

Abstract
The specificity of CD8+ T cell responses can vary dramatically between primary and secondary infections. For example, NP366–374/Db- and PA224–233/Db-specific CD8+ T cells respond in approximately equal numbers to a primary influenza virus infection in C57BL/6 mice, whereas NP366–374/Db-specific CD8+ T cells dominate the secondary response. To investigate the mechanisms underlying this changing pattern of immunodominance, we analyzed the role of antigen presentation in regulating the specificity of the T cell response. The data show that both dendritic and nondendritic cells are able to present the NP366–374/Db epitope, whereas only dendritic cells effectively present the PA224–233/Db epitope after influenza virus infection, both in vitro and in vivo. This difference in epitope expression favored the activation and expansion of NP366–374/Db-specific CD8+ memory T cells during secondary infection. The data also show that the immune response to influenza virus infection may involve T cells specific for epitopes, such as PA224–233/Db, that are poorly expressed at the site of infection. In this regard, vaccination with the PA224–233 peptide actually had a detrimental effect on the clearance of a subsequent influenza virus infection. Thus, differential antigen presentation impacts both the specificity of the T cell response and the efficacy of peptide-based vaccination strategies.
Keywords: antigen-presenting cells, antigen presentation, CD8-positive T lymphocytes, influenza, immunologic memory
Introduction
The CD8+ T cell responses to respiratory virus infections tend to be highly focused in terms of antigen specificity (1). In the case of influenza virus infection of mice, CD8+ T cell responses are typically directed at only a handful of specific epitopes. And in a particularly extreme example, the entire CD8+ T cell response to a mouse parainfluenza virus (Sendai virus) is directed at a single epitope (2, 3). The highly focused nature of CD8+ T cell responses to pathogens indicates that individual epitopes differ in their capacity to induce T cell responses (4). Indeed, depending on their relative contributions to the total T cell response, individual epitopes can be classified as dominant, codominant, or subdominant, thereby establishing an immunodominance hierarchy. Interestingly, the immunodominance status of any given epitope is generally only a relative, rather than intrinsic, characteristic. Thus, elimination of a dominant epitope from a pathogen usually results in the elevation of a previously subdominant epitope to a dominant status (4, 5). Such a switch in T cell specificity does not necessarily affect the magnitude of the total CD8 T cell response, suggesting that epitope immundominance does not directly regulate the extent of T cell expansion.
Understanding immunodominance patterns is critical for the development of effective vaccines designed to promote cellular immunity. Therefore, substantial effort has been directed at identifying dominant epitopes and understanding the mechanisms that regulate dominance hierarchies. These studies have identified several factors that appear to determine the immunodominance hierarchy of MHC class I–restricted epitopes. The most important factor appears to be the density of antigen that is effectively presented on antigen-presenting cells. Antigen density is controlled by the relative efficiency of distinct antigen processing steps such as route of antigen acquisition, protease/proteosome degradation, transport into the endoplasmic reticulum and the affinity of the peptide for class I molecules (or its ability to form stable peptide/class I complexes; references 5–12). The complexity of the T cell repertoire also influences immunodominance and epitope selection in the course of an infection (13). Thus, some epitopes may be subdominant if the number of T cells available to respond to that epitope is especially low.
Recently, it has been shown that T cell immunodominance patterns can differ substantially between primary and secondary responses to infection (14–16). For example, Belz et al. (15) have demonstrated a substantial shift in the relative contributions of T cells specific for two major epitopes, nucleoprotein (NP366–374/Db) and acidic polymerase (PA224–233/Db), between the primary and secondary response in influenza virus–infected C57BL/6 mice. Whereas T cells specific for both of these epitopes are present in equivalent numbers in the lung during the primary response to influenza virus infection, T cells specific for the NP366–374/Db epitope dominate the secondary response (15, 16). In this report, we investigated the mechanism underlying this changing pattern of immunodominance after influenza virus infection. The data show that the NP366–374/Db and PA224–233/Db epitopes of influenza virus are differentially processed by dendritic and nondendritic cells and that this difference in epitope expression favors the expansion of NP366–374/Db specific memory CD8+ T cells during secondary infection.
Materials and Methods
Viruses, Animals, and Infections.
The Enders strain of Sendai virus and influenza virus A/HK-x31 (x31, H3N2) and A/PR8/34 (PR8, H1N1) were grown, stored, and titrated as described previously (13, 17). Female C57BL/6 (CD45.2+) and B6.SJL-Ptprca Pep3b/BoyJ (CD45.2−) mice were purchased from The Jackson Laboratory. Mice (6–12 wk) were anesthetized by i.p. injection with 2,2,2 tribromoethanol and infected intranasally with 250 50% egg infectious doses (EID50) of Sendai virus or 300 EID50 of x31 influenza virus for primary infections and with 3,000 EID50 of PR8 for secondary infections.
Cell Lines and Culture Conditions.
JAWS II, an immature dendritic cell line (American Type Culture Collection CLR-11904), was grown in media containing 5 ng/ml rmGM-CSF. AM11 cells, a retrovirally transformed B6 alveolar macrophage line provided by Dr. Bill Walker (St. Jude's Children's Research Hospital, Memphis, TN) were grown as described previously (18). The T cell thymoma (EL4) and L929/Db cells were grown as described previously (19). L cells transected with the Db MHC genes have been described previously (20). The BWZ.36 fusion partner (21) was a gift of Dr. Nilabh Shastri (University of California, Berkeley, CA). For in vitro infections, the cells were infected with influenza virus, then irradiated at 3,000 rads.
Tissue Preparation.
Lymphocytes were collected from the broncho-alveolar lavage (BAL), spleens, lung, and mediastinal lymph nodes (MLNs)* as described previously (3). Dendritic cells and macrophages were isolated from 5 mm pieces of lungs and spleens that were incubated with 5 ml of a 5 mg/ml stock of Collagenase D (Roche). After digestion, the cells were washed and depleted of erythrocytes. For the isolation of epithelial cells, the lungs were digested and the B and T lymphocytes were removed by panning on goat anti–rat IgG coated flasks followed by complement-mediated cytotoxicity (22–24).
MHC Tetramer Reagents and Analysis.
MHC class I peptide tetramers specific for NP366–374/Db and PA224–233/Db were generated by the Molecular Biology Core Facility at the Trudeau Institute as described previously (25). Tetramer staining was performed for 1 h at room temperature, followed by incubation with anti-CD8-PerCP, and 200,000 events were collected on a Becton Dickinson FACSCalibur™ flow cytometer. Data was analyzed using FlowJo (TreeStar) software.
Cell Sorting and Adoptive Transfer.
For isolation of dendritic cells and macrophages, cells were stained with anti-CD45R-CyChrome (B220), anti-CD11c-FITC, and anti-CD11b-PE. Samples were then sorted on a FACSVantage™ flow cytometer with DiVa options into B220−CD11b+CD11c− (predominantly macrophages) and B220−CD11b+/−CD11c+ (predominantly dendritic cell) populations. For isolation of memory cells, spleen cells were stained with anti-CD8-PE and anti-CD44-FITC and sorted into a purified CD8+CD44+ (memory) population. The purity after sorting was 95% or greater. Isolated dendritic cells and macrophages were used in an antigen presentation assay and purified memory cells (2.3 × 105) were injected i.v. into naive B6.SJL-Ptprca Pep3b/BoyJ mice.
Generation of LacZ-inducible T Cell Hybridomas.
Splenocytes were harvested from C57BL/6 mice 28 d after intranasal challenge with A/HK-x31. 30 × 106 immune splenocytes were cultured with 30 × 106 irradiated (3,000 rad) peptide pulsed (1 μM specific peptide) syngeneic splenocytes for 5 d. Blast cells were enriched by Ficoll and then fused with BWZ.36 cells (26, 27). Resulting hybrids were stained for TCRβ expression and TCRβhi hybrids were cloned by single cell sorting using a MoFlo (DakoCytomation). The resulting clones were tested for specificity using peptide-pulsed L cells transected with the Db MHC genes. Influenza virus peptides (NP366–374, PA224–233, PB1703–711, and HA192–207) were purchased from New England Peptide Inc. and peptide purity was evaluated using reverse-phase HPLC analysis.
Antigen Presentation Assays.
Antigen presentation assays were performed as described previously (21, 27–29). Briefly, hybridomas (105) were cultured with virus-infected or peptide-pulsed cells in flat-bottomed microtiter plates. The plates were incubated overnight, washed with PBS and fixed with β-galactosidase fixative (2% formaldehyde/0.2% glutaraldehyde). Cells were washed again with PBS followed by the addition of 50 μl of a 1 mg/ml X-gal solution (5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, and 2 mM magnesium chloride). After 4 h, the hybridomas were examined under a light microscope for the presence of blue cells.
Bone Marrow–derived Dendritic Cells.
Bone marrow was flushed from the legs of C57BL/6 mice, depleted of erythrocytes, and 2 × 106 lymphocytes were placed into a bacteriological Petri dish with media supplemented with 20 ng/ml recombinant murine granulocyte/monocyte colony-stimulating factor (rmGM-CSF; PeproTech) and incubated at 37°C with 10% CO2 (30). On day 3, an additional 10 ml of CTM containing 20 ng/ml rmGM-CSF was added. On days 6 and 8, half of the cells were removed, centrifuged, and added back to the same plate in 10 ml of fresh media containing 20 ng/ml rmGM-CSF. On day 10 of the culture, the cells were used either in antigen presentation assays or for dendritic cell vaccination (30). For vaccination, the cells were resuspended at 5 × 106/ml and incubated at 37°C for 3 h with peptide at a concentration of 50 μg/ml (31). After pulsing, the dendritic cells were washed and injected i.v. into mice at a final concentration of 0.5 × 106 per mouse.
Results
Changing Patterns of Immunodominance during Influenza Virus Infection.
The CD8+ T cell response to intranasal influenza virus infection in C57BL/6 mice is predominantly directed against epitopes derived from the nucleoprotein and acid polymerase proteins (NP366–374/Db and PA224–233/Db; 15, 16). Whereas T cells specific for both of these epitopes are present in equivalent numbers in the lung airways during a primary response, T cells specific for the NP366–374/Db epitope dominate a secondary (memory) response (15). To further investigate these changing patterns of immunodominance, we undertook a detailed kinetic and phenotypic analysis of the NP366–374/Db- and PA224–233/Db-specific T cell response after primary and secondary influenza virus infection. As shown in Fig. 1 , NP366–374/Db- and PA224–233/Db-specific T cells were equally represented in the lung airways and parenchyma throughout the primary response to x31 infection. In contrast, NP366–374/Db-specific T cells dominated the secondary (memory) T cell response to a subsequent PR8 infection, during both the early and late stages of the secondary response. The dominance of NP366–374/Db-specific T cells could not be attributed to a higher precursor frequency of memory cells specific for the NP366–374/Db epitope before the secondary infection. Indeed, the number of memory cells specific for each epitope were the same in the lungs, MLN, and spleen at the time of secondary infection, as determined by tetramer staining and ELISPOT analysis (∼11,000 in the lungs, 8,000 in the MLN, and 80,000 in the spleen, data not depicted). Both populations of memory T cells were also indistinguishable on the basis of phenotype (using the CD44, CD43, Fas, NKG2, CD62L, CD11a markers), cytolytic activity, and ability to proliferate in vitro after restimulation (unpublished data). There were also no functional differences between NP366–374/Db and PA224–233/Db-specific effector T cells generated in either the primary or secondary T cell responses (unpublished data, 15, 16).
Figure 1.

Kinetics of the NP366–374/Db and PA224–233/Db-specific responses after primary and secondary infection with influenza virus. C57BL/6 mice (three mice per time point) were infected intranasally with 300 EID50 x31 on day 0 and given a secondary intranasal infection with 3,000 EID50 PR8 on day 42 (indicated by arrow). Tissues were removed at the indicated time points and stained with anti-CD8 and NP366–374/Db- or PA224–233/Db-specific tetramers. Shown are the number of NP- (closed) and PA- (open) specific T cells. The data shown are one of two complete kinetic experiments with similar results.
Changing Patterns of Immunodominance Are Not Dependent on Virus Strain, Dose, or Prior Infection.
To determine whether the change in T cell dominance during primary and secondary infections reflected intrinsic differences between the x31 and PR8 influenza virus strains, we reversed the order of viral infections (PR8 followed by x31). As shown in Table I, the pattern of NP366–374/Db and PA224–233/Db immunodominance in C57BL/6 mice was independent of the strain of virus used. Similar patterns were also obtained when antibody-deficient μMT mice were infected twice with the same virus (unpublished data).
Table I.
No. of Tetramer+ Cells in the Lung Airways After Influenza Virus Infection
| No. of cells (10−4)
|
||||||
|---|---|---|---|---|---|---|
| Mouse strain | 1° infectiona | 2° infectionb | Day of analysisc | NP366–374/Db | PA224–233/Db | NP/PA ratiod |
| C57BL/6 | x31 | – | 10 | 1.27 | 1.74 | 0.7 |
| x31 | PR8 | 7 | 6.27 | 1.33 | 4.7 | |
| PR8 | – | 10 | 4.91 | 5.70 | 0.9 | |
| PR8 | x31 | 7 | 3.07 | 0.76 | 4.0 | |
| C57BL/6 | Sendai | x31 | 10 | 3.6 | 4.8 | 0.8 |
The primary infection was 300 EID50 (all viruses) delivered intranasally.
The secondary infection was 3,000 EID50 (all viruses) delivered intranasally on day 42 after primary (or day 31 after primary for the Sendai virus primed group).
Cells were analyzed at the peak of infection (day 10 after primary or day 7 after secondary).
The NP/PA ratio was determined by dividing the absolute number of NP-tetramer positive cells by the number of PA-tetramer positive cells in the lung airways.
The changing patterns of immunodominance were also not due to a nonspecific conditioning of the lung by a prior infection (32). Thus, the ratio of NP366–374/Db- and PA224–233/Db-specific T cells after x31 infection in mice that had recovered from a heterologous intranasal Sendai virus infection was similar to that of a primary x31 infection (Table I). As Sendai virus and influenza virus induce very similar inflammatory responses in the lung, this suggests that changes in lung structure and cellular composition induced by a relatively recent respiratory virus infection do not affect the immunodominance of the subsequent T cell response to influenza virus. In addition, the changing patterns of immunodominance do not reflect differences in the inoculating dose of virus. Infection with different virus doses altered the magnitude, but not the composition of the T cell responses (unpublished data). Rather, the immunodominance of the T cell response depends simply on whether the response is primary or secondary in nature.
Differential Expression of NP366–374/Db and PA224–233/Db in Dendritic and Nondendritic Cells.
One mechanism that could account for changing patterns of immunodominance is that the NP366–374/Db and PA224–233/Db antigens are differentially expressed on distinct cell types during a primary and secondary influenza virus infection (15, 16, 33). To address this question, we generated panels of T cell hybridomas specific for the NP366–374/Db and PA224–233/Db epitopes that could be used as probes for detecting the presentation of these epitopes. The hybridomas were generated by fusion with BWZ.36, which contains the Lac Z reporter gene under the control of the NFAT element of the IL-2 enhancer (21, 27–29). TCR mediated recognition of the relevant antigen by the hybridoma results in β-galactosidase production, which is detected using an X-gal assay. The advantage of LacZ hybridomas is that they are independent of costimulatory requirements and provide a highly sensitive readout for assessing antigen presentation ex vivo.
Fig. 2 shows the specificity and sensitivity of two hybridomas that were selected for further studies. Using these hybridomas, we analyzed NP366–374/Db- and PA224–233/Db-presentation on H-2b cell lines that had been infected in vitro with various doses of either x31 or PR8 influenza virus. These included EL4 (a C57BL/6 derived T cell thymoma), AM11 (a C57BL/6 derived alveolar macrophage line), and JAWS II (an immature C57BL/6 dendritic cell line). Uninfected cell lines were used as negative controls. As shown in Fig. 3 , the EL4, AM11, and JAWS II cell lines all expressed the NP366–374/Db epitope after infection with various doses of live virus. In contrast, only the JAWS II dendritic cell line presented the PA224–233/Db epitope to the hybridoma.
Figure 2.

Specificity and sensitivity of Lac Z inducible hybridomas. Clones 53-A8 and 6291-B8 were screened using L-Db, AM11, or JAWS II cells incubated overnight with either the NP366–374, PA224–233, PB1703–711, or HA192–207 peptides at the indicated concentrations. The graphs show the number of positive Lac Z hybridomas per well (no more than 2,000 spots were counted per well).
Figure 3.

Presentation of the NP366–374/Db and PA224–233/Db epitopes after in vitro infection with influenza virus. The EL4, AM11, and JAWS II cell lines were infected with influenza x31 (top panels) or PR8 (bottom panels) for four hours. The cells were infected with three doses of influenza virus or left uninfected: multiplicity of infection (MOI) of 0 (open circle), 2 (closed circle), 10 (open triangle), or 50 (closed triangle). After infection, the cells were irradiated and then used in a standard antigen presentation assay with the 53-A8 (NP366–374/Db) or 6291-B8 (PA224–233/Db) hybridomas.
These data suggested that there was an intrinsic difference in the array of antigens presented by dendritic and nondendritic cells. However, we first considered and excluded several alternative explanations for these data. First, the failure of the nondendritic cell lines to express the PA224–233/Db epitope did not appear to be due to inefficient infection of the cells as the NP366–374/Db epitope was strongly presented by these cell lines. Second, the difference in antigen presentation was also not hybridoma clone dependent, as the same results were obtained with several other NP366–374/Db- and PA224–233/Db-specific hybridomas (unpublished data). Third, the difference in antigen presentation could not be explained by a fundamental inability of the nondendritic cell lines to present PA224–233/Db to the hybridomas or to low levels of class I molecules since both dendritic and nondendritic cells efficiently presented exogenously added PA224–233 peptide (Fig. 2). Fourth, the difference in antigen presentation was not virus strain specific nor dose-dependent since virtually identical results were obtained when the cell lines were infected with different doses of the PR8 virus (Fig. 3). Fifth, the failure of nondendritic cell lines to present the PA224–233/Db epitope did not simply reflect differences in the kinetics of infection and antigen presentation (Figs. 4 and 5) . Furthermore, we saw no detectable differences in cell viability after infection of either the macrophage (AM11) or dendritic (JAWS II) cell line (unpublished data). Finally, we considered the possibility that the pattern of NP366–374/Db and PA224–233/Db epitope expression in immortalized cell lines was not representative of cells in vivo. To address this issue, we isolated peritoneal macrophages, flow cytometrically sorted splenic B cells, and preparations of lung epithelial cells directly from mice (22, 23, 34). We also generated dendritic cells from the bone marrow of C57BL/6 mice by culturing the cells according to standard protocols (30, 31). These cells were then infected with x31 or PR8 in vitro and assayed for antigen presentation using the hybridomas. As shown in Fig. 6 (left panels), dendritic cells readily presented both the NP366–374/Db and PA224–233/Db epitopes suggesting that PA224–233/Db expression is a general characteristic of this cell type (it should be noted that the enhanced presentation of the PA224–233/Db epitope, relative to NP366–374/Db, in this experiment reflects experimental variation and was not observed in other experiments). In contrast, peritoneal macrophages, flow cytometrically sorted splenic B cells, and lung epithelial cells expressed only the NP366–374/Db epitope, confirming the patterns of epitope expression seen using cultured cell lines. Together, these data suggested that there was an intrinsic difference in antigen handling by dendritic cells compared with other cell types.
Figure 4.

Kinetics of antigen presentation after in vitro infection of the JAWS II cell line. The JAWS II cell line was infected with influenza (x31) for the times shown with a MOI of 0 (open circle), 2 (closed circle), 10 (open triangle), or 50 (closed triangle). After infection, the cells were irradiated and then used in a standard antigen presentation assay with the 53-A8 (NP366–374/Db) or 6291-B8 (PA224–233/Db) hybridomas.
Figure 5.

Kinetics of antigen presentation after in vitro infection of the AM11 cell line. The AM11 cell line was infected with influenza (x31) for the times shown with a MOI of 0 (open circle), 2 (closed circle), 10 (open triangle), or 50 (closed triangle). After infection, the cells were irradiated and then used in a standard antigen presentation assay with the 53-A8 (NP366–374/Db) or 6291-B8 (PA224–233/Db) hybridomas.
Figure 6.

Presentation of the NP366–374/Db and PA224–233/Db epitopes after in vitro infection with the x31 strain of influenza virus. Bone marrow–derived dendritic cells, peritoneal exudate cells (PECs), B cells, and lung epithelial cells were collected from uninfected mice. The cells were then infected in vitro with influenza virus (x31, top panels; PR8, bottom panels) at an MOI of 0 (open circle), 2 (closed circle), 10 (open triangle), or 50 (closed triangle) for 4 h, irradiated, and then used in a standard antigen presentation assay with the 53-A8 (NP366–374/Db) or 6291-B8 (PA224–233/Db) hybridomas.
Differential Expression of NP366–374/Db and PA224–233/Db in Dendritic and Nondendritic Cells in Influenza Virus-infected Mice.
The preceding studies show that in vitro infected cells differ in their capacity to present PA224–233/Db and NP366–374/Db epitopes to T cells. To determine whether this was also true in vivo, we analyzed antigen presentation by different cell types in influenza virus infected mice using T cell hybridomas as a read-out. As shown in Fig. 7 A, expression of the NP366–374/Db and PA224–233/Db epitopes was readily detected in both the MLN and BAL on day 6 post x31 infection (tissues from uninfected mice did not stimulate the hybridomas, unpublished data). Extrapolating the data in Fig. 7 A, we estimate that ∼1:6,000 cells in the MLN expressed the PA224–233/Db epitope during a primary infection. In contrast, the frequency of cells expressing the NP366–374/Db epitope in the MLN was in the order of 10 times higher. This general difference was observed in multiple experiments and is consistent with the idea that only a subset of NP366–374/Db-expressing cells also express the PA224–233/Db epitope in vivo.
Figure 7.

Ex vivo presentation of the NP366–374/Db and PA224–233/Db epitopes after infection with influenza virus. Mice were infected intranasally with x31 and cells from the lung airways (closed circle) and MLN (open triangle) were collected on day 6 after infection. (A) Twofold serial dilutions of cells were made in flat-bottom, 96-well plates starting at 105 cells/well and a standard antigen presentation assay was performed using the 53-A8 (NP366–374/Db) or 6291-B8 (PA224–233/Db) Lac Z-inducible hybridomas. (B) Cells from the lung were stained with anti-B220-CyChrome, anti-CD11c-FITC, and anti-CD11b-PE. The macrophage (B220−CD11b+) and dendritic cell (B220−CD11c+) populations were collected using a FACSVantage™ flow cytometer with DiVa options. Twofold serial dilutions of cells were made in flat-bottom, 96-well plates, and a standard Lac Z APC assay was performed using the 53-A8 (NP366–374/Db) or 6291-B8 (PA224–233/Db) Lac Z–inducible hybridomas. Cells from uninfected control mice did not elicit a response following incubation with either the NP366–374/Db or PA224–233/Db hybridomas.
We next analyzed antigen expression during a secondary infection of x31-immune mice that had been challenged with the PR8 virus. Again, expression of both the NP366–374/Db and PA224–233/Db epitopes were readily detected in the lung and MLN after secondary infection (unpublished data). As in the primary infection, a higher frequency of cells expressed the NP366–374/Db epitope (relative to PA224–233/Db) in the MLN. We have repeated these studies several times and at different times during the primary and secondary infection with very similar results. Together, these data show that the reduced T cell response to the PA224–233/Db epitope during a secondary influenza virus infection cannot be explained by a simple lack of PA224–233/Db expression by antigen-presenting cells.
The fact that there appeared to be different frequencies of cells expressing the PA224–233/Db and NP366–374/Db epitopes in vivo was consistent with the in vitro data suggesting that these antigens might be expressed on different subsets of cells. Thus, we analyzed the presentation of the NP366–374/Db and PA224–233/Db epitopes by different cell subsets after in vivo infection. C57BL/6 mice were intranasally infected with x31 and total lung tissue was isolated 6 d later. The cells were then stained with antibodies specific for B220, CD11b, and CD11c and then sorted by flow cytometry into predominantly macrophage (B220− CD11b+CD11c−) and predominantly dendritic cell (B220− CD11b+/−CD11c+) populations (Fig. 7 B). Purified cells were then analyzed for PA224–233/Db and NP366–374/Db expression using the hybridoma assay. As shown in Fig. 7 B, the B220−CD11b+CD11c− cells appeared to exclusively express the NP366–374/Db epitope, whereas the cells B220−CD11b+/−CD11c+ (predominantly dendritic cells) expressed both the PA224–233/Db and NP366–374/Db epitopes. These data were consistent with the in vitro infection studies and suggest that cell subpopulations differ in their expression of influenza virus antigens.
Naive and Memory T Cells Differ in Their Capacity to Detect Antigen Presented by Different Types of Antigen-presenting Cells.
Based on the previous data, we speculated that differences in the specificity of the primary and secondary (memory) T cell responses to influenza virus might be regulated by differences in the capacity of naive and memory T cells to detect antigen presented by dendritic versus nondendritic cells. For example, naive T cells may respond exclusively to dendritic cells (expressing both the PA224–233/Db and NP366–374/Db epitopes), whereas memory T cells may preferentially respond to nondendritic cells (expressing only the NP366–374/Db epitope). An alternative hypothesis is that the primary infection induces a fundamental change in the characteristics, distribution, or numbers of antigen presenting cells and it is this change that affects the specificity of the secondary (memory) T cell response. To distinguish these possibilities, we used a transfer model to compare the specificity of concurrent naive and memory T cell responses to influenza virus infection in the same animal (33, 35). This approach allowed us to compare naive and memory T cell responses under conditions that excluded differences in antigen handling. Thus, 2.5 × 105 CD44+/CD8+ memory spleen cells (with equivalent numbers of NP366–374/Db and PA224–233/Db specific T cells, unpublished data) were sorted from C57BL/6 (CD45.2+) mice by flow cytometry and transferred into naive congenic (CD45.2−) mice. 1 d later, the mice were intranasally infected with influenza virus and host and donor T cells specific for the NP366–374/Db and PA224–233/Db epitopes were assessed by tetramer staining on day 10 after infection. As shown in Fig. 8 , equal numbers of host naive (CD45.2−, panel B) T cells in the lung airways were specific for the NP366–374/Db and PA224–233/Db epitopes, whereas the majority of donor memory (CD45.2+, panel A) T cells were specific for the NP366–374/Db epitope. As these distinct responses occurred in the same animals, these data rule out the possibility that changes in the characteristics, distribution, or numbers of antigen-presenting cells between primary and secondary infections regulate the specificity of the T cell response. Rather, the differences reflect the differential capacity of naive and memory T cells to detect antigen on different sets of antigen-presenting cells. Taken together these data indicate that the dominance of the NP366–374/Db response during a secondary infection is regulated by differential antigen presentation by dendritic and nondendritic cells as well as the differential capacity of these cell types to activate naive and memory CD8+ T cells.
Figure 8.

Simultaneous detection of influenza virus-specific naive and memory cells. Cells were collected from the spleens of C57BL/6 mice infected 32 d previously with 300 EID50 x31. The cells were made into a single-cell suspension, enriched for CD8+ cells and stained with anti-CD8-PE and anti-CD44-FITC. The memory cells (CD8+CD44+) were collected using a FACSVantage™ flow cytometer with DiVa options. 2.5 × 105 purified memory cells were then injected into naive PepBoy/J mice. 1 d after transfer, the recipient mice were infected with 300 EID50 x31. 10 d after infection (11 d after transfer), cells from the lung airways and spleen were collected and stained with anti-CD8-PerCP, anti-CD45.2-FITC and anti- NP366–374/Db-APC or anti- PA224–233/Db-APC. A and B show the percentage of NP366–374/Db- and PA224–233/Db-specific CD8 cells, respectively, within the donor (CD45.2+) or host (CD45.2−) lung airway populations. C shows the average (± standard deviation) percent NP366–374/Db- (solid bars) and PA224–233/Db- (striped bars) specific CD8 cells in the lung airways or spleen.
Vaccination with Peptide-pulsed Dendritic Cells Results in Increased Numbers of PA224–233/Db-specific Cells but Delayed Viral Clearance.
Our data have demonstrated that the PA224–233/Db epitope is not, or poorly, expressed on virally infected lung epithelial cells. As T cell interaction with virally infected epithelial cells is believed to be required for effective viral control, PA224–233/Db-specific T cells may be ineffective at clearing an influenza virus infection (36). Based on previous vaccination studies (19), we hypothesized that mice primed with the PA224–233 peptide would mediate a response to influenza biased toward nonprotective PA224–233/Db-specific T cells resulting in poor viral clearance. To test this hypothesis, we vaccinated mice with dendritic cells pulsed with either the PA224–233 peptide or a control Sendai virus peptide. 2 wk after dendritic cell vaccination, the mice were infected with 300 50% egg infectious dose (EID50) x31 and the number of tetramer positive cells and viral load was determined on day 10 after infection. As shown in Fig. 9 A, vaccination with PA224–233 pulsed dendritic cells resulted in a significant increase in the frequency of PA224–233/Db-specific T cells (61.7% in vaccinated versus 13.9% in controls) and a significant reduction in the frequency of NP366–374/Db specific T cells (4.7% in vaccinated versus 9.1% in controls) in the lung airways. Despite the fact that PA224–233 peptide vaccinated mice made an enhanced PA224–233/Db-specific T cell response to influenza virus infection, they were less effective at clearing the virus. Vaccinated mice were still harboring virus at day 10 post infection whereas control mice had completely cleared virus at this time (Fig. 9 B). These data indicate that vaccination with peptide defining epitopes expressed exclusively on dendritic cells (and not at the site of infection) can have a negative impact on antiviral immunity.
Figure 9.
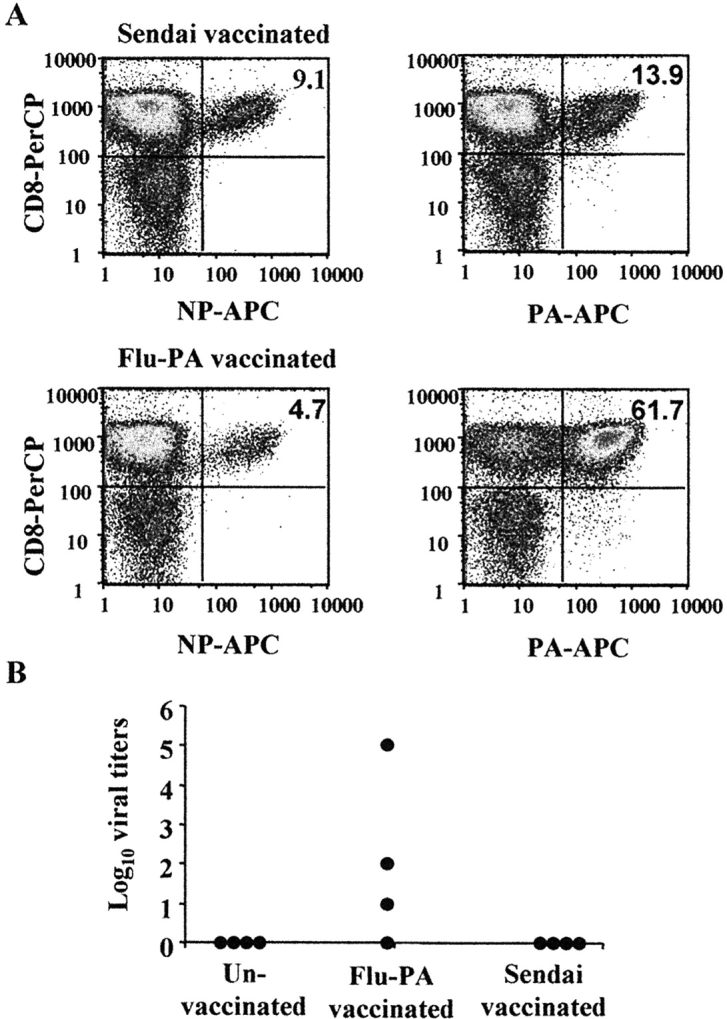
Vaccination with the PA224–233 peptide results in delayed viral clearance after influenza virus challenge. Mice were injected i.v. with 0.5 × 106 dendritic cells pulsed with either the PA224–233 peptide of influenza or a Sendai virus peptide. As a further control, one group of mice did not receive any dendritic cells (un-vaccinated). 2 wk after vaccination, the mice were infected with 300 EID50 x31. 10 d after infection, cells from the lung airways were collected and stained with anti-CD8-PerCP, and anti-NP366–374/Db-APC or anti-PA224–233/Db-APC. Panel A shows the percentage of NP366–374/Db- and PA224–233/Db-specific cells of total CD8s after infection of the Sendai peptide or PA224–233 peptide vaccinated mice. B shows the viral load in the lungs of individual mice on day 10 after infection.
Discussion
We have investigated the mechanisms underlying the changing contributions of NP366–374/Db- and PA224–233/Db-specific T cells to the primary and secondary response to influenza virus infection in C57BL/6 mice (15, 16). The data show that the dominance of NP366–374/Db-specific T cells in the secondary response cannot be attributed to the virus strain, nonspecific conditioning of the lungs after viral infection, or permanent changes in the characteristics, distribution, or numbers of antigen presenting cells after the primary infection. Rather, the specificity of the T cell response in this system is regulated by differences in the presentation of the NP366–374/Db and PA224–233/Db epitopes by dendritic and nondendritic cells and the capacity of naive and memory CD8+ T cells to be activated by these cells. Based on these data, we speculate that the change in the specificity of the T cell response reflects the capacity of memory T cells (in contrast to naive T cells) to respond to nondendritic cells (Fig. 10) . During a primary infection, naive T cells can only be activated by dendritic cells (4, 37–39). As dendritic cells express both the NP366–374/Db and PA224–233/Db antigens, both NP366–374/Db- and PA224–233/Db-specific T cell responses are elicited. In contrast, during a secondary infection, dendritic and nondendritic cells can activate memory T cells. As there is a preponderance of nondendritic cells that express only the NP366–374/Db antigen, this results in a competitive bias toward reactivation of NP366–374/Db-specific memory CD8+ T cells. Importantly, the model predicts that the T cell response to the PA224–233/Db epitope should be diminished, but not completely absent from the secondary T cell response since the PA224–233/Db antigen is still being presented by dendritic cells (Fig. 10). Consistent with this, PA224–233/Db-specific memory T cells do contribute to the recall response at a reduced level compared with the NP366–374/Db-specific response (Fig. 1). The model also predicts that the specificity of the T cell response is independent of conditioning or alteration of the antigen-presenting cell pool by prior infection. In support of this, both primary and secondary patterns of T cell specificity were simultaneously induced in a transfer model in which the host and its antigen presenting cells were naive with respect to influenza virus infection. Finally, the model predicts that memory T cell responses to the PA224–233/Db epitope should be poorly competitive with all epitopes that are expressed on both dendritic and nondendritic cells (such as NP366–374/Db). Indeed, recent studies using influenza virus mutants that lack the NP366–374/Db epitope but contain other class I–restricted epitopes indicate that the PA224–233/Db-specific responses are not amplified after secondary viral infection (15, 40). Thus, taken together, the data indicate that differential antigen presentation by dendritic and nondendritic cells and the capacity of the T cells to perceive the antigens presented by these cells regulate the patterns of T cell specificity in this system (Fig. 10).
Figure 10.

A model of antigen presentation and T cell responses after influenza virus infection. After infection with influenza virus, the NP366–374/Db epitope is processed and presented by both macrophages (MΦ) and dendritic cells (DC). The PA224–233/Db epitope, however, is only presented by the dendritic cells. During the primary response, the naive T cells respond only to dendritic cells resulting in an equivalent NP366–374/Db- and PA224–233/Db-specific T cell response. After secondary infection, the memory T cells are able to respond to both macrophages and dendritic cells resulting in a predominance of NP366–374/Db specific T cells.
A surprising feature of these studies is the observation that antigen-specific T cells can be elicited during a primary response that are potentially unable to recognize antigen at the site of infection. The failure to detect the PA224–233/Db antigen on infected lung epithelial cells is of particular interest, as T cell lysis of the epithelial layer is believed to be required for effective viral control (36). Why might ineffective cells be generated in an infection? One possibility is that dendritic cells are simply superior presenting cells compared with other cell types and that PA224–233/Db presentation to T cells is just above a threshold in dendritic, but not nondendritic cells. Enhanced presentation of epitopes by dendritic cells would normally benefit the response by ensuring optimal activation of antigen-specific T cells. However, a negative consequence might be that weakly presented epitopes result in the induction of T cells that are not able to recognize antigen at the site of infection. The idea that ineffective T cells might be generated during an influenza virus infection has important implications for the development of peptide-based vaccines designed to promote cellular immunity. The choice of relevant epitopes is usually made based on data regarding responses to natural infection. However, some epitopes identified in this manner might actually be detrimental in terms of their ability to elicit protective immunity if the epitope targeted were inadequately expressed at the site of infection. Indeed, in the current studies, the specific priming of PA224–233/Db-specific T cells by vaccination actually resulted in impaired clearance of a subsequent influenza virus infection (Fig. 9). In this situation, we speculate that primed (memory) PA224–233/Db-specific T cells and naive NP366–374/Db-specific T cells are both activated by antigen exclusively expressed on dendritic cells. However, the higher frequency and higher activation status of the PA224–233/Db-specific memory T cells resulted in a dominance of nonprotective PA224–233/Db-specific T cells and concomitant reduction in protective NP366–374/Db-specific T cells (19). Clearly the selection of appropriate epitopes for vaccination must take into account the pattern of antigen presentation on different cell types.
A critical question raised by these studies is the frequency with which differential antigen presentation might affect the specificity of T cell responses. Several recent studies suggest that this may be a very frequent occurrence. For example, Selin et al. (14) have shown that T cells responding to two major epitopes (LCMV-NP396–404/Db and LCMV-GP33–43/Db) of lymphocytic choriomeningitis virus (LCMV) in C57BL/6 mice differentially expand during primary and secondary infections. Thus, the percentage of LCMV-GP33–43/Db-specific T cells is significantly increased in the secondary response (relative to the primary), whereas the percentage of LCMV-NP396–404/Db-specific T cells remains constant between the primary and secondary responses. Furthermore, when LCMV infected fibroblasts or dendritic cells were used to restimulate CTLs, the resulting CTL lines differentially presented the same epitopes (41). In addition, following HSV infection the primary CTL response to two immunodominant epitopes (gB498–505 and ICP27445–452) have similar kinetics and precursor frequencies. After reinfection, CTLs specific for the ICP27445–452 epitope are present at higher frequencies than CTLs specific for the gB498–505 epitope (42).
There are several possible mechanisms that would explain the differential antigen processing by dendritic and nondendritic cells after influenza virus infection. One possibility is that it depends on the degree of infection established in the antigen-presenting cells. It has previously been shown that whereas ∼90% of both dendritic cells and macrophages can be infected with influenza virus, the infection in dendritic cells does not result in cell death and few progeny virions are produced (43, 44). In contrast, macrophages die within 10–12 h after infection and produce 10-fold higher progeny than dendritic cells (43). However, increased levels of virus in macrophages would tend to favor the processing of both antigens, whereas the data show that macrophages fail to present detectable levels of the PA224–233/Db epitope. A related possibility is that the presentation of the PA224–233/Db antigen may not depend on viral infection of the antigen presenting cells (45). However, AM11, JAWS II, and bone marrow–derived dendritic cells were unable to present the PA224–233/Db or NP366–374/Db epitopes after administration of high doses of β-propriolactone inactivated virus (unpublished data, reference 46). While this indicates that viral infection was necessary for antigen presentation of these epitopes, it does not specifically rule out cross-priming as a mechanism for epitope presentation. Another potential mechanism to explain the differential processing of the NP366–374/Db and PA224–233/Db epitopes is that the processing of the PA224–233/Db epitope depends on immunoproteosomes (47). Thus, we considered the possibility that the failure of AM11 cells to present the PA224–233/Db epitope might be reversed through the induction of immunoproteosomes by IFN-γ (48, 49). However, addition of various doses of recombinant IFN-γ had no impact on the patterns of PA224–233/Db and NP366–374/Db presentation by JAWS II or AM11 (unpublished data) indicating that immunoproteosomes may not absolutely required for processing the NP366–374/Db and PA224–233/Db epitopes.
Taken together, the data in this report show that differential antigen presentation by dendritic and nondendritic T cells can have a significant impact on the specificity of the T cell responses. This phenomenon may play a role in determining immunodominance hierarchies and the efficiency of T cell responses at effector sites. Additionally, we have shown that vaccination with an epitope presented predominantly by infected dendritic cells resulted in delayed control of a subsequent viral infection. Understanding the impact of differential antigen processing is therefore essential for the development of effective peptide-based vaccines.
Acknowledgments
We would like to thank Simon Monard, the Trudeau Institute Molecular Biology Core Facility, Sherri Surman, and Twala Hogg for their technical assistance.
This work was supported by funds from the Trudeau Institute, National Institutes of Health grants HL63925, HL69502, AI29579, AI55154, AI04982, and CA21765, and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Abbreviations used in this paper: EID50, 50% egg infectious dose; LCMV, lymphocytic choriomeningitis virus; MLN, mediastinal lymph nodes, MOI, multiplicity of infection.
References
- 1.Doherty, P.C., and R.M. Zinkernagel. 1976. Specific immune lysis of paramyxovirus-infected cells by H-2-compatible thymus-derived lymphocytes. Immunology. 31:27–32. [PMC free article] [PubMed] [Google Scholar]
- 2.Kast, W.M., L. Roux, J. Curren, H.J. Blom, A.C. Voordouw, R.H. Meloen, D. Kolakofsky, and C.J. Melief. 1991. Protection against lethal Sendai virus infection by in vivo priming of virus-specific cytotoxic T lymphocytes with a free synthetic peptide. Proc. Natl. Acad. Sci. USA. 88:2283–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole, G.A., T.L. Hogg, and D.L. Woodland. 1994. The MHC class I-restricted T cell response to Sendai virus infection in C57BL/6 mice: a single immunodominant epitope elicits an extremely diverse repertoire of T cells. Int. Immunol. 6:1767–1775. [DOI] [PubMed] [Google Scholar]
- 4.Yewdell, J.W., and J.R. Bennink. 1999. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 17:51–88. [DOI] [PubMed] [Google Scholar]
- 5.Chen, W., L.C. Anton, J.R. Bennink, and J.W. Yewdell. 2000. Dissecting the multifactorial causes of immunodominance in class I-restricted T cell responses to viruses. Immunity. 12:83–93. [DOI] [PubMed] [Google Scholar]
- 6.Eisenlohr, L.C., J.W. Yewdell, and J.R. Bennink. 1992. Flanking sequences influence the presentation of an endogenously synthesized peptide to cytotoxic T lymphocytes. J. Exp. Med. 175:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yewdell, J.W., and J.R. Bennink. 1992. Cell biology of antigen processing and presentation to major histocompatibility complex class I molecule-restricted T lymphocytes. Adv. Immunol. 52:1–123. [DOI] [PubMed] [Google Scholar]
- 8.Bergmann, C.C., L. Tong, R. Cua, J. Sensintaffar, and S. Stohlman. 1994. Differential effects of flanking residues on presentation of epitopes from chimeric peptides. J. Virol. 68:5306–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, W., J.R. Bennink, P.A. Morton, and J.W. Yewdell. 2002. Mice deficient in perforin, CD4+ T cells, or CD28-mediated signaling maintain the typical immunodominance hierarchies of CD8+ T-cell responses to influenza virus. J. Virol. 76:10332–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belz, G.T., P.G. Stevenson, and P.C. Doherty. 2000. Contemporary analysis of MHC-related immunodominance hierarchies in the CD8+ T cell response to influenza A viruses. J. Immunol. 165:2404–2409. [DOI] [PubMed] [Google Scholar]
- 11.Mullbacher, A., M. Lobigs, J.W. Yewdell, J.R. Bennink, R. Tha Hla, and R.V. Blanden. 1999. High peptide affinity for MHC class I does not correlate with immunodominance. Scand. J. Immunol. 50:420–426. [DOI] [PubMed] [Google Scholar]
- 12.Sette, A., A. Vitiello, B. Reherman, P. Fowler, R. Nayersina, W.M. Kast, C.J. Melief, C. Oseroff, L. Yuan, J. Ruppert, et al. 1994. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. 153:5586–5592. [PubMed] [Google Scholar]
- 13.Daly, K., P. Nguyen, D.L. Woodland, and M.A. Blackman. 1995. Immunodominance of major histocompatibility complex class I-restricted influenza virus epitopes can be influenced by the T-cell receptor repertoire. J. Virol. 69:7416–7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selin, L.K., M.Y. Lin, K.A. Kraemer, D.M. Pardoll, J.P. Schneck, S.M. Varga, P.A. Santolucito, A.K. Pinto, and R.M. Welsh. 1999. Attrition of T cell memory: selective loss of LCMV epitope-specific memory CD8 T cells following infections with heterologous viruses. Immunity. 11:733–742. [DOI] [PubMed] [Google Scholar]
- 15.Belz, G.T., W. Xie, J.D. Altman, and P.C. Doherty. 2000. A previously unrecognized H-2Db-restricted peptide prominent in the primary influenza A virus-specific CD8+ T-cell response is much less apparent following secondary challenge. J. Virol. 74:3486–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belz, G.T., W. Xie, and P.C. Doherty. 2001. Diversity of epitope and cytokine profiles for primary and secondary influenza a virus-specific CD8+ T cell responses. J. Immunol. 166:4627–4633. [DOI] [PubMed] [Google Scholar]
- 17.Hou, S., P.C. Doherty, M. Zijlstra, R. Jaenisch, and J.M. Katz. 1992. Delayed clearance of Sendai virus in mice lacking class I MHC-restricted CD8+ T cells. J. Immunol. 149:1319–1325. [PubMed] [Google Scholar]
- 18.Deckhut, A.M., W. Allan, A. McMickle, M. Eichelberger, M.A. Blackman, P.C. Doherty, and D.L. Woodland. 1993. Prominent usage of V beta 8.3 T cells in the H-2Db-restricted response to an influenza A virus nucleoprotein epitope. J. Immunol. 151:2658–2666. [PubMed] [Google Scholar]
- 19.Cole, G.A., T.L. Hogg, M.A. Coppola, and D.L. Woodland. 1997. Efficient priming of CD8+ memory T cells specific for a subdominant epitope following Sendai virus infection. J. Immunol. 158:4301–4309. [PubMed] [Google Scholar]
- 20.Cole, G.A., V.K. Clements, E.P. Garcia, and S. Ostrand-Rosenberg. 1987. Allogeneic H-2 antigen expression is insufficient for tumor rejection. Proc. Natl. Acad. Sci. USA. 84:8613–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanderson, S., and N. Shastri. 1994. LacZ inducible, antigen/MHC-specific T cell hybrids. Int. Immunol. 6:369–376. [DOI] [PubMed] [Google Scholar]
- 22.Dobbs, L.G., R. Gonzalez, and M.C. Williams. 1986. An improved method for isolating type II cells in high yield and purity. Am. Rev. Respir. Dis. 134:141–145. [DOI] [PubMed] [Google Scholar]
- 23.Dobbs, L.G. 1990. Isolation and culture of alveolar type II cells. Am. J. Physiol. 258:L134–L147. [DOI] [PubMed] [Google Scholar]
- 24.Wilson, J.S., J.A. Steinkamp, and B.E. Lehnert. 1986. Isolation of viable type II alveolar epithelial cells by flow cytometry. Cytometry. 7:157–162. [DOI] [PubMed] [Google Scholar]
- 25.Altman, J.D., P.H. Moss, P.R. Goulder, D.H. Barouch, M.G. McHeyzer-Williams, J.I. Bell, A.J. McMichael, and M.M. Davis. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science. 274:94–96. [DOI] [PubMed] [Google Scholar]
- 26.White, J., M. Blackman, J. Bill, J. Kappler, P. Marrack, D.P. Gold, and W. Born. 1989. Two better cell lines for making hybridomas expressing specific T cell receptors. J. Immunol. 143:1822–1825. [PubMed] [Google Scholar]
- 27.Usherwood, E.J., T.L. Hogg, and D.L. Woodland. 1999. Enumeration of antigen-presenting cells in mice infected with Sendai virus. J. Immunol. 162:3350–3355. [PubMed] [Google Scholar]
- 28.Liu, L., E. Flano, E.J. Usherwood, S. Surman, M.A. Blackman, and D.L. Woodland. 1999. Lytic cycle T cell epitopes are expressed in two distinct phases during MHV-68 infection. J. Immunol. 163:868–874. [PubMed] [Google Scholar]
- 29.Karttunen, J., and N. Shastri. 1991. Measurement of ligand-induced activation in single viable T cells using the lacZ reporter gene. Proc. Natl. Acad. Sci. USA. 88:3972–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lutz, M.B., N. Kukutsch, A.L. Ogilvie, S. Rossner, F. Koch, N. Romani, and G. Schuler. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 223:77–92. [DOI] [PubMed] [Google Scholar]
- 31.Liu, L., E.J. Usherwood, M.A. Blackman, and D.L. Woodland. 1999. T-Cell vaccination alters the course of murine herpesvirus 68 infection and the establishment of viral latency in mice. J. Virol. 73:9849–9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen, H.D., A.E. Fraire, I. Joris, M.A. Brehm, R.M. Welsh, and L.K. Selin. 2001. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat. Immunol. 2:1067–1076. [DOI] [PubMed] [Google Scholar]
- 33.Turner, S.J., R. Cross, W. Xie, and P.C. Doherty. 2001. Concurrent naive and memory CD8(+) T cell responses to an influenza A virus. J. Immunol. 167:2753–2758. [DOI] [PubMed] [Google Scholar]
- 34.Massey, T.E., B.A. Geddes, and P.G. Forkert. 1987. Isolation of nonciliated bronchiolar epithelial (Clara) cells and alveolar type II cells from mouse lungs. Can. J. Physiol. Pharmacol. 65:2368–2372. [DOI] [PubMed] [Google Scholar]
- 35.White, D.W., A. MacNeil, D.H. Busch, I.M. Pilip, E.G. Pamer, and J.T. Harty. 1999. Perforin-deficient CD8+ T cells: in vivo priming and antigen-specific immunity against Listeria monocytogenes. J. Immunol. 162:980–988. [PubMed] [Google Scholar]
- 36.Topham, D.J., R.A. Tripp, and P.C. Doherty. 1997. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 159:5197–5200. [PubMed] [Google Scholar]
- 37.Norbury, C.C., D. Malide, J.S. Gibbs, J.R. Bennink, and J.W. Yewdell. 2002. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat. Immunol. 3:265–271. [DOI] [PubMed] [Google Scholar]
- 38.Jenkins, M.K., A. Khoruts, E. Ingulli, D.L. Mueller, S.J. McSorley, R.L. Reinhardt, A. Itano, and K.A. Pape. 2001. In vivo activation of antigen-specific CD4 T cells. Annu. Rev. Immunol. 19:23–45. [DOI] [PubMed] [Google Scholar]
- 39.Hamilton-Easton, A., and M. Eichelberger. 1995. Virus-specific antigen presentation by different subsets of cells from lung and mediastinal lymph node tissues of influenza virus-infected mice. J. Virol. 69:6359–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webby, R.J., S. Andreansky, J. Stambas, J.E. Rehg, R.G. Webster, P.C. Doherty, and S.J. Turner. Protection and compensation in the influenza virus-specific CD8+ T cell response. Proc. Natl. Acad. Sci. USA. In press. [DOI] [PMC free article] [PubMed]
- 41.Butz, E.A., and M.J. Bevan. 1998. Differential presentation of the same MHC class I epitopes by fibroblasts and dendritic cells. J. Immunol. 160:2139–2144. [PubMed] [Google Scholar]
- 42.Nugent, C.T., J.M. McNally, R. Chervenak, R.M. Wolcott, and S.R. Jennings. 1995. Differences in the recognition of CTL epitopes during primary and secondary responses to herpes simplex virus infection in vivo. Cell. Immunol. 165:55–64. [DOI] [PubMed] [Google Scholar]
- 43.Bender, A., M. Albert, A. Reddy, M. Feldman, B. Sauter, G. Kaplan, W. Hellman, and N. Bhardwaj. 1998. The distinctive features of influenza virus infection of dendritic cells. Immunobiology. 198:552–567. [DOI] [PubMed] [Google Scholar]
- 44.Lopez, C.B., A. Fernandez-Sesma, S.M. Czelusniak, J.L. Schulman, and T.M. Moran. 2000. A mouse model for immunization with ex vivo virus-infected dendritic cells. Cell. Immunol. 206:107–115. [DOI] [PubMed] [Google Scholar]
- 45.Heath, W.R., and F.R. Carbone. 1999. Cytotoxic T lymphocyte activation by cross-priming. Curr. Opin. Immunol. 11:314–318. [DOI] [PubMed] [Google Scholar]
- 46.Budowsky, E.I., E.A. Friedman, N.V. Zheleznova, and F.S. Noskov. 1991. Principles of selective inactivation of viral genome. VI. Inactivation of the infectivity of the influenza virus by the action of beta-propiolactone. Vaccine. 9:398–402. [DOI] [PubMed] [Google Scholar]
- 47.Van Kaer, L., P.G. Ashton-Rickardt, M. Eichelberger, M. Gaczynska, K. Nagashima, K.L. Rock, A.L. Goldberg, P.C. Doherty, and S. Tonegawa. 1994. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity. 1:533–541. [DOI] [PubMed] [Google Scholar]
- 48.Chen, W., C.C. Norburg, Y. Cho, J.W. Yewdell, and J.R. Bennink. 2001. Immunoproteosomes shape immunodominance hierarchies of antiviral CD8+ T cells at the levels of T cell repertoire and presentation of viral antigens. J. Exp. Med. 193:1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van den Eynde, B.J., and S. Morel. 2001. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr. Opin. Immunol. 13:147–153. [DOI] [PubMed] [Google Scholar]