 106 = 7.7 × 1011 bp/ml = 1.54 × 1012 nucleotide/ml. 1.54 × 1012 nucleotide/ml ÷ 6.02 × 1023 molecules (nucleotide)/mol = 2.56 × 10−12 mol/ml. 2.56 × 10−12 mol/ml × 323 g/mol = 8.26 × 10−10 g/ml or 0.826 ng/ml. Thus, the purified HSV-2 DNA, used at the concentration of 547.2 ng/ml, corresponded to roughly 600-fold higher levels than that found in the virions at 5 × 106 PFU/ml (547.2 ng/ml ÷ 0.826 ng/ml = 662.4 or roughly 600-fold).
The amount of immunostimulatory CpG oligonucleotide needed to induce equivalent levels of IFN-α in pDCs stimulated with 5 × 106 PFU/ml of HSV-2 was 5 μg/ml (see Fig. 2 A). The amount of virus-associated DNA present in 5 × 106 PFU/ml is 0.826 ng/ml (see calculation above). Therefore, virion-associated DNA was ∼6,000-fold more effective in IFN-α induction compared with synthetic soluble CpG oligonucleotide.
Figure 2.
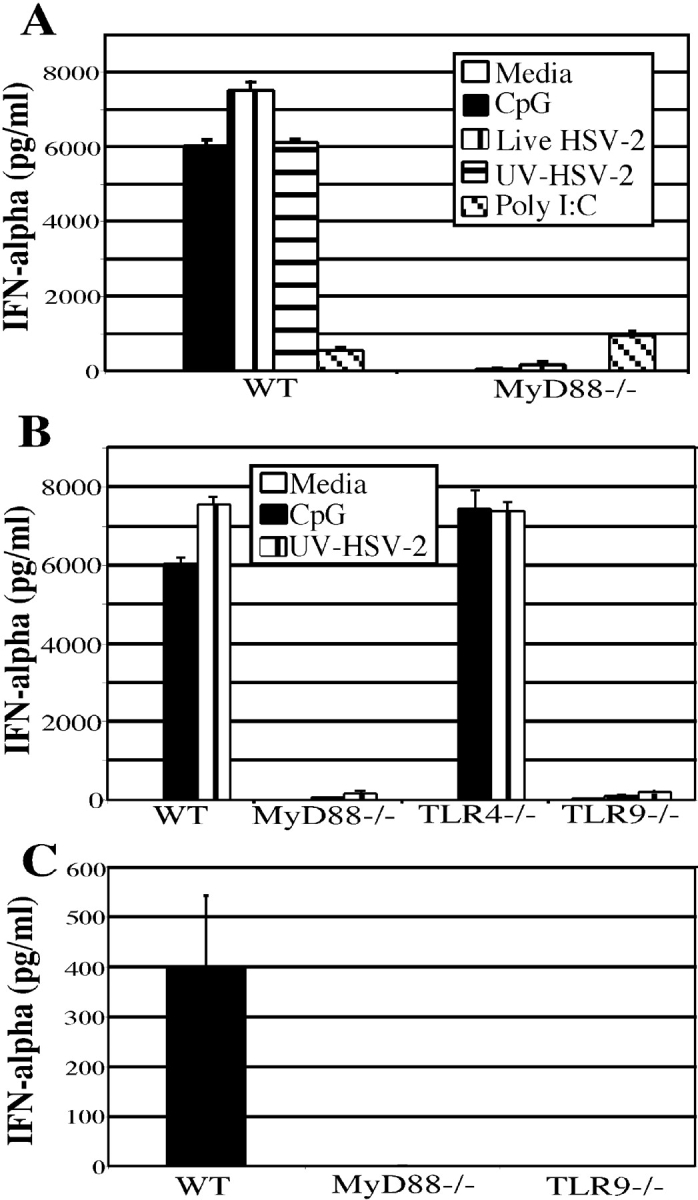
IFN-α secretion requires MyD88 and TLR9 signaling in vitro and in vivo. (A) Bone marrow cells from wild-type or MyD88−/− mice were cultured at 5 × 106 cells/ml for 18 h with either media alone, CpG (5 μg/ml) or live HSV-2 (5 × 106 PFU/ml), UV-HSV-2 (5 × 106 PFU/ml), or Poly I:C (25 μg/ml) and IFN-α was measured by ELISA. (B) IFN-α levels were measured from bone marrow cells harvested from wild-type (B6 129F2), MyD88−/−, TLR4−/−, and TLR9−/− mice stimulated for 18 h with media alone, CpG (5 μg/ml), or UV-HSV-2 (5 × 106 PFU/ml) at 5 × 106 cells/ml. (C) Wild-type, MyD88−/−, and TLR9−/− mice received i.v. injections of 107 PFU of UV-HSV-2. 6 h after injection, serum was collected by cardiac puncture and measured for IFN-α by ELISA. Results are representative of three independent experiments.
The amount of iCpG required to completely block virus-induced IFN-α was 100 μg/ml. As the amount of virus-associated DNA present in 5 × 106 PFU/ml is 0.826 ng/ml, this amount of iCpG represents 120,000-fold higher level compared with the level of CpG motifs present in the HSV genome. As 10 μg/ml of iCpG was required to completely inhibit 100 ng/ml of stimulatory CpG (see Fig. 3 A), or 100-fold, HSV-2–associated DNA is 120,000 ÷ 100 = 1,200 times more difficult to inhibit than stimulatory CpG oligonucleotide.
Figure 3.

HSV-2 genomic DNA activates IFN-α secretion from pDCs via TLR9. Bone marrow cells from wild-type, MyD88−/−, and TLR9−/− mice were stimulated with (A) stimulatory CpG alone (100 ng/ml) or with 1 μg/ml or 10 μg/ml inhibitory CpG (iCpG) or (B) UV-HSV-2 (5 × 106 PFU/ml) with or without iCpG at 1 μg/ml, 10 μg/ml, or 100 μg/ml. Cells were cultured at 5 × 106 cells/ml with stimuli for 18 h and IFN-α was measured by ELISA. (C) Bone marrow cells harvested from B6 129F2 mice were cultured for 18 h in the presence of either 5 × 106 PFU/ml UV-HSV-2, purified HSV-2 DNA (547 ng/ml or 600× more DNA than that present in 5 × 106 PFU/ml of virions) as well as 10-fold dilutions (54.72 ng/ml or 60×, 5.42 ng/ml or 6×), HSV-2 DNA (547 ng/ml) digested with DNase, or DNase alone. 18 h later, supernatants were harvested and IFN-α levels were measured by ELISA.
Results
Bone Marrow Contains a High Frequency of pDCs That Secrete IFN-α upon HSV-2 Stimulation.
We first examined the distribution of the pDCs capable of secreting IFN-α upon HSV-2 stimulation. Total lymph node, spleen, and bone marrow cells were stimulated with HSV-2 in vitro, and IFN-α levels were measured after 18 h. The lymph node was found to contain very few cells capable of secreting IFN-α in response to UV-inactivated HSV-2 (UV-HSV-2) or Poly I:C stimulation, whereas the spleen and bone marrow contained cells that secreted IFN-α (Fig. 1 A). In particular, the bone marrow cells were found to secrete considerably higher levels of IFN-α in response to UV-HSV-2 than did splenic cells (Fig. 1 A). Next, the presence of pDCs in the bone marrow was analyzed by FACS®. The total bone marrow cell population was found to contain ∼2.6% CD11c+ cells (Fig. 1 B, left panel; CD11c+ cells = R2 gate), which displayed the characteristic pDC phenotype, namely slightly larger than lymphocytes with a low granularity (Fig. 1 B, right panel). In addition, these cells were found to be B220+ and expressed low levels of CD11b (unpublished data) and varying levels of Gr-1 (Fig. 1 B, center panel), also consistent with the previously described mouse pDC phenotype (4, 9, 10).
Figure 1.

Bone marrow contains a high frequency of pDCs that secrete IFN-α. (A) Total lymph node, spleen, and bone marrow cell suspensions from B6 129F2 mice were cultured with either media alone, Poly I:C, or UV-HSV-2, and IFN-α secretion was measured by ELISA. (B) Bone marrow cells were analyzed by flow cytometry. CD11c+ cells in gate R2 were examined for FSC vs. SSC as well as their expression of Gr-1 and B220. (C) Total bone marrow cells depleted of either CD11c+, CD11b+, or B220+ cells were stimulated for 18 h with either media alone, live HSV-2 (5 × 106 PFU/ml), or UV-HSV-2 (5 × 106 PFU/ml). IFN-α was detected by ELISA. (D) Total bone marrow cells were sorted into CD11c+B220+ (pDC), CD11c−B220+(B cells), and CD11c−B220− fractions and cultured with either media alone, live HSV-2 (5 × 106 PFU/ml), UV-HSV-2 (5 × 106 PFU/ml), UV-HSV-1 (5 × 106 PFU/ml), CpG (5 μg/ml), or Poly I:C (25 μg/ml) for 18 h. Cells were cultured at 106 cells/ml and IFN-α was measured by ELISA.
To determine the precise phenotype of cells capable of secreting IFN-α in response to HSV-2 stimulus, bone marrow cells were depleted of either CD11c+, CD11b+, or B220+ cells using a magnetic depletion column and incubated with live or UV-HSV-2. IFN-α secretion was completely diminished in the population that was depleted of CD11+ or B220+ cells (Fig. 1 C). CD11b-depleted bone marrow cells were able to secrete levels of IFN-α higher than an equivalent number of total bone marrow cells, likely due to an ∼40% enrichment of pDCs caused by the CD11b-depletion (Fig. 1 C, and unpublished data). The complete block of IFN-α secretion in CD11c- or B220-depleted bone marrow cells suggested that pDCs are the cell type responsible for IFN-α secretion. To positively identify the phenotype of cells that secrete IFN-α in the bone marrow, CD11c+B220+ cells, as well as the rest of the nonpDC bone marrow populations (CD11c−B220+ and CD11c−B220−) were sorted by FACS® and stimulated with HSV-2. The CD11c+B220+ cells were the only sorted population capable of secreting IFN-α in response to live or UV-HSV-2 (Fig. 1 D), thereby identifying CD11c+B220+ pDCs as the IFN-α producing cells in the bone marrow. An equivalent number of total (unsorted) bone marrow cells were also found to secrete IFN-α, albeit to a lesser extent, likely due to the lower proportion of pDCs contained within total bone marrow cells. Similar levels of IFN-α were secreted by the sorted pDCs in response to live or UV-inactivated HSV-2, indicating that virus replication was not required for this stimulation (Fig. 1 D). Moreover, UV-inactivated HSV-2 and HSV-1 elicited similar levels of IFN-α secretion from pDCs (Fig. 1 D). These pDCs also secreted IFN-α in response to CpG (Fig. 1 D), suggesting that they express functional TLR9.
MyD88 Is Required for HSV-2–induced IFN-α Secretion from pDCs.
To determine if a toll-like receptor is involved in the recognition of HSV-2 by pDCs, the requirement of MyD88 in HSV-2–induced IFN-α secretion was examined. MyD88 is an obligate adaptor molecule that interacts with the intracellular domains of all known TLRs and is necessary for subsequent signaling events. Although MyD88 deficiency did not affect the phenotype or the frequency of the bone marrow pDCs as determined by FACS® (data not shown), MyD88−/− bone marrow cells were not capable of secreting IFN-α in response to live or inactivated HSV-2 (Fig. 2 A). In addition, sorted pDCs from MyD88−/− mice were unable to secrete IFN-α in response to HSV-2 (unpublished data), thereby demonstrating that a pure population of pDCs lacking MyD88 is also unable to secrete IFN-α in response to HSV-2. The lack of IFN-α secretion from MyD88−/− pDCs was specific to HSV-2 and CpG, as the IFN-α response to Poly I:C by MyD88−/− bone marrow was similar to that seen in wild-type bone marrow (Fig. 2 A), presumably owing to the MyD88-independent pathway for type I IFN secretion for TLR3 (24, 25). Therefore, an intact MyD88-dependent pathway is required for HSV-2–induced IFN-α secretion from pDCs.
TLR9 Is Required for HSV-induced IFN-α Secretion from pDCs.
The finding that HSV-2–induced IFN-α secretion depended on MyD88 led us to examine the specific role of TLR9 in this process. TLR9 was chosen as a target of investigation because pDCs are known to express high levels of TLR9 and the natural ligand for this receptor, CpG, is found abundantly in HSV-2 genomic DNA (18). Using real-time PCR, we observed that the sorted pDCs expressed TLR9 (unpublished data). To investigate this hypothesis, the bone marrow cells of wild-type, MyD88−/−, and TLR9−/− mice were stimulated in vitro with CpG or UV-HSV-2. As a control, bone marrow cells from TLR4−/− mice were also examined in parallel. Strikingly, TLR9−/− cells were unable to secrete IFN-α in response to HSV-2, whereas TLR4−/− or wild-type bone marrow cells secreted similar high levels of IFN-α (Fig. 2 B). The ability of the TLR4−/− bone marrow cells to secrete IFN-α to HSV-2 at the normal levels eliminated the possibility that LPS contamination accounted for the observed IFN-α secretion by pDCs. These data clearly demonstrate the requirement for TLR9 in HSV-2 recognition by pDCs, and that this interaction is critical in the ability of pDCs to secrete IFN-α in response to HSV-2.
TLR9 Is Required for In Vivo HSV-2–mediated IFN-α Secretion.
In a previous study, pDCs were identified as the primary source of IFN-α secretion after i.v. inoculation of viruses including HSV-1 (26). Using a similar approach, we examined the in vivo relevance of HSV-2 recognition by TLR9 in IFN-α secretion. Mice were injected i.v. with either PBS or 107 PFU of UV-HSV-2 and 6 h later, serum was collected for measurement of IFN-α levels by ELISA. Serum from wild-type mice injected with UV-HSV-2 was found to contain significant levels of IFN-α, whereas no IFN-α was detected in the serum of MyD88−/− or TLR9−/− mice injected with UV-HSV-2 (Fig. 2 C). Thus, secretion of IFN-α upon in vivo inoculation of HSV-2 also requires an intact TLR9 signaling pathway.
HSV-2 Genomic DNA Provides the Ligand for TLR9 Expressed by pDCs.
Our data indicated that TLR9 is required for the HSV-mediated induction of IFN-α secretion from pDCs. The only known ligand for TLR9 is the unmethylated CpG motif, which is found in bacterial and viral DNA. To examine whether CpG motifs within the HSV-2 genome were responsible for binding TLR9 and inducing IFN-α secretion, TLR9 signaling was specifically blocked using an oligonucleotide containing the inhibitory CpG motif (17). The activity of inhibitory CpG (iCpG; CpG 2088) was first confirmed by coincubation with stimulatory CpG (CpG 2084). As shown in Fig. 3 A, when stimulatory CpG and 10 or 100 times that amount of iCpG was used to treat bone marrow cells, IFN-α secretion was completely diminished. Similarly, when iCpG was used with HSV-2, there was a dose-dependent reduction in the levels of IFN-α secreted that correlated with increasing the amount of iCpG used (Fig. 3 B). The amount of iCpG (100 μg/ml) that was required to completely inhibit virion-mediated IFN-α secretion from pDCs was ∼120,000-fold higher than the amount of CpG motifs present within the added virions (see Materials and Methods for calculation). Thus, compared with the synthetic CpG oligonucleotide, the virion-associated DNA was 1,200 times more difficult to inhibit with iCpG. The bone marrow cells of the MyD88−/− and TLR9−/− mice failed to induce an IFN-α response under the same conditions (Fig. 3 B). Thus, these results suggested that CpG motifs present in the HSV-2 genome are highly capable of activating TLR9 and inducing IFN-α secretion from pDCs.
Finally, the viral ligand responsible for TLR9 activation in pDCs was examined. First, stimulation with DNA isolated from purified HSV-2 virions resulted in IFN-α secretion from pDCs (Fig. 3 C). Roughly 600-fold higher amounts of purified naked viral DNA was required to produce similar IFN-α secretion from pDCs stimulated with 5 × 106 PFU/ml of purified UV-inactivated virions (Fig. 3 C; see Materials and Methods for calculation). Digestion of purified HSV-2 DNA by deoxyribonuclease (DNase) completely diminished its ability to stimulate pDC secretion of IFN-α (Fig. 3 C). This effect was not due to the toxicity mediated by DNase since DNase treatment of R848, a ligand for TLR7 that is not digested by DNase, did not alter cytokine secretion from pDCs (unpublished data). Thus, the genomic DNA of HSV-2 presented in the virion-encapsulated form represents a potent activator of TLR9 expressed by pDCs.
Recognition of HSV-2 by TLR9 Requires an Intact Endocytic Pathway.
In an effort to understand the intracellular mechanism of HSV-2 recognition by pDCs, we examined the requirement for the endocytic pathway. As TLR9 must be recruited to the lysosomal compartment upon synthetic CpG oligonucleotide-driven stimulation (27), we hypothesized that HSV-2 recognition also requires the maturation of endosomal vesicles in order to activate the TLR9 pathway. To test this hypothesis, bone marrow cells from wild-type mice were first treated with inhibitors of acidification of the endosomes, chloroquine or bafilomycin A1. These inhibitors of endosomal acidification reduced HSV-2–induced secretion of IFN-α from pDCs in a dose dependent manner (Fig. 4, A and C) . The reduction in IFN-α secretion was not due to the toxicity of these reagents, as IL-12p40 secretion from pDCs stimulated with R848, a ligand for TLR7, was not affected by pretreatment with these inhibitors (Fig. 4, B and D). Thus, HSV-2–mediated activation of TLR9 also requires endocytosis and acidification of endosomes within the pDCs.
Figure 4.

Endosomal acidification is required for pDC activation by HSV-2. Wild-type bone marrow cells were pretreated with either media alone, chloroquine (A and B), or with bafilomycin A1 (C and D) at the indicated concentrations. Either stimulatory CpG oligonucleotide (5 μg/ml), UV-HSV-2 (5 × 106 PFU/ml), or R848 (1 μg/ml) were added to the pretreated cells and incubated for 18 h at 37°C. Supernatants were collected and IFN-α (A and C) or IL-12p40 (B and D) levels were measured using ELISA.
Discussion
The TLRs have been shown to play a crucial role in the innate recognition of microbial pathogens in mammals and insects (3). Stimulation through the TLR molecules provides a vital signal for the activation of DCs and macrophages in response to bacterial and fungal stimuli including zymosan (TLR2), lipopolysaccharide (TLR4), lipoteichoic acid (TLR4), flagellin (TLR5), and unmethylated CpG DNA (TLR9) (3). However, little is known with respect to the role of TLRs in antiviral immunity in vivo. The pDCs have been known for their exquisite ability to secrete high levels of type I IFNs in response to virus infection. Both the expression of TLR7 and TLR9, as well as the responsiveness to their respective ligands, have been documented in both human and mouse pDCs (7, 14, 28, 29). Because the HSV-2 genome contains a high frequency of CpG motifs (18), we hypothesized that TLR9 may be involved in the recognition of HSV-2 by pDCs.
The signaling pathways of TLRs have been extensively studied. All known TLRs have been shown to utilize the MyD88 signaling pathway. The MyD88 adaptor protein associates with the TLRs via interaction with the Toll/IL-1 receptor (TIR) domains. Upon stimulation, MyD88 recruits a death domain–containing serine/threonine kinase, the IL-1R–associated kinase (IRAK). Upon IRAK phosphorylation, it in turn associates with TRAF6, leading to activation of two separate signaling pathways, JNK and NF-κB (3). Recently, MyD88-independent TLR signaling events involving TIR domain-containing adaptor protein (TIRAP) (30) and TIR domain-containing adaptor inducing IFN-β (TRIF; reference 25) have been described. However, our results demonstrated a complete dependency for MyD88 and TLR9 in HSV-induced IFN-α secretion from pDCs. Therefore, pDCs seem to possess a unique signaling pathway whereby TLR9 recognition of HSV-2 DNA leads to MyD88 activation that ultimately results in IFN-α secretion, which is not shared by other DC subsets. Future studies are needed to examine the precise mechanism by which MyD88-dependent downstream signaling events result in the activation of the IFN-α promoter after HSV-recognition by pDCs.
In this study, we have demonstrated that the pDCs from mouse bone marrow secrete high levels of IFN-α in response to HSV-2. Remarkably, this recognition does not require virus replication and was blocked by the addition of inhibitory CpG which specifically blocks the activation of TLR9 (17) in a dose-dependent fashion. Further, pDCs from MyD88 or TLR9 deficient mice, but not TLR4-knockout mice, failed to secrete IFN-α. The TLR9-mediated recognition of HSV-2 required acidification of the endosomes similar to that described for synthetic CpG oligonucleotide (27). Finally, in vivo injection of HSV-2 resulted in serum IFN-α secretion only in mice that had intact TLR9 signaling. Here, we propose a model by which viral DNA is recognized by pDCs through TLR9. We have demonstrated that virion-coated viral DNA is at least 600 times more potent than linear naked viral DNA or CpG synthetic oligonucleotide in inducing IFN-α secretion from pDCs. The efficiency of virion-associated viral DNA in triggering TLR9 may be explained by two mechanisms: (a) the virion associated DNA is resistant to degradation by enzymatic digestion by DNase compared with naked viral DNA or synthetic oligonucleotide containing CpG motifs, and (b) the virion-encapsulated DNA may be endocytosed much more efficiently than free DNA in solution. A large body of evidence exists for efficient endocytosis of viruses involving caveolae-, clathrin-, nonclathrin/noncaveolae-mediated endocytic mechanisms (for a review, see reference 31). Collectively, these results provide the first evidence that pDCs recognize CpG motifs present in the genomic DNA of viruses via TLR9, and that this recognition is required for IFN-α secretion by these cells. The TLR9-mediated recognition of HSV implicates the importance of this recognition pathway for other DNA viruses by the pDCs.
Acknowledgments
We wish to thank Dr. Charles Dela Cruz for critical discussion.
A. Iwasaki was supported by a Burroughs Wellcome Career Award in Biomedical Sciences.
References
- 1.Cates, W., Jr. 1999. Estimates of the incidence and prevalence of sexually transmitted diseases in the United States. American Social Health Association Panel. Sex. Transm. Dis. 26:S2–S7. [DOI] [PubMed] [Google Scholar]
- 2.Zhao, X.Y., E. Deak, K. Soderberg, M. Linehan, D. Spezzano, J. Zhu, D.M. Knipe, and A. Iwasaki. 2003. Vaginal submucosal dendritic cells, but not Langerhans' cells, induce protective Th1 responses to herpes simplex virus-2. J. Exp. Med. 197:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–145. [DOI] [PubMed] [Google Scholar]
- 4.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 5.Brawand, P., D.R. Fitzpatrick, B.W. Greenfield, K. Brasel, C.R. Maliszewski, and T. De Smedt. 2002. Murine plasmacytoid pre-dendritic cells generated from flt3 ligand-supplemented bone marrow cultures are immature APCs. J. Immunol. 169:6711–6719. [DOI] [PubMed] [Google Scholar]
- 6.Gilliet, M., A. Boonstra, C. Paturel, S. Antonenko, X.L. Xu, G. Trinchieri, A. O'Garra, and Y.J. Liu. 2002. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 195:953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito, T., R. Amakawa, T. Kaisho, H. Hemmi, K. Tajima, K. Uehira, Y. Ozaki, H. Tomizawa, S. Akira, and S. Fukuhara. 2002. Interferon-alpha and interleukin-12 are induced differentially by Toll-like receptor 7 ligands in human blood dendritic cell subsets. J. Exp. Med. 195:1507–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siegal, F.P., N. Kadowaki, M. Shodell, P.A. Fitzgerald-Bocarsly, K. Shah, S. Ho, S. Antonenko, and Y.J. Liu. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science. 284:1835–1837. [DOI] [PubMed] [Google Scholar]
- 9.Nakano, H., M. Yanagita, and M.D. Gunn. 2001. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 194:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Keeffe, M., H. Hochrein, D. Vremec, I. Caminschi, J.L. Miller, E.M. Anders, L. Wu, M.H. Lahoud, S. Henri, B. Scott, et al. 2002. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J. Exp. Med. 196:1307–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalod, M., T.P. Salazar-Mather, L. Malmgaard, C. Lewis, C. Asselin-Paturel, F. Briere, G. Trinchieri, and C.A. Biron. 2002. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 195:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller, U., U. Steinhoff, L.F. Reis, S. Hemmi, J. Pavlovic, R.M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science. 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 13.Katze, M.G., Y. He, and M. Gale, Jr. 2002. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2:675–687. [DOI] [PubMed] [Google Scholar]
- 14.Hornung, V., S. Rothenfusser, S. Britsch, A. Krug, B. Jahrsdorfer, T. Giese, S. Endres, and G. Hartmann. 2002. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 168:4531–4537. [DOI] [PubMed] [Google Scholar]
- 15.Boonstra, A., C. Asselin-Paturel, M. Gilliet, C. Crain, G. Trinchieri, Y.J. Liu, and A. O'Garra. 2003. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J. Exp. Med. 197:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745. [DOI] [PubMed] [Google Scholar]
- 17.Stunz, L.L., P. Lenert, D. Peckham, A.K. Yi, S. Haxhinasto, M. Chang, A.M. Krieg, and R.F. Ashman. 2002. Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur. J. Immunol. 32:1212–1222. [DOI] [PubMed] [Google Scholar]
- 18.Karlin, S., W. Doerfler, and L.R. Cardon. 1994. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 68:2889–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng, M., D.M. Klinman, M. Gierynska, and B.T. Rouse. 2002. DNA containing CpG motifs induces angiogenesis. Proc. Natl. Acad. Sci. USA. 99:8944–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones, C.A., T.J. Taylor, and D.M. Knipe. 2000. Biological properties of herpes simplex virus 2 replication-defective mutant strains in a murine nasal infection model. Virology. 278:137–150. [DOI] [PubMed] [Google Scholar]
- 21.Eloranta, M.L., and G.V. Alm. 1999. Splenic marginal metallophilic macrophages and marginal zone macrophages are the major interferon-alpha/beta producers in mice upon intravenous challenge with herpes simplex virus. Scand. J. Immunol. 49:391–394. [DOI] [PubMed] [Google Scholar]
- 22.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 9:143–150. [DOI] [PubMed] [Google Scholar]
- 23.Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, and S. Akira. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749–3752. [PubMed] [Google Scholar]
- 24.Oshiumi, H., M. Matsumoto, K. Funami, T. Akazawa, and T. Seya. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 4:161–167. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto, M., S. Sato, K. Mori, K. Hoshino, O. Takeuchi, K. Takeda, and S. Akira. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 169:6668–6672. [DOI] [PubMed] [Google Scholar]
- 26.Barchet, W., M. Cella, B. Odermatt, C. Asselin-Paturel, M. Colonna, and U. Kalinke. 2002. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J. Exp. Med. 195:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmad-Nejad, P., H. Hacker, M. Rutz, S. Bauer, R.M. Vabulas, and H. Wagner. 2002. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 32:1958–1968. [DOI] [PubMed] [Google Scholar]
- 28.Gibson, S.J., J.M. Lindh, T.R. Riter, R.M. Gleason, L.M. Rogers, A.E. Fuller, J.L. Oesterich, K.B. Gorden, X. Qiu, S.W. McKane, et al. 2002. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell. Immunol. 218:74–86. [DOI] [PubMed] [Google Scholar]
- 29.Jarrossay, D., G. Napolitani, M. Colonna, F. Sallusto, and A. Lanzavecchia. 2001. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 31:3388–3393. [DOI] [PubMed] [Google Scholar]
- 30.Horng, T., G.M. Barton, and R. Medzhitov. 2001. TIRAP: an adapter molecule in the Toll signaling pathway. Nat. Immunol. 2:835–841. [DOI] [PubMed] [Google Scholar]
- 31.Sieczkarski, S.B., and G.R. Whittaker. 2002. Dissecting virus entry via endocytosis. J. Gen. Virol. 83:1535–1545. [DOI] [PubMed] [Google Scholar]