

Abstract
Allergic asthma is characterized by airway hyperresponsiveness, eosinophilia, and mucus accumulation and is associated with increased IgE concentrations. We demonstrate here that peroxisome proliferator–activated receptors (PPARs), PPAR-α and PPAR-γ, which have been shown recently to be involved in the regulation of various cell types within the immune system, decrease antigen-induced airway hyperresponsiveness, lung inflammation, eosinophilia, cytokine production, and GATA-3 expression as well as serum levels of antigen-specific IgE in a murine model of human asthma. In addition, we demonstrate that PPAR-α and -γ are expressed in eosinophils and their activation inhibits in vitro chemotaxis and antibody-dependent cellular cytotoxicity. Thus, PPAR-α and -γ (co)agonists might be of therapeutic interest for the regulation of allergic or inflammatory reactions by targeting both regulatory and effector cells involved in the immune response.
Keywords: nuclear receptors, asthma, eosinophils, IgE, ADCC
Introduction
Allergic asthma is typically associated with airway hyperresponsiveness (AHR),* recruitment of inflammatory cells, in particular eosinophils, and mucus accumulation due to goblet cell metaplasia in the lung epithelium (1). These functional and anatomical changes are also associated with increased IgE production due to the development of a strong Th2 response, mainly characterized by the production of IL-4, IL-5, and IL-13 (1, 2).
Eosinophils are recruited from bone marrow and differentiated within tissues upon activation by IL-5 and eotaxins (CCL11, 24 and 26) binding to their specific receptors (IL-5 R and CCR3). After their recruitment at inflammatory sites, eosinophils are activated through various receptors, among them immunoglobulin receptors, in particular for IgE and monomeric or secretory IgA. Activation leads to the release of cytotoxic mediators, such as eosinophil peroxidase, eosinophil cationic protein, and major basic protein, generating radical oxygen species, which together are able to damage nonself targets such as parasites, larvae, and bacteria but also self targets such as pulmonary epithelial, cardiac, or nervous cells (3).
Most of inflammatory reactions lead to the activation of NF-κB through several interconnected pathways. Among the various regulators affecting these signaling cascades, some nuclear receptors such as the peroxisome proliferator–activated receptors (PPARs) have been identified. PPARs are ligand-activated transcription factors, belonging to the nuclear receptor superfamily, that form dimers with the retinoid X receptors (for review see reference 4). Three PPAR subtypes (α, β/δ, and γ) have been identified (4). These receptors are activated by fatty acid and metabolites such as leukotriene B4 (LTB4), which is preferentially bound by PPAR-α (5). PPAR-β/δ binds prostacyclin (prostaglandin I2 or PGI2; reference 6), whereas PPAR-γ is activated by some eicosanoids: 13-hydroxyoctadecadienoic acid (7), 15-hydroxyeicosatetraenoic acid and 15-deoxy-Δ12,14-prostaglandinJ2 (15d-PGJ2; reference 8), a prostaglandin D2 metabolite. Both PPAR-α and PPAR-γ have been shown to negatively regulate inflammation and to play a role within the immune system, (9–11) acting on T lymphocytes (12, 13), monocytes/macrophages (14–18), dendritic cells (19), and mast cells (20). Besides its role in skin wound healing and keratinocyte proliferation, little is known about the role of PPAR-β/δ in the control of inflammation (21).
In vivo, PPAR-γ has been shown to decrease the severity of colitis in two experimental models (22, 23): arthritis and IgM production (24). Furthermore, PPAR-γ agonists have been tested with some success as a potential therapy for ulcerative colitis (25). In humans, PPAR-γ expression is increased in bronchial submucosa, airway epithelium, and smooth muscle cells from asthmatic patients (26). PPAR-α–deficient mice display prolonged LTB4-induced ear swelling (5), absence of regulation of spontaneous proinflammatory cytokine production upon aging (27), and exacerbated response of aortic explants to proinflammatory stimuli (28).
In this paper, we aimed at determining whether PPAR-α and -γ also act as modulators of asthma and lung inflammation associated with prominent eosinophilia, and whether they would directly affect eosinophil function.
Materials and Methods
Animals.
PPAR-α–deficient mice, originally obtained from Dr. F.J. Gonzalez (National Institutes of Health, Bethesda, MD; reference 29), and corresponding wild-type (WT) 129S1/SVImJ were purchased from Jackson ImmunoResearch Laboratories and bred within the specific pathogen-free facility from the Institut Pasteur de Lille. Balb/c animals were purchased from Iffa-Credo. Hypereosinophilic IL-5 × hFcɛRIαTg mice have been produced in our laboratory and described previously (30). 8–12-wk-old female mice were used for all the experiments. LouM rats were produced at the Institut Pasteur de Lille.
Asthma Model.
Mice were sensitized with OVA (50 μg in 100 μl Alum–Imject™; Pierce Chemical Co.) or received Alum only and were challenged for 20 min on days 14, 15, 18, 19, and 20 by aerosol nebulization with OVA (1% in PBS) using an ultrasonic nebulizer (Systam). For some experiments, 5 × 10−5 M ciglitazone (Qbiogene) or 5 × 10−5 ciglitazone M and 5 × 10−5 M GW9662 (Cayman) in PBS (or vehicle) were administered to Balb/c mice by nebulization for 20 min immediately before and during each OVA sensitization. Serum was collected on day 21 and AHR (enhanced Pause) to increasing concentrations of methacholine (1.5–12 mg/ml) was measured by whole-body plethysmography (Emka) on day 22 (31). Lungs were used either for bronchoalveolar lavages (BALs) and protein extraction or for histological analyses and protein extraction.
For histology, lung samples were fixed with Immunohistofix™ and embedded in Immunohistowax™ (Universite Libre de Bruxelles). 4-μm sections were stained either with May Grünwald Giemsa or periodic acid schiff (PAS) for detection of mucopolysaccharide accumulation. Cellular content of BAL was analyzed on cytospin preparations after staining with May Grünwald Giemsa.
Quantification of cytokines in lung tissue was performed as described previously (32, 33). In brief, samples were homogenized, using an Ultra-turrax, in 400 μl of buffer (per right lung) containing 1% NP-40, 150 mM NaCl, 50 mM Hepes, PMSF, and Complete protease inhibitor cocktail (Roche). IL-4, IL-5, IL-6, IL-13, eotaxin/CCL11, and soluble VCAM-I were measured using specific ELISA kits (R&D Systems).
Serum anti-OVA IgG1 was measured by ELISA, using OVA-coated plates and HRP-conjugated anti–mouse IgG1 (Southern Biotechnology Associates, Inc.). Serum anti-OVA IgE was measured by ELISA using anti-IgE (BD Biosciences) as capture antibody and biotinylated OVA and HRP-conjugated streptavidin (Amersham Biosciences) for detection. Twofold serial dilutions were prepared for each serum (starting dilution 1:25 for IgE and 1:5,000 for IgG1 titrations). Antibody titers were calculated as the dilution corresponding to twice the mean absorbance value obtained for nonsensitized mouse sera.
Eosinophil Purification.
Human eosinophils were isolated, from the venous blood of hypereosinophilic patients (eosinophilia associated with skin diseases or allergy), by negative selection using anti–CD16- and anti–CD3-coated magnetic beads and MACS system (Miltenyi Biotec), as described previously (34). Eosinophil purity >97%.
Mouse eosinophils were obtained from spleens from naive IL-5 × hFcɛRIα Tg mice and purified by negative selection, using CD90 (Thy1.2), CD45R (B220), and CD8α (Ly-2) magnetic beads for depletion, as described previously (35). Typical purity was 95–97%.
Rat eosinophils were obtained by lavage of the peritoneal cavity from naive LouM rats. Eosinophils were purified by negative selection using anti-CD45R, anti-CD8α, and antimononuclear phagocyte (1C7) antibodies to remove B cells, T cells, and macrophages. Typical purity was 90%.
Reverse Transcriptase (RT)–PCR.
Total RNA was isolated using RNAplus (Qbiogene). Reverse transcription was performed with 2 μg RNA using SuperScript™ RT (GIBCO BRL, Life Technologies). cDNA was amplified using various primers as follows: human PPAR-α (5′-ATGAGGCCATATTCGCCATGC-3′ and 5′-GTTGCTCTGCAGGTGGAGTCT-3′); mouse/rat PPAR-α (5′-CGGCCTGGCCTTCTAAACATAGGC-3′ and 5′-CAGTGGGTGCAGCGCTGCGTCGGACTCGGTC-3′); human PPAR-γ (5′-GCCTTGCAGTGGGGATGTCTCA-3′ and 5′-GATGCGGATGGCCACCTCTTT-3′); mouse PPAR-γ (5′-TCTCTCCGTAATGGAAGACC-3′ and 5′-GCATTATGAGACATCCCCAC-3′); rat PPAR-γ (5′-GGCGAGGGCGATCTTGACAGG-3′ and 5′-AGGGCTTCCGCAGGCTTTTGA-3′); human β-actin (5′-GGGTCAGAAGGATTCCTATG-3′ and 5′-GGTCTCAAACATGATCT-GGG-3′); and mouse β-actin (5′-GTCGGGCGCCCCAGGCACCA-3′ and 5′-CTCCTTAATGTCACGCACGAT-TTC-3′).
PCR was run for 38–40 cycles (annealing: 54°C) using Taq polymerase (Bioprobe). PCRs were performed on each sample using β-actin–specific primers and analyzed on 2% agarose gels after ethidium bromide staining. Gel loadings of the various amplified products were normalized according to the signal intensity provided by the β-actin amplification. RNA prepared from mouse liver and spleen, rat spleen, and HepG2 cells were used as positive controls.
Western Blot.
Protein extracts were prepared as described in the Asthma Model section for the quantification of cytokines in lung tissues by ELISA. 10-μg proteins were loaded on a 10% SDS-PAGE gel. Material was transferred onto nitrocellulose and probed with rabbit anti–PPAR-α (RDI), anti–PPAR-γ (BIOMOL Research Laboratories, Inc.), and anti–GATA-3 (Santa Cruz Biotechnology, Inc.) specific antisera. Signals were detected using peroxidase-conjugated antibody (Jackson ImmunoResearch Laboratories) and Renaissance Western Plus (NEN Life Science Products). Extracts from mouse liver and human adipose tissue were used as positive controls for PPAR-α and -γ, respectively. The pool of mediastinal LNs from OVA-sensitized and -challenged PPAR-α−/− and Balb/c mice were used as positive controls for GATA-3.
Flow Cytometry.
2 × 105 cells were fixed with 2% paraformaldehyde and permeabilized with 0.1% saponin in PBS, before incubation in PBS-saponin with rabbit anti–PPAR-α (RDI), anti–PPAR-γ (Biomol), or corresponding control antisera for 30 min in the presence of saponin. Nonspecific binding was blocked with 5 μl goat serum for 10 min and FITC-conjugated goat anti–rabbit (Jackson ImmunoResearch Laboratories) was added to the cells. Samples were analyzed on a FACSCalibur™ using the CELLQuest™ software (Becton Dickinson).
Chemotaxis.
Human eosinophil chemotaxis was assessed by a modification of the Boyden micropore filter technique (36). Eosinophils (5 × 104 cells/well) were incubated in quadruplicate with WY14653 (Qbiogene), ciglitazone, rosiglitazone, GW9662, or rosiglitazone and GW9662 (concentration range 10−8–10−5 M; Cayman Chemical) or DMSO in the upper chamber separated from the chemoattractant 10 ng/ml IL-5 or 1 ng/ml eotaxin by a 5-μm pore size polycarbonate membrane filter (Nucleopore Co.). The numbers of eosinophils that had migrated after 2 h were enumerated microscopically. Results for each dose of agonist were expressed as percentage of inhibition. Three to five independent experiments using cells from different donors were performed for each agonist.
ADCC toward Schistosoma mansoni Larvae.
Antibody-dependent cellular cytoxicity (ADCC) experiments were performed as described previously (37). Human or rat eosinophils (3 × 105 cells/well) were incubated overnight in medium containing PPAR-α and -γ agonists (concentration range 10−9–10−5 M) or vehicle. Schistosomula (∼100 per well) were added to human or rat eosinophils together with serum from S. mansoni–infected patients or rats, respectively. Schistosomula mortality was assessed microscopically 48 h later on duplicated wells. Results for each dose of agonist were expressed as percentage of inhibition. Three to eight independent experiments using cells from different donors or from different pools of rats were performed for each agonist.
Statisitical Analysis.
Statistical significance was determined using the Wilcoxon-signed rank test for paired groups for ADCC and chemotaxis experiments. For animal experiments, unpaired Student's t tests were used except for Penh determination for which ANOVA for repeated measures were used. P < 0.05 were considered as significant. Results of statistical analyses from in vivo experiments are summarized in Table I.
Table I.
Statistical Analysis of In Vivo Experiments
| P value | PPARα2/− OVA versus WT OVA |
PPARα2/− OVA versus PPARα2/− Alum |
WT OVA versus WT Alum |
Cigli. versus OVA |
Cigli. versus Cigli. + GW |
OVA versus control |
|---|---|---|---|---|---|---|
| Penh | <0.0001 | <0.0001 | <0.0001 | 0.003 | 0.014 | 0.007 |
| BAL (total cells) | <0.0001 | <0.0001 | 0.009 | 0.004 | 0.05 | 0.0007 |
| BAL (eosinophils) | <0.0001 | <0.0001 | 0.006 | 0.003 | 0.019 | 0.0005 |
| Anti-OVA IgE | 0.033 | ND | ND | 0.003 | 0.048 | ND |
| Anti-OVA IgG1 | NS | ND | ND | 0.004 | NS | ND |
| IL-4 | NS | NS | NS | 0.021 | 0.012 | 0.0002 |
| IL-5 | NS | NS | NS | 0.006 | 0.0006 | <0.0001 |
| IL-6 | 0.034 | NS | NS | 0.0007 | 0.002 | <0.0001 |
| IL-13 | 0.05 | NS | NS | 0.009 | 0.003 | 0.007 |
| Eotaxin | 0.011 | <0.0001 | 0.007 | NS | NS | NS |
| sVCAM-1 | NS | NS | NS | NS | NS | NS |
Unpaired t test was used for all parameters except Penh, for which ANOVA for repeated measures was used. NS, not significant; ND, not determined.
Results
PPAR-α–deficient Mice Exhibit Increased AHR and Eosinophilia.
The effect of PPAR-α–deficiency on AHR was studied in a murine model closely mimicking human asthma. Indeed, upon sensitization by intraperitoneal injection of OVA in the presence of alum and challenge by repeated OVA nebulizations, mice develop lung inflammation characterized by eosinophilia, IgE production, and increased AHR in response to methacholine provocation. In this model, responses of WT (PPAR-α+/+) 129S1/SVImJ and PPAR-α–deficient (PPAR-α−/−) animals were compared. AHR was measured by whole body plethysmography upon challenge with increasing doses of methacholine (results were expressed as the enhanced pause-Penh values). OVA-sensitized and -challenged PPAR-α–deficient animals displayed a 2.4-fold higher response (for 6 mg/ml methacholine) compared with their WT counterparts (Fig. 1 a). Indeed, sensitization led to a 6.8-fold Penh increase in PPAR-α–deficient animals, whereas it only led to a 2.2-fold increase in WT animals. After sensitization and airway antigen challenge, the overall inflammatory infiltrate in the BALs and the total number of eosinophils were greatly increased (4.7- and 5.3-fold, respectively) in PPAR-α–deficient animals, compared with WT animals (Fig. 1 b).
Figure 1.
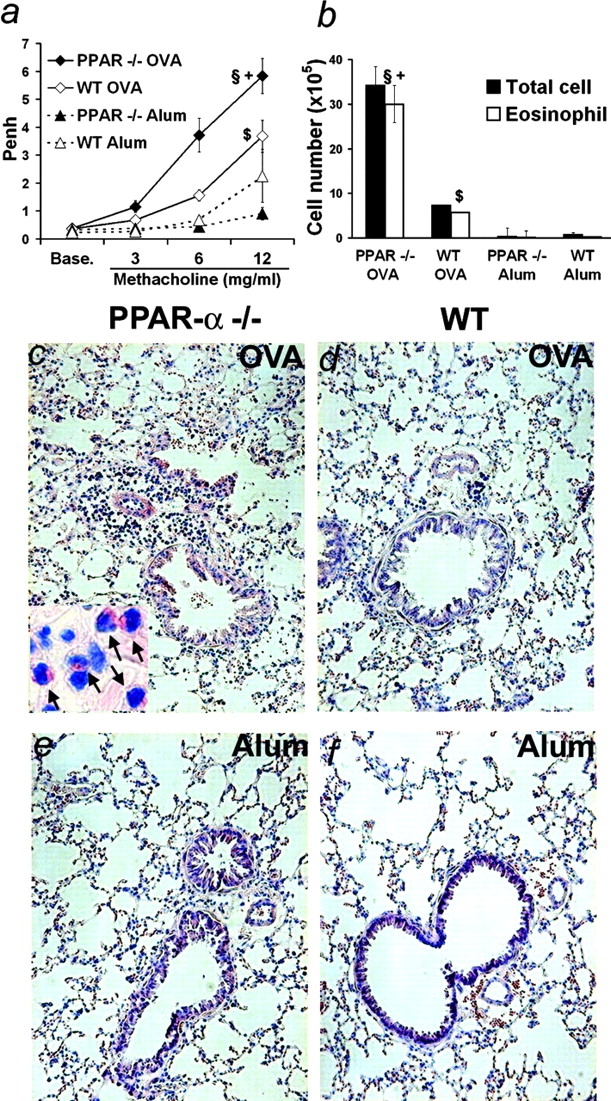
Increased asthma-like reactions in PPAR-α−/− mice. (a) AHR of OVA-sensitized and -challenged or unsensitized but challenged PPAR-α−/− or corresponding WT animals to increasing methacholine concentrations 48 h after the last OVA nebulization. (b) Cellularity and eosinophilia in BALs at the time of sacrifice. (n = 4–13 animals per group. Data expressed as mean ± SEM; some bars may fall within mark). §, Statistically different from OVA-sensitized and aerosol-challenged WT animals. +, Statistically different from unsensitized but aerosol challenged PPAR-α−/− mice. $, Statistically different from unsensitized but aerosol challenged WT mice (see Table I for P values). (c–f) May Grünwald Giemsa staining of lung sections from OVA-sensitized and aerosol-challenged (c and d) or unsensitized but aerosol-challenged (e and f) PPAR-α−/− (c and e) or WT (d and f) mice (original magnification 100). Inset: arrows indicate eosinophils (original magnification 400).
Histological examination of the lung confirmed that, upon sensitization and challenge, PPAR-α–deficient animals displayed more pronounced inflammation with conspicuous peribronchial and perivascular eosinophilia (Fig. 1 c and inset) than the corresponding WT animals (Fig. 1 d). As reported previously for this asthma model (38), eosinophils did not show evidence of degranulation. PAS staining also revealed increased mucus accumulation in the bronchial lumen from PPAR-α–deficient mice compared with WT animals (unpublished data). Neither strain was exhibiting an inflammatory phenotype in the absence of sensitization (Fig. 1, e and f).
The humoral Th2 response characteristic of asthmatic reactions was also affected in PPAR-α–deficient mice. Indeed, serum levels of OVA-specific IgE were increased 3.5-fold in PPAR-α–deficient mice compared with WT animals (Fig. 2 a, left). Levels of specific IgG1, also associated with a Th2 response, were increased 1.5-fold in PPAR-α–deficient mice (not statistically significant; Fig. 2 a, right).
Figure 2.

Increased lung inflammation and humoral response in PPAR-α−/− mice. (a) Serum OVA-specific IgE (left) and IgG1 (right) from animals treated as in Fig. 1 24 h after the last OVA nebulization. (b) IL-6, -13, and eotaxin content from lung extracts from animals treated as in Fig. 1. (n = 4–13 animals per group. Data expressed as mean ± SEM; some bars may fall within mark.) §, Statistically different from OVA-sensitized and aerosol-challenged WT animals. +, Statistically different from unsensitized but aerosol-challenged PPAR-α−/− mice. $, Statistically different from unsensitized but aerosol-challenged WT mice (see Table I for P values). (c) Western blot analysis of GATA-3 expression in lung extracts (and lymph nodes [L. Node]) from individual animals treated as in Fig. 1.
Increased lung inflammation, eosinophilia, and IgE production from PPAR-α–deficient mice were locally associated with higher levels of proinflammatory cytokine IL-6, eosinophil chemoattractant eotaxin, and IgE-inducing IL-13 in lung extracts compared with tissue from WT animals (Fig. 2 b). By contrast, no differences were observed for IL-4 and IL-5 nor for sVCAM-1 (unpublished data). Immunohistological detection of VCAM-1 and ICAM-1 in the lung of PPAR-α–deficient and WT animals did not reveal any differences (unpublished data). Furthermore, after antigen sensitization and challenge, expression of GATA-3, a transcription factor essential for Th2 polarization (39) and expressed in lung eosinophils upon antigen sensitization (40), was greatly increased in lung extracts from PPAR-α–deficient mice compared with tissues from WT animals, where expression is virtually nondetectable (Fig. 2 c). This reflects an enhanced recruitment of cells involved in the Th2 response at the tissular inflammatory site. Together, these results demonstrate that the absence of PPAR-α exacerbates asthmalike reactions and eosinophilia.
PPAR-γ Activation Reduces AHR and Eosinophilia.
Because the complete inactivation of the PPAR-γ gene leads to embryonic lethality, the PPAR-γ agonist ciglitazone was used to investigate the potential role of PPAR-γ as a regulator of airway inflammation. Ciglitazone was targeted to the airways by nebulization immediately before and during OVA sensitization. Agonist administration to Balb/c mice led to a decreased AHR compared with untreated OVA-sensitized animals (Fig. 3 a), which was accompanied by a decreased eosinophilia in BAL (1.9-fold; Fig. 3 b). Eosinophilia was also reduced in lung tissue after ciglitazone treatment (Fig. 3 c). Mucus accumulation in the airways evidenced by PAS staining was likewise diminished by ciglitazone (unpublished data). Importantly, the inhibitory effects of ciglitazone treatment on AHR were abrogated when GW9662, a PPAR-γ–specific antagonist, was administered concomitantly with the agonist (Fig. 3 a), thus demonstrating that ciglitazone was mainly acting through PPAR-γ in this model. Indeed, GW9662 also significantly, albeit only partially, reverted inhibition of cellular recruitment and eosinophilia in BAL and lung tissue after ciglitazone treatment (Fig. 3, b and c).
Figure 3.
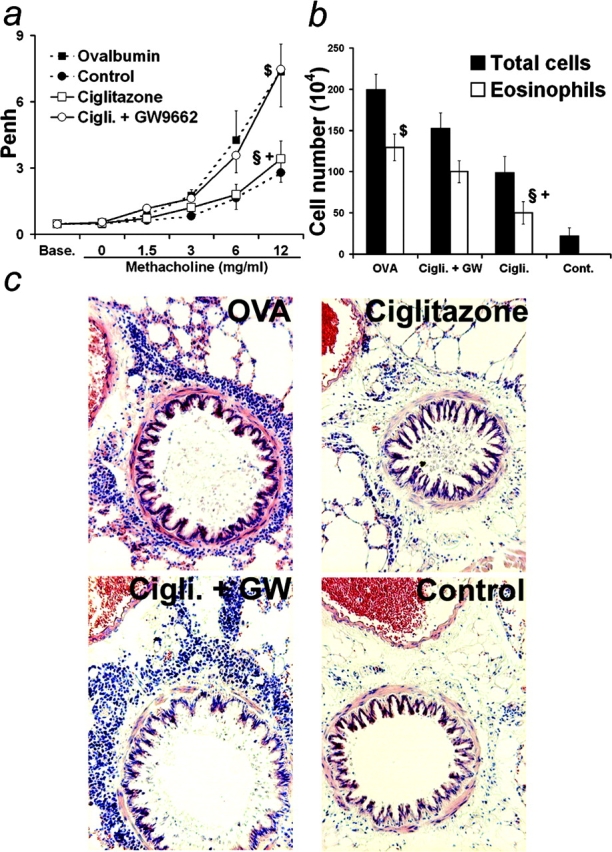
Regulation of asthma-like reactions by PPAR-γ. Mice were sensitized by intraperitoneal injection of OVA in alum and challenged by repeated nebulizations of OVA together with nebulization with 5 × 10−5 M ciglitazone, 5 × 10−5 M ciglitazone and 5 × 10−5 M GW9662 or vehicle. Unsensitized control animals received alum only and were challenged with OVA as for sensitized animals. (a) AHR to increasing methacholine concentrations 48 h after the last nebulization. (b) Cellularity and eosinophilia in BALs at the time of sacrifice. (n = 4–8 animals per group; data expressed as mean ± SEM, some bars may fall within mark). §: Statistically different from OVA-sensitized and aerosol-challenged animals. +: Statistically different from OVA-sensitized and aerosol-challenged mice treated with both ciglitazone and GW9662. $: Statistically different from unsensitized but aerosol challenged mice (see Table I for P values). (c) May Grünwald Giemsa staining of lung sections from sensitized mice nebulized with OVA together with vehicle (upper left), with 5 × 10−5 M ciglitazone (upper right) or with 5 × 10−5 M ciglitazone and 5 × 10−5 M GW9662 (lower left) and from unsensitized animals (lower right) (original magnification 100).
Furthermore, ciglitazone reduced serum levels of OVA-specific IgE and IgG1 2.5- and 9.8-fold, respectively (Fig. 4 a). Cytokines associated with inflammation (IL-6), eosinophilia (IL-5), and Th2-driven humoral immune responses (IL-4 and IL-13) were also decreased in lung extracts (Fig. 4 b), whereas eotaxin and sVCAM-1 were not affected (unpublished data). Likewise, ciglitazone treatment very effectively decreased the amount of GATA-3 protein present in lung extracts, reflecting inhibition of local Th2 response (Fig. 4 c). With the exception of OVA-specific IgG1 production, GW9662 treatment reverted the inhibitory effects of ciglitazone on these parameters, in particular on IL-5 and IgE production (Fig. 4, a–c). Thus, administration of a PPAR-γ agonist during antigen challenge periods decreased most of the characteristic parameters of airway inflammation.
Figure 4.

Regulation of pulmonary inflammation and humoral response by PPAR-γ. (a) Serum OVA-specific IgE (left) and IgG1 (right) from animals treated as in Fig. 3 24 h after the last OVA nebulization. (b) IL-4, -5, -6, and -13 content of lung extracts from animals treated as in Fig. 3. (n = 4–19 animals per group. Data expressed as mean ± SEM; some bars may fall within mark.) §, Statistically different from OVA-sensitized and aerosol-challenged animals. +, Statistically different from OVA-sensitized and aerosol-challenged mice treated with both ciglitazone and GW9662. $, Statistically different from unsensitized but aerosol-challenged mice (see Table I for P values). (c) Western blot analysis of GATA-3 expression in lung extracts (and lymph nodes [L. Node]) from individual animals treated as in Fig. 3.
PPAR-α and -γ Are Expressed by Eosinophils.
To determine whether the observed effects of PPAR-α and -γ on eosinophilia in vivo may be in part related to a direct modulation of eosinophil activation, PPAR expression was analyzed in eosinophils in vitro. PPAR-α and -γ expression was assessed in highly purified eosinophils from three different species (human, mouse, and rat) using three independent techniques.
Specific transcripts for PPAR-α (Fig. 5 a, top) and, at a lower level, for PPAR-γ (Fig. 5 a, middle) were detected by RT-PCR in mouse (purity > 95%), rat (purity > 90%), and human (purity > 97%) eosinophils. These results were confirmed at the protein level by Western blot analysis (Fig. 5 b). To further exclude that the signal obtained by RT-PCR or Western blot was due to a minor contamination with another cell type highly expressing these PPARs, we performed flow cytometry analyses on permeabilized cells using a narrow gating on eosinophil populations, which display a very characteristic scatter pattern (small highly granular cells). Indeed, both PPAR-α and -γ were expressed in the gated cells from mouse, rat, and human eosinophil preparations (Fig. 5 c, top left and bottom). Finally, PPAR-α and -γ expression levels were investigated in eight eosinophilic patients. PPAR-α expression was homogenous, whereas PPAR-γ expression was present at more variable levels between individuals (Fig. 5 c top right), which suggests, as reported previously (26), that PPAR-γ may indeed be regulated by some factors associated with the pathology. No correlation was found between PPAR expression and the activation state of eosinophils, as evaluated by serum eosinophil cationic protein and CD25 measurements (unpublished data).
Figure 5.
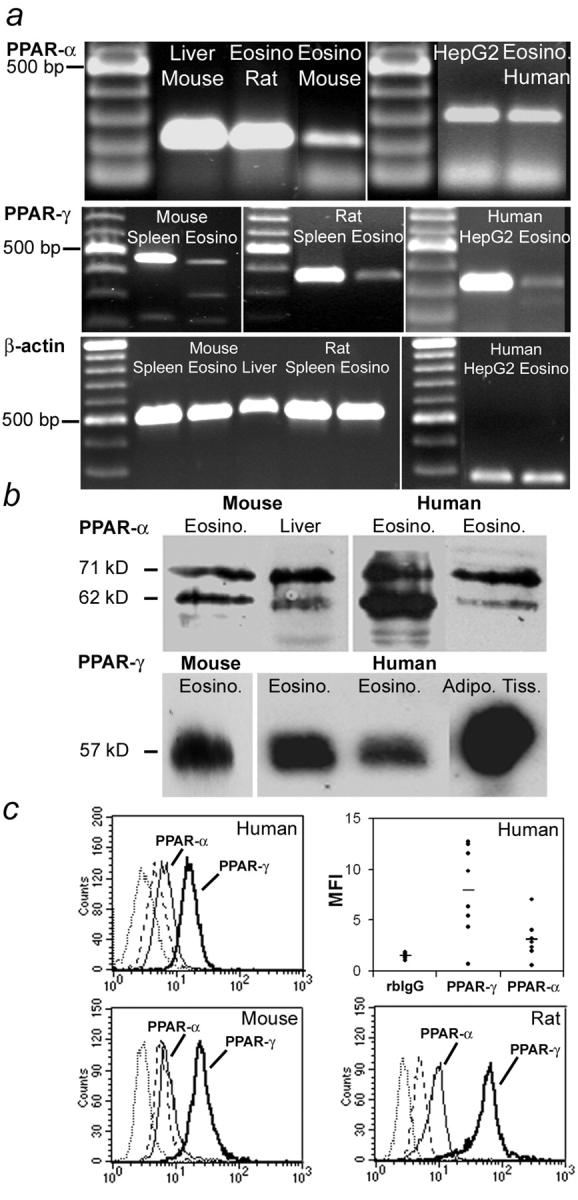
PPAR expression in eosinophils. (a) RT-PCR amplification of PPAR-α (top), PPAR-γ (middle), and β-actin (bottom) mRNA from human, mouse, and rat eosinophils. (top) Total WT mouse liver, rat peritoneal eosinophils, and eosinophils from IL-5 Tg mouse spleen (amplicon size: 215 bp; left). HepG2 cells and human peripheral blood eosinophils (amplicon size: 304 bp; right). (middle) Total WT mouse spleen and eosinophils from IL-5 Tg mouse spleen (amplicon size: 473 bp; left). Total rat spleen and rat peritoneal eosinophils (amplicon size: 343 bp; center). HepG2 cells and human peripheral blood eosinophils (amplicon size: 337 bp; right). (bottom) Total WT mouse spleen, eosinophils from IL-5 Tg mouse spleen, total WT mouse liver, total rat spleen, and peritoneal rat eosinophils (amplicon size: 532 bp; left). Human peripheral blood eosinophils and HepG2 cells (amplicon size: 238 bp; right). (b) Identification of PPAR-α (top) and PPAR-γ (bottom) proteins in human and mouse eosinophil lysates by Western blot analysis. Immunodetection of PPAR-α and PPAR-γ in cell lysates from IL-5 Tg mouse eosinophils and WT mouse liver or human peripheral blood eosinophils from two different donors and adipose tissue after SDS-PAGE and transfer on membrane. (c) Detection of PPAR-α and PPAR-γ by flow cytometry on permeabilized human (top), mouse (bottom left), and rat eosinophils (bottom right). Relative expression levels of PPAR-α and PPAR-γ in eosinophils from eight donors with hypereosinophilia (mean fluorescence intensity [MFI]; control rabbit IgG (rbIgG); average value is represented by the horizontal bar in each group). On histogram plots, anti–PPAR-α (thin line), anti–PPAR-γ (thick line), control rabbit IgG (dotted line), and FITC-conjugated secondary antibody (dashed line).
PPAR-α and -γ Activation Inhibits Eosinophil Chemotaxis.
To establish whether PPAR-α and -γ agonists directly modulate eosinophil chemotactic response to IL-5 or eotaxin, two potent physiological chemoattractants, we performed in vitro chemotaxis experiments with human eosinophils. PPAR-α (WY14653) or PPAR-γ (rosiglitazone) agonists were able to inhibit IL-5– and eotaxin-induced eosinophil migration (Fig. 6 , a and b). Similar results were obtained with ciglitazone (unpublished data). The requirement of PPAR-γ for this inhibitory effect was confirmed by using GW9662. Indeed, eosinophil incubation with GW9662 in conjunction with rosiglitazone led to a virtually complete loss of chemotaxis inhibition (Fig. 6 a, inset). Thus, PPAR-α and -γ activation by specific agonists can act directly on eosinophils and regulate their movement, at least in vitro.
Figure 6.

Regulation of eosinophil function by PPAR in vitro. (a and b) Inhibition of eosinophil chemotaxis by PPAR. Dose-dependent inhibition of IL-5– and eotaxin-induced chemotaxis of human peripheral blood eosinophils by rosiglitazone (a) and WY14653 (b). P < 0.0001 for each agonist with both chemoattractants versus untreated cells. Inset: Abrogation of rosiglitazone inhibition of IL-5–induced chemotaxis by GW9662 (concentration of each compound 10−5 M). (c and d) Inhibition of eosinophil-mediated ADCC by PPAR agonists. Dose-dependent inhibition of human (c) or rat (d) eosinophil-mediated ADCC toward S. mansoni larvae by WY14653, ciglitazone, and rosiglitazone (n = 3–8 independent experiments; data expressed as mean ± SEM). (1) and (5) P < 0.0001 for rosiglitazone-treated versus vehicle-treated cells; (2) P = 0.034 and (6) P < 0.0001 for ciglitazone-treated versus vehicle-treated cells; (3) P = 0.037 and (4) P < 0.0001 for WY14653-treated versus vehicle-treated cells.
PPAR-α and -γ Activation Inhibits Eosinophil-mediated Cytotoxicity.
Upon activation with various antibody isotypes, including IgA and IgE, both human and rat eosinophils are able to develop cytotoxic properties against parasitic targets in vitro associated with a protective immune response in vivo. Measurement of eosinophil-dependent cytotoxicity is thus a good in vitro correlate of their effector function in vivo (41, 42). Therefore, the effects of PPAR-α and -γ agonists on eosinophil-mediated ADCC were investigated. A dose-dependent inhibition of human eosinophil-mediated ADCC was observed with rosiglitazone, ciglitazone, and WY14653, with respective maximal inhibition of 45, 33, and 28% (at 10−5 M; Fig. 6 c). Similar results were obtained with rat eosinophils (Fig. 6 d). However, maximal inhibitory effects for the three agonists were obtained at lower concentrations (41, 44, and 39% at 10−7 M). Our results demonstrate that both PPAR-α and PPAR-γ agonists significantly inhibit ADCC reactions mediated by human or rat eosinophils.
Discussion
The regulatory role of PPAR-α and -γ on allergic lung inflammation and eosinophilia was investigated in vivo using a classical animal model for human asthma. This model, in which both CD4+ and CD8+ T lymphocytes play an important role (43, 44), is characterized by increased airway hyperreactivity, eosinophilia, and high IgE concentrations.
Use of PPAR-α–deficient mice allowed us to unequivocally demonstrate that the absence of PPAR-α led to increased lung eosinophilia and AHR, which were accompanied by increased antigen-specific IgE concentrations compared with WT animals. This increased functional, cellular, and humoral response was correlated to increased lung expression of GATA-3, associated with Th2 polarization (39), and to increased levels of the proinflammatory cytokine IL-6 as well as IL-13 and eotaxin, two major factors promoting AHR, eosinophilia, and IgE production (1, 45).
LTB4, a natural PPAR-α ligand (5), is abundantly produced, among other cell types, by mast cells in inflammatory reactions such as asthma (46) and is highly chemotactic for eosinophils that express a specific membrane LTB4 receptor: BLTR (35). Because PPAR-α activation by LTB4 leads to an anti-inflammatory response, the absence of PPAR-α expression (in eosinophils and other cell types from PPAR-α–deficient mice) would exacerbate the chemotactic response by creating an imbalance between stimulatory and inhibitory effects.
PPAR-α has been recently shown to negatively regulate the Th1-inducing transcription factor T-bet (47). It might also affect directly or indirectly GATA-3, the T-bet Th2 counterpart, which is not only expressed in Th2 cells (39) but also in lung eosinophils (40). Thus, GATA-3 decreased expression could be due to an effect on both cell types. Although an inhibitory effect of PPAR-α has been reported for IL-6 production by vascular tissues (28), this represents the first paper demonstrating a regulation by PPAR-α of eotaxin, an eosinophil chemotactic factor, and IL-13, an IgE-inducing factor for B cells. IL-13 is produced mainly by T cells, which express PPAR-α (13), and by eosinophils themselves (48). This production is regulated by both IL-5, another cytokine preferentially produced by Th2 cells, and by eotaxin (49). Besides B cells, IL-13 affects airway epithelial cells, where it is able to induce eotaxin production (50) and to regulate AHR and mucus overproduction (51), smooth muscle cells, and fibroblasts, which can also be triggered to produce eotaxin, IL-6, and VCAM-1 (52). Additionally, PPAR-α might regulate IgE production through a direct action on B cells, in which it is expressed (13). Finally, eotaxin is produced by endothelial cells (53), in which PPAR-α is expressed (54). Hence, PPAR-α agonists, at least in this latter cell type, might be able to modulate its expression.
In the same model of antigen sensitization and airway challenge, we demonstrated that delivery of a PPAR-γ agonist to the lung, through nebulization, profoundly decreased AHR, IgE (and IgG1) production, and eosinophilia, with a concomitant decrease of IL-4, IL-5, IL-6, IL-13, and GATA-3 expression. According to the parameter measured, this inhibitory effect was totally or partially abrogated by coadministration of a GW9662 an irreversible PPAR-γ antagonist, thus demonstrating that most ciglitazone-mediated effects indeed occur through PPAR-γ stimulation in this model.
PPAR-γ appears to regulate IL-4 and IL-5, which promote AHR in conjunction with IL-13 (1), but not eotaxin production. PPAR-γ activation might also influence lung infiltration by inflammatory cells and, among them, eosinophils, which are responsive to IL-5. Furthermore, in addition to T cell function, PPAR-γ is expressed in airway epithelial, submucosal and smooth muscle cells and regulates their function (26, 55, 56). Other likely targets for PPAR-γ are dendritic cells, whose activity is modulated by PPAR-γ (19, 57), and which play a key role in the induction of the pulmonary response to antigen inhalation (58). Inhibition of dendritic cell migration by PPAR-γ agonists, as reported previously for prostaglandin D2 (59), a precursor of 15d-PGJ2 (a natural PPAR-γ ligand), would contribute to decreased eosinophilia as well as to decreased T cell proliferation, which is also directly inhibited by PPAR-γ (24, 60). Finally, it has been shown in an arthritis model, that PPAR-γ hemizygous mice displayed increased B cell and T cell proliferation and IgM production (24). A regulation by PPAR-γ agonists of IgE germline transcript synthesis in vitro by a human B cell line was also reported (61). In our in vivo model, PPAR-γ agonists also regulate IgE and IgG1 production in a secondary immune response.
The further demonstration that both PPAR-α and -γ are expressed by mouse, human, and rat eosinophils and the ability for specific PPAR-α and -γ agonists to regulate two of their physiological functions in vitro strongly support the hypothesis that their inhibitory effects on lung inflammation observed in vivo in an asthma model might, in part, be due to a direct action on eosinophils. Indeed, PPAR-α and -γ agonists induced a dose-dependent inhibition of IL-5 or eotaxin-mediated eosinophil chemotaxis, whereas the effects of the PPAR-γ agonist were fully reversed by cotreatment in vitro with a specific antagonist. Eosinophil chemotaxis requires signaling through IL-5R (βc chain) or CCR3, the G protein-coupled seven transmembrane domain receptor for eotaxin, and leads to MAP kinase (ERK, p38, and JNK) as well as c-jun and c-fos activation (62, 63).These factors are known targets for PPAR-α and -γ regulation (9, 23). In a similar way, ADCC reactions require activation of some Fc receptors, which vary according to the immunoglobulin isotype and the cell type. In eosinophil-mediated ADCC toward S. mansoni larvae, IgE and IgA are mainly involved. FcɛRI and FcαRI/CD89, their cognate receptors, share a common subunit, FcRγ, whose complex signal transduction cascade leads to MAP kinase, c-jun, c-fos (64–66), and NF-κB activation (67), which are potential PPAR targets (9). It is perhaps not surprising that inhibition of ADCC by PPAR agonists is weaker than for chemotaxis. Indeed, cytotoxicity is a more complex biological process not only involving chemotaxis but also cell–cell contacts and granule release, each of which would only be partially inhibited by agonists.
Together, our results demonstrate that both PPAR-α and -γ directly affect eosinophil functions in vitro and are able to regulate eosinophilia in vivo, in a murine model of asthma, where complex interactions occur. Due to recently reported failure of monotherapies targeted to eosinophils, such as anti–IL-5, in the treatment of eosinophilia, asthma, and AHR (68), these nuclear receptors, which are expressed in many immune (including T cells) and nonimmune cell types in the inflammatory airways, might thus represent attractive alternative therapeutic targets for these pathologies.
Acknowledgments
We thank A.-M. Lefèvre for her scientific contribution, and S. Loiseau (U547-IFR17) and P. Marquillies (U416-IFR17) for their skillful technical assistance.
This work was supported by grants from INSERM, CNRS, Institut Pasteur de Lille, Hôpitaux Universitaires de Strasbourg, and the European Union Research Technology Development program (GLGICT-1999-00674 and GLRT-2001-00930).
Footnotes
Abbreviations used in this paper: ADCC, antibody-dependent cellular cytoxicity; AHR, airway hyperresponsiveness; BAL, bronchoalveolar lavage; LTB4, leukotriene B4; PAS, periodic acid schiff; PPAR, peroxisome proliferator–activated receptor; RT, reverse transcriptase; WT, wild-type.
References
- 1.Wills-Karp, M. 1999. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 17:255–281. [DOI] [PubMed] [Google Scholar]
- 2.Maddox, L., and D.A. Schwartz. 2002. The pathophysiology of asthma. Annu. Rev. Med. 53:477–498. [DOI] [PubMed] [Google Scholar]
- 3.Dombrowicz, D., and M. Capron. 2001. Eosinophils, allergy and parasites. Curr. Opin. Immunol. 13:716–720. [DOI] [PubMed] [Google Scholar]
- 4.Desvergne, B., and W. Wahli. 1999. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 20:649–688. [DOI] [PubMed] [Google Scholar]
- 5.Devchand, P.R., H. Keller, J.M. Peters, M. Vazquez, F.J. Gonzalez, and W. Wahli. 1996. The PPAralpha-leukotriene B4 pathway to inflammation control. Nature. 384:39–43. [DOI] [PubMed] [Google Scholar]
- 6.Hatae, T., M. Wada, C. Yokoyama, M. Shimonishi, and T. Tanabe. 2001. Prostacyclin-dependent apoptosis mediated by PPARdelta. J. Biol. Chem. 276:46260–46267. [DOI] [PubMed] [Google Scholar]
- 7.Nagy, L., P. Tontonoz, J.G. Alvarez, H. Chen, and R.M. Evans. 1998. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 93:229–240. [DOI] [PubMed] [Google Scholar]
- 8.Forman, B.M., P. Tontonoz, J. Chen, R.P. Brun, B.M. Spiegelman, and R.M. Evans. 1995. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 83:803–812. [DOI] [PubMed] [Google Scholar]
- 9.Delerive, P., J.C. Fruchart, and B. Staels. 2001. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 169:453–459. [DOI] [PubMed] [Google Scholar]
- 10.Clark, R.B. 2002. The role of PPARs in inflammation and immunity. J. Leukoc. Biol. 71:388–400. [PubMed] [Google Scholar]
- 11.Daynes, R.A., and D.C. Jones. 2002. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2:748–759. [DOI] [PubMed] [Google Scholar]
- 12.Clark, R.B., D. Bishop-Bailey, T. Estrada-Hernandez, T. Hla, L. Puddington, and S.J. Padula. 2000. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J. Immunol. 164:1364–1371. [DOI] [PubMed] [Google Scholar]
- 13.Jones, D.C., X. Ding, and R.A. Daynes. 2001. Nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) is expressed in resting murine lymphocytes: the PPARalpha in T and B lymphocytes is both transactivation and transrepression competent. J. Biol. Chem. 277:6838–6845. [DOI] [PubMed] [Google Scholar]
- 14.Chinetti, G., S. Griglio, M. Antonucci, I.P. Torra, P. Delerive, Z. Majd, J.C. Fruchart, J. Chapman, J. Najib, and B. Staels. 1998. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 273:25573–25580. [DOI] [PubMed] [Google Scholar]
- 15.Ricote, M., A.C. Li, T.M. Willson, C.J. Kelly, and C.K. Glass. 1998. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 391:79–82. [DOI] [PubMed] [Google Scholar]
- 16.Tontonoz, P., L. Nagy, J.G. Alvarez, V.A. Thomazy, and R.M. Evans. 1998. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 93:241–252. [DOI] [PubMed] [Google Scholar]
- 17.Huang, J.T., J.S. Welch, M. Ricote, C.J. Binder, T.M. Willson, C. Kelly, J.L. Witztum, C.D. Funk, D. Conrad, and C.K. Glass. 1999. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 400:378–382. [DOI] [PubMed] [Google Scholar]
- 18.Moore, K.J., E.D. Rosen, M.L. Fitzgerald, F. Randow, L.P. Andersson, D. Altshuler, D.S. Milstone, R.M. Mortensen, B.M. Spiegelman, and M.W. Freeman. 2001. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat. Med. 7:41–47. [DOI] [PubMed] [Google Scholar]
- 19.Faveeuw, C., S. Fougeray, V. Angeli, J. Fontaine, G. Chinetti, P. Gosset, P. Delerive, C. Maliszewski, M. Capron, B. Staels, et al. 2000. Peroxisome proliferator-activated receptor gamma activators inhibit interleukin-12 production in murine dendritic cells. FEBS Lett. 486:261–266. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama, H., T. Nonaka, T. Kishimoto, K. Komoriya, K. Tsuji, and T. Nakahata. 2000. Peroxisome proliferator-activated receptors are expressed in human cultured mast cells: a possible role of these receptors in negative regulation of mast cell activation. Eur. J. Immunol. 30:3363–3370. [DOI] [PubMed] [Google Scholar]
- 21.Tan, N.S., L. Michalik, N. Noy, R. Yasmin, C. Pacot, M. Heim, B. Fluhmann, B. Desvergne, and W. Wahli. 2001. Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 15:3263–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su, C.G., X. Wen, S.T. Bailey, W. Jiang, S.M. Rangwala, S.A. Keilbaugh, A. Flanigan, S. Murthy, M.A. Lazar, and G.D. Wu. 1999. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J. Clin. Invest. 104:383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desreumaux, P., L. Dubuquoy, S. Nutten, M. Peuchmaur, W. Englaro, K. Schoonjans, B. Derijard, B. Desvergne, W. Wahli, P. Chambon, et al. 2001. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator–activated receptor γ (PPARγ) heterodimer. A basis for new therapeutic strategies. J. Exp. Med. 193:827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Setoguchi, K., Y. Misaki, Y. Terauchi, T. Yamauchi, K. Kawahata, T. Kadowaki, and K. Yamamoto. 2001. Peroxisome proliferator-activated receptor-gamma haploinsufficiency enhances B cell proliferative responses and exacerbates experimentally induced arthritis. J. Clin. Invest. 108:1667–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis, J.D., G.R. Lichtenstein, R.B. Stein, J.J. Deren, T.A. Judge, F. Fogt, E.E. Furth, E.J. Demissie, L.B. Hurd, C.G. Su, et al. 2001. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis. Am. J. Gastroenterol. 96:3323–3328. [DOI] [PubMed] [Google Scholar]
- 26.Benayoun, L., S. Letuve, A. Druilhe, J. Boczkowski, M.C. Dombret, P. Mechighel, J. Megret, G. Leseche, M. Aubier, and M. Pretolani. 2001. Regulation of peroxisome proliferator-activated receptor gamma expression in human asthmatic airways: relationship with proliferation, apoptosis, and airway remodeling. Am. J. Respir. Crit. Care Med. 164:1487–1494. [DOI] [PubMed] [Google Scholar]
- 27.Poynter, M.E., and R.A. Daynes. 1998. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J. Biol. Chem. 273:32833–32841. [DOI] [PubMed] [Google Scholar]
- 28.Delerive, P., K. De Bosscher, S. Besnard, W. Vanden Berghe, J.M. Peters, F.J. Gonzalez, J.C. Fruchart, A. Tedgui, G. Haegeman, and B. Staels. 1999. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 274:32048–32054. [DOI] [PubMed] [Google Scholar]
- 29.Lee, S.S., T. Pineau, J. Drago, E.J. Lee, J.W. Owens, D.L. Kroetz, P.M. Fernandez-Salguero, H. Westphal, and F.J. Gonzalez. 1995. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol. 15:3012–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kayaba, H., D. Dombrowicz, G. Woerly, J.P. Papin, S. Loiseau, and M. Capron. 2001. Human eosinophils and human high affinity IgE receptor transgenic mouse eosinophils express low levels of high affinity IgE receptor, but release IL-10 upon receptor activation. J. Immunol. 167:995–1003. [DOI] [PubMed] [Google Scholar]
- 31.Hamelmann, E., J. Schwarze, K. Takeda, A. Oshiba, G.L. Larsen, C.G. Irvin, and E.W. Gelfand. 1997. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care Med. 156:766–775. [DOI] [PubMed] [Google Scholar]
- 32.Standiford, T.J., S.L. Kunkel, N.W. Lukacs, M.J. Greenberger, J.M. Danforth, R.G. Kunkel, and R.M. Strieter. 1995. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J. Immunol. 155:1515–1524. [PubMed] [Google Scholar]
- 33.Angeli, V., C. Faveeuw, P. Delerive, J. Fontaine, Y. Barriera, N. Franchimont, B. Staels, M. Capron, and F. Trottein. 2001. Schistosoma mansoni induces the synthesis of IL-6 in pulmonary microvascular endothelial cells: role of IL-6 in the control of lung eosinophilia during infection. Eur. J. Immunol. 31:2751–2761. [DOI] [PubMed] [Google Scholar]
- 34.Woerly, G., N. Roger, S. Loiseau, D. Dombrowicz, A. Capron, and M. Capron. 1999. Expression of CD28 and CD86 by human eosinophils and role in the secretion of type 1 cytokines (interleukin 2 and interferon γ): inhibition by immunoglobulin A complexes. J. Exp. Med. 190:487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang, W.W., E.A. Garcia-Zepeda, A. Sauty, H.C. Oettgen, M.E. Rothenberg, and A.D. Luster. 1998. Molecular and biological characterization of the murine leukotriene B4 receptor expressed on eosinophils. J. Exp. Med. 188:1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capron, M., S. Loiseau, J.P. Papin, S. Robertson, and A. Capron. 1998. Inhibitory effects of lodoxamide on eosinophil activation. Int. Arch. Allergy Immunol. 116:140–146. [DOI] [PubMed] [Google Scholar]
- 37.Dombrowicz, D., B. Quatannens, J.P. Papin, A. Capron, and M. Capron. 2000. Expression of a functional Fc epsilon RI on rat eosinophils and macrophages. J. Immunol. 165:1266–1271. [DOI] [PubMed] [Google Scholar]
- 38.Malm-Erjefalt, M., C.G. Persson, and J.S. Erjefalt. 2001. Degranulation status of airway tissue eosinophils in mouse models of allergic airway inflammation. Am. J. Respir. Cell Mol. Biol. 24:352–359. [DOI] [PubMed] [Google Scholar]
- 39.Zheng, W., and R.A. Flavell. 1997. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 89:587–596. [DOI] [PubMed] [Google Scholar]
- 40.Justice, J.P., M.T. Borchers, J.J. Lee, W.H. Rowan, Y. Shibata, and M.R. Van Scott. 2002. Ragweed-induced expression of GATA-3, IL-4, and IL-5 by eosinophils in the lungs of allergic C57BL/6J mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L302–L309. [DOI] [PubMed] [Google Scholar]
- 41.Capron, M., and A. Capron. 1994. Immunoglobulin E and effector cells in schistosomiasis. Science. 264:1876–1877. [DOI] [PubMed] [Google Scholar]
- 42.Grezel, D., M. Capron, J.M. Grzych, J. Fontaine, J.P. Lecocq, and A. Capron. 1993. Protective immunity induced in rat schistosomiasis by a single dose of the Sm28GST recombinant antigen: effector mechanisms involving IgE and IgA antibodies. Eur. J. Immunol. 23:454–460. [DOI] [PubMed] [Google Scholar]
- 43.Gavett, S.H., X. Chen, F. Finkelman, and M. Wills-Karp. 1994. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am. J. Respir. Cell Mol. Biol. 10:587–593. [DOI] [PubMed] [Google Scholar]
- 44.Hamelmann, E., A. Oshiba, J. Paluh, K. Bradley, J. Loader, T.A. Potter, G.L. Larsen, and E.W. Gelfand. 1996. Requirement for CD8+ T cells in the development of airway hyperresponsiveness in a marine model of airway sensitization. J. Exp. Med. 183:1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foster, P.S., A.W. Mould, M. Yang, J. Mackenzie, J. Mattes, S.P. Hogan, S. Mahalingam, A.N. McKenzie, M.E. Rothenberg, I.G. Young, et al. 2001. Elemental signals regulating eosinophil accumulation in the lung. Immunol. Rev. 179:173–181. [DOI] [PubMed] [Google Scholar]
- 46.Hart, P.H. 2001. Regulation of the inflammatory response in asthma by mast cell products. Immunol. Cell Biol. 79:149–153. [DOI] [PubMed] [Google Scholar]
- 47.Jones, D.C., B.M. Manning, and R.A. Daynes. 2002. A role for the peroxisome proliferator-activated receptor alpha in T-cell physiology and ageing immunobiology. Proc. Nutr. Soc. 61:363–369. [DOI] [PubMed] [Google Scholar]
- 48.Woerly, G., P. Lacy, A.B. Younes, N. Roger, S. Loiseau, R. Moqbel, and M. Capron. 2002. Human eosinophils express and release IL-13 following CD28-dependent activation. J. Leukoc. Biol. 72:769–779. [PubMed] [Google Scholar]
- 49.Mattes, J., M. Yang, S. Mahalingam, J. Kuehr, D.C. Webb, L. Simson, S.P. Hogan, A. Koskinen, A.N. McKenzie, L.A. Dent, et al. 2002. Intrinsic defect in T cell production of interleukin (IL)-13 in the absence of both IL-5 and eotaxin precludes the development of eosinophilia and airways hyperreactivity in experimental asthma. J. Exp. Med. 195:1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu, Z., R.J. Homer, Z. Wang, Q. Chen, G.P. Geba, J. Wang, Y. Zhang, and J.A. Elias. 1999. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 103:779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuperman, D.A., X. Huang, L.L. Koth, G.H. Chang, G.M. Dolganov, Z. Zhu, J.A. Elias, D. Sheppard, and D.J. Erle. 2002. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8:885–889. [DOI] [PubMed] [Google Scholar]
- 52.Lee, J.H., N. Kaminski, G. Dolganov, G. Grunig, L. Koth, C. Solomon, D.J. Erle, and D. Sheppard. 2001. Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am. J. Respir. Cell Mol. Biol. 25:474–485. [DOI] [PubMed] [Google Scholar]
- 53.Rothenberg, M.E., A.D. Luster, and P. Leder. 1995. Murine eotaxin: an eosinophil chemoattractant inducible in endothelial cells and in interleukin 4-induced tumor suppression. Proc. Natl. Acad. Sci. USA. 92:8960–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee, H., W. Shi, P. Tontonoz, S. Wang, G. Subbanagounder, C.C. Hedrick, S. Hama, C. Borromeo, R.M. Evans, J.A. Berliner, and L. Nagy. 2000. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ. Res. 87:516–521. [DOI] [PubMed] [Google Scholar]
- 55.Wang, A.C., X. Dai, B. Luu, and D.J. Conrad. 2001. Peroxisome proliferator-activated receptor-gamma regulates airway epithelial cell activation. Am. J. Respir. Cell Mol. Biol. 24:688–693. [DOI] [PubMed] [Google Scholar]
- 56.Patel, H.J., M.G. Belvisi, D. Bishop-Bailey, M.H. Yacoub, and J.A. Mitchell. 2003. Activation of peroxisome proliferator-activated receptors in human airway smooth muscle cells has a superior anti-inflammatory profile to corticosteroids: relevance for chronic obstructive pulmonary disease therapy. J. Immunol. 170:2663–2669. [DOI] [PubMed] [Google Scholar]
- 57.Gosset, P., A.S. Charbonnier, P. Delerive, J. Fontaine, B. Staels, J. Pestel, A.B. Tonnel, and F. Trottein. 2001. Peroxisome proliferator-activated receptor gamma activators affect the maturation of human monocyte-derived dendritic cells. Eur. J. Immunol. 31:2857–2865. [DOI] [PubMed] [Google Scholar]
- 58.Lambrecht, B.N., M. De Veerman, A.J. Coyle, J.C. Gutierrez-Ramos, K. Thielemans, and R.A. Pauwels. 2000. Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J. Clin. Invest. 106:551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Angeli, V., C. Faveeuw, O. Roye, J. Fontaine, E. Teissier, A. Capron, I. Wolowczuk, M. Capron, and F. Trottein. 2001. Role of the parasite-derived prostaglandin D2 in the inhibition of epidermal Langerhans' cell migration during schistosomiasis infection. J. Exp. Med. 193:1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang, X.Y., L.H. Wang, T. Chen, D.R. Hodge, J.H. Resau, L. DaSilva, and W.L. Farrar. 2000. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J. Biol. Chem. 275:4541–4544. [DOI] [PubMed] [Google Scholar]
- 61.Miyazaki, Y., H. Tachibana, and K. Yamada. 2002. Inhibitory effect of peroxisome proliferator-activated receptor-gamma ligands on the expression of IgE heavy chain germline transcripts in the human B cell line DND39. Biochem. Biophys. Res. Commun. 295:547–552. [DOI] [PubMed] [Google Scholar]
- 62.Adachi, T., and R. Alam. 1998. The mechanism of IL-5 signal transduction. Am. J. Physiol. 275:C623–C633. [DOI] [PubMed] [Google Scholar]
- 63.Kampen, G.T., S. Stafford, T. Adachi, T. Jinquan, S. Quan, J.A. Grant, P.S. Skov, L.K. Poulsen, and R. Alam. 2000. Eotaxin induces degranulation and chemotaxis of eosinophils through the activation of ERK2 and p38 mitogen-activated protein kinases. Blood. 95:1911–1917. [PubMed] [Google Scholar]
- 64.Tsai, M., S.Y. Tam, and S.J. Galli. 1993. Distinct patterns of early response gene expression and proliferation in mouse mast cells stimulated by stem cell factor, interleukin-3, or IgE and antigen. Eur. J. Immunol. 23:867–872. [DOI] [PubMed] [Google Scholar]
- 65.Razin, E., Z. Szallasi, M.G. Kazanietz, P.M. Blumberg, and J. Rivera. 1994. Protein kinases C-beta and C-epsilon link the mast cell high-affinity receptor for IgE to the expression of c-fos and c-jun. Proc. Natl. Acad. Sci. USA. 91:7722–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ishizuka, T., K. Chayama, K. Takeda, E. Hamelmann, N. Terada, G.M. Keller, G.L. Johnson, and E.W. Gelfand. 1999. Mitogen-activated protein kinase activation through Fc epsilon receptor I and stem cell factor receptor is differentially regulated by phosphatidylinositol 3-kinase and calcineurin in mouse bone marrow-derived mast cells. J. Immunol. 162:2087–2094. [PubMed] [Google Scholar]
- 67.Pelletier, C., N. Varin-Blank, J. Rivera, B. Iannascoli, F. Marchand, B. David, A. Weyer, and U. Blank. 1998. Fc epsilonRI-mediated induction of TNF-alpha gene expression in the RBL-2H3 mast cell line: regulation by a novel NF-kappaB-like nuclear binding complex. J. Immunol. 161:4768–4776. [PubMed] [Google Scholar]
- 68.Leckie, M.J., A. ten Brinke, J. Khan, Z. Diamant, B.J. O'Connor, C.M. Walls, A.K. Mathur, H.C. Cowley, K.F. Chung, R. Djukanovic, et al. 2000. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 356:2144–2148. [DOI] [PubMed] [Google Scholar]