

Abstract
Hypoxia-induced VEGF governs both physiological retinal vascular development and pathological retinal neovascularization. In the current paper, the mechanisms of physiological and pathological neovascularization are compared and contrasted. During pathological neovascularization, both the absolute and relative expression levels for VEGF164 increased to a greater degree than during physiological neovascularization. Furthermore, extensive leukocyte adhesion was observed at the leading edge of pathological, but not physiological, neovascularization. When a VEGF164-specific neutralizing aptamer was administered, it potently suppressed the leukocyte adhesion and pathological neovascularization, whereas it had little or no effect on physiological neovascularization. In parallel experiments, genetically altered VEGF164-deficient (VEGF120/188) mice exhibited no difference in physiological neovascularization when compared with wild-type (VEGF+/+) controls. In contrast, administration of a VEGFR-1/Fc fusion protein, which blocks all VEGF isoforms, led to significant suppression of both pathological and physiological neovascularization. In addition, the targeted inactivation of monocyte lineage cells with clodronate-liposomes led to the suppression of pathological neovascularization. Conversely, the blockade of T lymphocyte–mediated immune responses with an anti-CD2 antibody exacerbated pathological neovascularization. These data highlight important molecular and cellular differences between physiological and pathological retinal neovascularization. During pathological neovascularization, VEGF164 selectively induces inflammation and cellular immunity. These processes provide positive and negative angiogenic regulation, respectively. Together, new therapeutic approaches for selectively targeting pathological, but not physiological, retinal neovascularization are outlined.
Keywords: retina, angiogenesis, VEGF, leukocyte, immunity
Introduction
Retinal neovascular diseases such as retinopathy of prematurity and proliferative diabetic retinopathy are the major causes of neonatal and adult blindness. During both physiological retinal vascular development (1) and pathological proliferative retinopathy (2), new blood vessel growth is regulated by vascular endothelial growth factor (VEGF), a hypoxia-induced endothelial cell–specific mitogen produced in ischemic retinal cells. Although both types of neovascularization stem from retinal ischemia, there is an essential difference in the direction of vessel growth during physiological and pathological neovascularization. In the former, new vessels extend from the optic disc toward the peripheral avascular retina, and follow the guidance of VEGF-expressing retinal astrocytes (2). During pathological neovascularization, although ischemic retinal astrocytes and neurons express VEGF (1), the new vessels invade the vitreous cavity. This ectopic neovascularization leads to fibrovascular proliferation, resulting in vision-threatening complications, such as vitreous hemorrhage and traction retinal detachment. Coincident with the pathological neovascularization, the once-vascularized and newly ischemic central areas of the retina are reinvested with normal-appearing blood vessels, a process termed “revascularization.” When reagents that block all VEGF isoforms are used, this compensatory intraretinal revascularization is suppressed together with the pathological neovascularization (3). Thus, ophthalmologists await the establishment of a new therapy that selectively targets pathological neovascularization, while sparing compensatory revascularization.
The current paper demonstrates that the differential expression of VEGF164 results in leukocyte adhesion at the leading edge of pathological, but not physiological, neovascularization. Furthermore, differential functions for the various leukocyte subpopulations recruited to the sites of neovascularization are demonstrated here. As a result, new mechanistic differences are uncovered at the molecular and cellular levels between the physiological and pathological neovascularization.
Materials and Methods
Rat Model of Proliferative Retinopathy.
All animal experiments followed the Association for Research in Vision and Ophthalmology guidelines and were approved by the Animal Care Committee of Massachusetts Eye and Ear Infirmary. Long-Evans rats (Charles River Laboratories) were used. Postnatal day zero (P0) rats with their nursing mothers were maintained for 10 full days in 80% oxygen, interrupted daily by 30 min in room air followed by a progressive return to 80% oxygen, to induce an avascular retina. On P10 (D0), they were placed in room air for an additional 7 d (D7) to induce retinal neovascularization.
ELISA for VEGF.
After sacrifice with an overdose of anesthesia, the eyes were immediately enucleated. The retina was carefully isolated, placed into 150 μl of lysis buffer, and sonicated. The lysate was centrifuged at 14,000 rpm for 15 min at 4°C, and the VEGF levels in the supernatant were determined with the Quantikine mouse VEGF ELISA kit (R&D Systems) according to manufacturer's protocol. The assay recognizes all VEGF isoforms.
Reverse Transcription (RT)–PCR for VEGF.
Total RNA was isolated from the retina and peripheral monocytes with TRIzol reagent (Life Technologies), and cDNA was produced using reverse transcriptase (SuperScript II; Invitrogen). The primer sequences for GAPDH and rat VEGF were 5′-CCATGGAGAAGGCTGGGG-3′ (sense) and 5′-CAAAGTTGTCATGGATGACC-3′ (antisense) for GAPDH and 5′-ACCTCCACCATGCCAAGT-3′ (sense) and 5′-TAGTTCCCGAA-ACCCTGA-3′ (antisense) for VEGF. Analysis of RT-PCR data was performed using the intensity ratios of VEGF164/VEGF120 in each lane.
Lectin Labeling of Retinal Vasculature and Adherent Leukocytes.
The retinal vasculature and adherent leukocytes were imaged by perfusion-labeling with FITC-coupled Concanavalin A lectin (Con A; Vector Laboratories) as described previously (4). The flat mounts were imaged using two epifluorescence microscopes (models DM RXA and MZ FLIII; Leica), each possessing a different range of magnification.
Intravitreous Injection of Anti-VEGF165 Aptamer (EYE001).
After deep anesthesia, an eyelid fissure was created with a fine blade to expose the eyeball. Intravitreous injections were performed by inserting a 33-gauge double caliber needle (Ito Corporation) under an operating microscope. Animals received intravitreous injections of 1 μl of sterile PBS containing 0.5 nmol of a pegylated anti-VEGF165 aptamer (EYE001; Eyetech Pharmaceuticals) or polyethylene glycol (PEG) control. Retinal neovascularization and leukocyte adhesion were evaluated 7 d after administration in D0 (P10) retinopathy rats and P3 normal neonates.
The anti-VEGF165 aptamer is an oligonucleotide that binds to the exon 7–encoded domain of human VEGF165 protein with high specificity and affinity. The oligonucleotide is conjugated to a 40-kD PEG moiety to increase its half-life. The aptamer does not bind to VEGF120(121) and efficiently neutralizes VEGF164 in rats.
Intravitreous Injection of a VEGFR-1/Fc Fusion Protein.
Animals received 1-μl intravitreous injections of sterile PBS containing 1 μg of a mouse VEGFR-1/Fc chimera (R&D Systems) or an isotype Fc control (R&D Systems). The fusion protein blocks all VEGF isoforms. A maximal effect dosage (1 μg per eye) was determined and used for comparison with the maximal effect dosage of the anti-VEGF165 aptamer.
Generation of VEGF164-deficient (VEGF120/188) Mice.
VEGF+/120 male and VEGF+/188 female mice, generated previously (5) via targeted mutagenesis with Cre/loxP–mediated site-specific recombination in embryonic stem cells, were mated to each other. Sired neonates were genotyped as described previously (5) to select VEGF164-deficient (VEGF120/188) mice and age-matched wild-type control (VEGF+/+) mice for analysis at P10.
CD13, CD8, and CD25 Immunofluorescence.
Adherent leukocytes were labeled with rhodamine-coupled Con A as described above in the fourth paragraph of Materials and Methods. Retinal flat mounts were permeabilized with 0.5% Triton X (Sigma-Aldrich) in PBS for 24 h and nonspecific binding was blocked with 1% BSA. The retinas were incubated with a FITC-conjugated mouse monoclonal antibody against CD13 (clone WM15, 1:100; Research Diagnostics), CD8 (clone OX-8, 1:100; Research Diagnostics), or CD25 (clone OX-39, 1:100; Research Diagnostics) overnight at 4°C.
Isolation and Hypoxic Stimulation of Monocytes.
Blood was drawn from D7 rats with retinopathy before sacrifice. The monocytes were purified by positive selection via magnetic cell sorting (MACS) using MicroBeads conjugated with a monoclonal anti–rat MHC-II antibody (clone OX-6; Miltenyi Biotec) according to the manufacturer's instructions. The isolated monocytes were seeded at a concentration of 2.5 × 106 cells per dish and stimulated for 8 h at 37°C with 1 or 21% oxygen in a humidified air-tight chamber (Modular Incubator Chamber; Billups-Rothenberg). The incubated cells were collected and processed for RT-PCR for VEGF as described above in the third paragraph of Materials and Methods.
Intravitreous Administration of Clodronate-liposomes.
Clodronate (dichloromethylene diphosphonate) was a gift from Roche Diagnostics. Animals with retinopathy received 1-μl intravitreous injections of clodronate- or PBS-liposomes on D0 (P10). The degree of retinal neovascularization was evaluated 7 d later.
Systemic CD2 Blockade.
Rats with retinopathy received intraperitoneal injections of 5 mg/kg/day of a mouse anti-rat CD2 neutralizing antibody (clone OX-34; Research Diagnostics) or a mouse isotype nonimmune antibody (R&D Systems). The reagents were injected for seven consecutive days before evaluation on D7 (P17).
Endothelial and Lymphocyte Cocultures.
Blood was drawn from D7 rats with retinopathy and age-matched (P17) controls before sacrifice. Cytotoxic T lymphocytes (CTLs) were purified by positive selection via MACS using MicroBeads conjugated with a monoclonal anti–rat CD8a antibody (clone G-28; Miltenyi Biotec) according to the manufacturer's instructions. Human microvascular endothelial cells (Cascade Biologics, Inc.) at passage 6–8 were seeded at a concentration of 2 × 105 cells per well and stimulated with 30 ng/ml recombinant human tissue necrosis factor-α (TNF-α; R&D Systems). Isolated CTLs were incubated for 15 min at 37°C with 50 μM carboxyfluorescein diacetate succinimidyl ester (CFDASE; Molecular Probes). The fluorescent cells were washed and incubated (8 × 105 cells/ml, 100 μl per well) with endothelial monolayers for 4 h, after which nonadherent lymphocytes were removed and the endothelial monolayer was washed. To examine whether the apoptosis is FasL-mediated, an anti–mouse FasL antibody (clone MFL4; BD Biosciences) or an isotype control antibody (BD Biosciences) was applied at 10 μg/ml for 10 min at 37°C to the T cell suspension before the coculture. Cell death was assayed using the TUNEL procedure according to the manufacturer's instructions (Intergen). Apoptotic cells were detected using a CD-330 charge-coupled device camera (Dage-MIT) attached to an epifluorescence microscope (model MZ FLIII; Leica). A minimum of eight fields each in three separate experiments was analyzed per condition.
Morphometric and Statistical Analysis.
All results were expressed as mean ± SD. The number of leukocytes in each flat mount was counted independently by two investigators under an epifluorescence microscope (DM RXA; Leica). The morphology of the pathological neovascularization was readily discerned from the intraretinal extension of physiological vessels. The neovascular retinal areas were photographed with an epifluorescence microscope (MZ FLIII; Leica) and measured using NIH Image. The values were processed for statistical analyses (Mann-Whitney U test). Differences were considered statistically significant when the P values were <0.05.
Results
VEGF Expression in Physiological and Pathological Retinal Neovascularization.
Absolute and relative VEGF isoform expression levels were studied during both physiological vascular development (Fig. 1, A and C) and pathological neovascularization (Fig. 1, B and D). ELISA results showed that retinal VEGF protein levels were approximately twofold higher in the pathologically neovascularizing retina (Fig. 1 B) than in the developing retina undergoing physiological neovascularization (Fig. 1 A). RT-PCR showed a substantial relative difference in VEGF isoform expression pattern between pathological (Fig. 1 D) and physiological (Fig. 1 C) neovascularization. The VEGF164/VEGF120 expression ratio was calculated to be 2.2 ± 1.1 in the physiologically developing retinas, and 25.3 ± 8.7 in the pathologically neovascularized retinas.
Figure 1.

Differential expression of VEGF isoforms during physiological and pathological retinal neovascularization. (A) Retinal VEGF protein levels (all isoforms) during postnatal development. (B) Retinal VEGF protein levels after the induction (D0) of pathological neovascularization. Dotted lines in A and B indicate the constitutive VEGF levels in normal adult rats. (C) Retinal VEGF mRNA expression during postnatal development. (D) Retinal VEGF mRNA expression after the induction of pathological neovascularization. The relative expression levels of VEGF164 increased dramatically during pathological neovascularization.
Existence of Adherent Leukocytes at the Leading Edge of Pathological, but Not Physiological, Retinal Neovascularization.
Leukocyte adhesion was examined at the leading edge of both physiological and pathological neovascularization. Adherent leukocytes were largely absent at the leading edge of physiological postnatal retinal vascular development (Fig. 2 A). In the retinopathy model of pathological neovascularization (Fig. 2, B–F), a vast area of avascular retina was formed (Fig. 2 B) during exposure to the hyperoxic environment (D0). Adherent leukocytes were found in the revascularizing retina (Fig. 2 C) on D2. These leukocytes were associated with the incipient pathological neovascular fronds (Fig. 2 D) on D3. By D7, leukocytes were observed in the areas of revascularization (Fig. 2 E), but were most prominent in the pathological neovascular fronds extending into the vitreous (Fig. 2 F).
Figure 2.

Leukocytes are present at the leading edge of pathological, but not physiological, retinal neovascularization. (A) No leukocyte adhesion at the leading edge of physiological retinal development on P6. (B–F) Leukocyte adhesion during the onset and progression of pathological neovascularization. (B) The formation of extensive retinal ischemia on D0, immediately after exposure to relative hypoxia (room air). (C) Abundant inflammatory cells (arrows) randomly adhered to the intraretinal vessels just before the onset of pathological neovascularization. (D) The leukocyte adhesion (arrows) associated with the incipient vascular budding on D3. (E) Compared with D0 (B), revascularization proceeded in concert with pathological neovascularization (arrows) on D7. (F) Leukocyte adhesion (arrows) spatially and temporally associated with vascular budding. Bars: (A, C, D, and F) 50 μm; (B and E) 0.5 mm.
Effect of VEGF164-specific Blockade on Physiological and Pathological Retinal Neovascularization.
A VEGF164(165) isoform-specific neutralizing aptamer (EYE001) was used to determine the role of VEGF164(165) in pathological neovascularization. During pathological neovascularization (Fig. 3, A–F) , VEGF164 blockade (Fig. 3 B) led to a significant inhibition of leukocyte adhesion (Fig. 3 D) and pathological neovascularization (Fig. 3 E). In contrast, little or no suppression of revascularization (Fig. 3 F) and physiological neovascularization (Fig. 3, G, H, and J) was observed. When a VEGFR-1/Fc chimeric protein was used to compare the neutralizing effects of the single VEGF164 isoform versus all VEGF isoforms, VEGF pan-isoform blockade (Fig. 3 C) resulted in a significant suppression of leukocyte adhesion (Fig. 3 D) and pathological neovascularization (Fig. 3 E). The result was similar to that seen with VEGF164-specific blockade. However, in contrast to the VEGF164 inhibition result, a significant suppression of revascularization (Fig. 3 F) and physiological neovascularization (Fig. 3, I and J) was observed with pan-isoform blockade. No difference was detected in physiological and pathological neovascularization among the following control groups: nontreated, PEG-treated, and Fc-treated rats (unpublished data). To further confirm the VEGF164 neutralizing effect on retinal vascular development, VEGF164-deficient mice were generated and evaluated. No difference in physiological neovascularization was detected between wild-type (VEGF+/+) mice and VEGF164-deficient (VEGF120/188) mice (Fig. 3, K–M).
Figure 3.
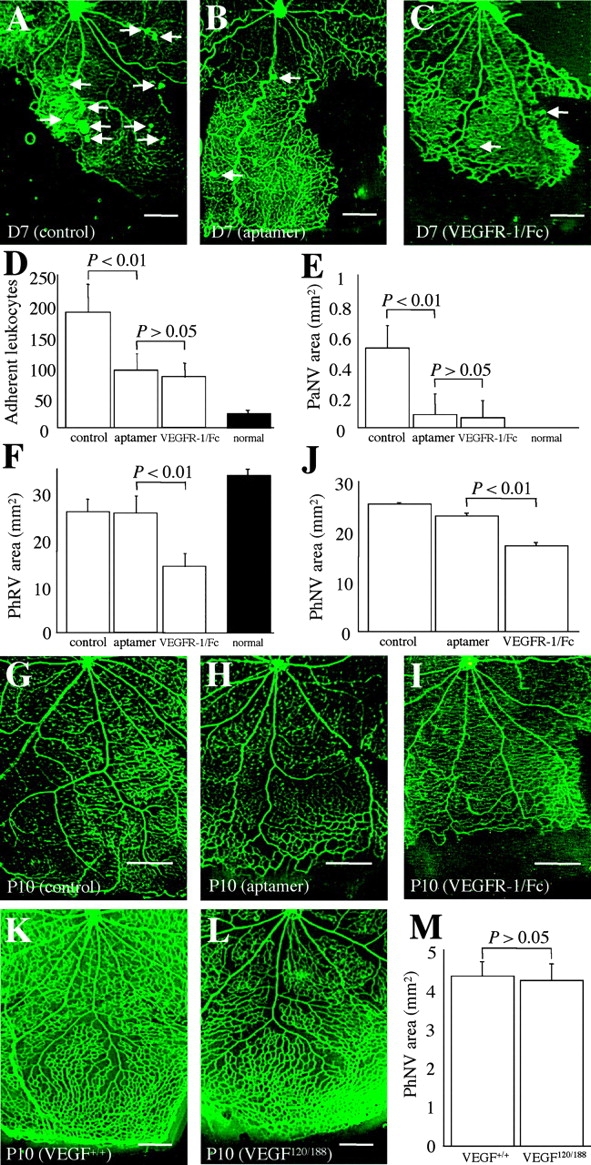
Effect of VEGF164-specific blockade on physiological and pathological retinal neovascularization. (A–F) VEGF164-specific blockade versus VEGF pan-isoform blockade in pathological neovascularization. (A) Pathological neovascularization (arrows, D7) treated with PEG control (n = 12) was not inhibited. (B) Pathological neovascularization (arrows, D7) treated with the anti-VEGF165 aptamer (n = 13). (C) Pathological neovascularization (arrows, D7) treated with the VEGFR-1/Fc chimera (n = 10). In addition to the inhibition of leukocyte adhesion to the retinal vasculature (D, P < 0.01), pathological neovascular budding into the vitreous (E, PaNV) was significantly suppressed (P < 0.01) via the anti-VEGF164 aptamer or the VEGFR-1/Fc. In contrast, the effect of VEGF164 inhibition on physiological revascularization (F, PhRV) was negligible (P > 0.05), but pan-isoform inhibition led to significant suppression of revascularization (F, P < 0.01). Shaded bars indicate comparable values of age-matched (P17) normal rat neonates (n = 8). (G–J) VEGF164-specific blockade versus VEGF pan-isoform blockade in retinal vascular development. (G) Developing retinal vasculature (P10) treated with PEG control (n = 7). (H) Developing retinal vasculature (P10) treated with anti-VEGF165 aptamer (n = 8). (I) Developing retinal vasculature (P10) treated with the VEGFR-1/Fc chimera (n = 10). (J) Note the mild suppression of physiological neovascularization during retinal development via VEGF164 inhibition, but the substantial suppression via pan-isoform inhibition (P < 0.01). (K–M) VEGF164-specific deficiency versus wild type in retinal vascular development. (K) Developing retinal vasculature (P10) in wild-type control (VEGF+/+) mice (n = 13). (L) Developing retinal vasculature (P10) in VEGF164-deficient (VEGF120/188) mice (n = 13). (M) VEGF164 deficiency had no significant effect on physiological neovascularization (P > 0.05). Bars: (K and L) 0.2 mm; (A–C and G–I) 0.5 mm.
Monocytes Are Positive Regulators of Pathological Retinal Neovascularization.
To investigate the role of monocytes in pathological neovascularization, clodronate-liposomes were used to inactivate monocyte lineage cells. Compared with PBS-liposomes (Fig. 4 A), monocyte-selective depletion (Fig. 4 B) led to a significant suppression of pathological neovascularization (Fig. 4 C), but had negligible effect on revascularization (Fig. 4 D). Immunohistochemistry confirmed that a subset of the adherent leukocytes in the vasculature (Fig. 4, E–J) were positive for CD13, aminopeptidase N coexpressed with MHC-II for monocyte-surface antigen processing. RT-PCR showed that circulating monocytes isolated from rats with proliferative retinopathy expressed VEGF120 and VEGF164, which was markedly induced after hypoxic stimulation (Fig. 4 K).
Figure 4.

Role of monocytes in pathological retinal neovascularization. (A) Pathological neovascularization (arrows, D7) treated with PBS control-liposomes (n = 8) was not inhibited. (B) Pathological neovascularization (arrows, D7) treated with clodronate-liposomes (n = 8). Notably, the pathological neovascular budding (C, PaNV) was suppressed (P < 0.01), whereas revascularization (D, phRV) was not (P > 0.05). (E–J) Monocyte adhesion was observed just before and during pathological neovascularization (H–J). Green fluorescence from the anti-CD13 antibody (E and H) and red fluorescence from the rhodamine-coupled Con A (F and I) identifies the Con A–stained cells as being CD13-positive leukocytes (arrows) when the images were superimposed (G and J). (K) Monocyte VEGF mRNA expression in normoxia (21% oxygen) and hypoxia (1% oxygen). VEGF levels were markedly increased in response to hypoxic stimulation. Bars: (E–J) 50 μm; (A and B) 0.5 mm.
T Lymphocytes Are Negative Regulators of Pathological Retinal Neovascularization.
To investigate the role of T lymphocytes in pathological neovascularization, a neutralizing antibody against CD2 was used. CD2 is an important adhesion molecule for T lymphocyte-mediated immune responses, and is also called lymphocyte function-associated antigen-2 (LFA-2). Notably, compared with nonimmune control IgG (Fig. 5 A), CD2 blockade (Fig. 5 B) led to significant increase in the pathological neovascularization (Fig. 5 C). Immunohistochemistry confirmed that adherent leukocytes at the vascular fronds were positive for CD8 (Fig. 5, D–F) and CD25 (Fig. 5, G–I; IL-2 receptor), indicative of cytotoxic and activated T lymphocytes, respectively. To determine whether CD8-positive cytotoxic T lymphocytes (CTLs) isolated from rats with retinopathy cause a FasL-mediated endothelial cell apoptosis, cell death was evaluated using a leukocyte–endothelial coculture system. Compared with CTLs isolated from age-matched normal control rats (Fig. 5 J), CTLs from rats with retinopathy (Fig. 5 K) increased endothelial cell apoptosis (Fig. 5 N). Administration of a FasL neutralizing antibody (Fig. 5 L), but not a control nonimmune antibody (Fig. 5 M), led to significant suppression of the apoptosis caused by the CTLs isolated from the rats with retinopathy (Fig. 5 N).
Figure 5.
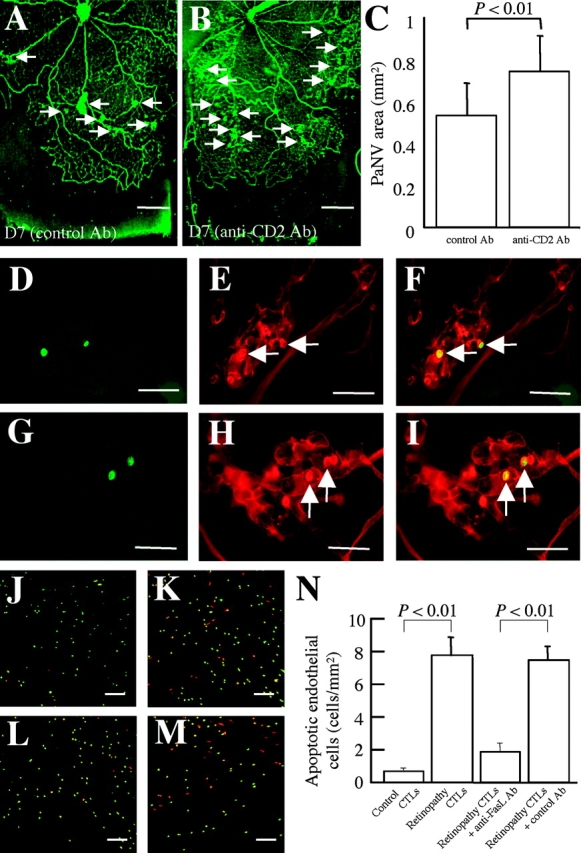
Role of T lymphocytes in pathological retinal neovascularization. (A) Pathological neovascularization (arrows, D7) treated with nonimmune isotype control (n = 9) showing a similar degree of pathological vascular budding (arrows) compared with Fig. 2 E. (B) Pathological neovascularization (arrows, D7) treated with anti-CD2 antibody (n = 11). Notably, the pathological neovascular budding (C, PaNV) was worsened (P < 0.01). (D–I) T cell subtypes in pathological neovascular buds. Green fluorescence from the antibody against CD8 or CD25 (D and G) and red fluorescence from the rhodamine-coupled Con A (E and H) identifies the Con A–stained cells as being CD8- and CD25-positive leukocytes (arrows) when the images were superimposed (F and I). (J–N) Rhodamine-labeled apoptotic cells in the leukocyte–endothelial cocultures were detected via TUNEL staining (red). CTLs were labeled with CFDASE (green). Compared with control CTLs (J), retinopathy CTLs (K) significantly increased the number of TUNEL-positive endothelial cells (N, P < 0.01). The process was significantly inhibited via FasL blockade (L–N, n = 18–24 in each condition, P < 0.01). Bars: (D–I) 50 μm; (J–M) 200 μm; and (A and B) 0.5 mm.
Discussion
The current data demonstrate that pathological, but not physiological, retinal neovascularization is characterized by the overexpression, both in relative and absolute terms, of the VEGF164 isoform. The hypoxia-induced VEGF164 isoform expression leads to the recruitment of leukocytes to sites of pathological neovascularization. These data demonstrate that ischemia-induced neovascularization, classically thought to be noninflammatory in nature, is characterized by the influx of inflammatory cells.
Monocyte/macrophage lineage cells express VEGF receptor (R)-1 through which VEGF exerts its chemotactic actions (6). VEGF also up-regulates the expression of ICAM-1 on vascular endothelial cells in vitro (7). VEGF-induced blood–retinal barrier breakdown was demonstrated to be leukocyte-dependent, in part, when the inhibition of ICAM-1 prevented blood–retinal barrier breakdown in VEGF165-injected rat eyes (8). In separate studies, VEGF-induced endothelial ICAM-1 expression (9) and vascular leakage (10) were shown to be mediated by VEGFR-2. Thus, VEGF functions as a proinflammatory cytokine targeting both leukocytes and endothelial cells. Taking into account the differential affinity of the VEGF isoforms for VEGF receptor binding (11, 12), VEGF165 seemed likely to be more responsible for the induction of inflammation than VEGF121.
The present work is the first to show that adherent leukocytes are associated with the leading edge of pathological, but not physiological, neovascularization (Fig. 2). The recruitment of leukocytes was largely suppressed via VEGF164 isoform-specific blockade (Fig. 3), directly implicating VEGF164 in the process. Additionally, VEGF164, a more proinflammatory isoform than VEGF120, was preferentially induced in pathological neovascularization (Fig. 1). Together, these observations indicate that leukocyte adhesion in pathological, but not the physiological, neovascularization is properly regarded as a VEGF164-induced pathology.
The current data also show that the selective depletion of monocyte lineage cells in proliferative retinopathy suppresses pathological neovascularization, but not revascularization (Fig. 4). The results mirror those seen after the VEGF164-specific blockade (Fig. 3). Thus, inflammatory monocytes appear to facilitate vitreous invasion by the aberrant vasculature. The VEGF164-mediated recruitment of monocytes just before the appearance of pathological neovascularization suggests that the monocytes may serve to mark the sites of future neovascularization (Fig. 2). Indeed, circulating monocytes have a pro-angiogenic potential expressing VEGF164, the production of which is markedly enhanced by hypoxia (Fig. 4). The positive feedback loop of VEGF164 and monocytes is a potential mechanism leading to the amplification of pathological neovascularization. Recent in situ hybridization data showing increased VEGF expression in the ischemic retinal cells (3) and macrophages (13) in the proliferative retinopathy model further support our conclusions.
VEGF164(165), an isoform more potently mitogenic (11) as well as more proinflammatory than VEGF120(121), appears to be sufficient for the physiological neovascularization of retinal development (5). However, VEGF isoforms other than VEGF164, in combination, may be sufficient to promote normal physiological neovascularization. Indeed, the complete inhibition of intracellular VEGF signaling was shown previously to result in substantial suppression of normal vascular development (3) and was reproduced here (Fig. 3), whereas VEGF164-selective blockade did not affect normal vascular development (Fig. 3). Consistent with these results, retinal vascular development was normal in genetically VEGF164-deficient mice expressing VEGF120 and VEGF188 (Fig. 3; VEGF120/188 mice) in agreement with the aptamer-based VEGF164 neutralization data (Fig. 3). Consequently, the expression pattern of the various VEGF isoforms, strictly regulated during normal development but disrupted in disease, is likely to contribute to an angiogenic switch, converting the orderly intraretinal extension of vessels into an inflammation-associated invasion of the vitreous.
The present paper shows that IL-2 receptor (CD25)-positive cells and CD8-positive cells adhere to the pathological neovascular fronds (Fig. 5), indicative of the presence of activated CTLs. In a murine tumor model, the systemic administration of IL-12 and IL-2, both known to activate cellular immunity, led to the recruitment of FasL-positive CTLs and the inhibition of tumor neovascularization and growth (14). Conversely, an angiogenic role of T lymphocytes, a source of VEGF, has been suggested in human prostate cancer (15). The present paper demonstrates that the inhibition of lymphocyte function results in the aggravation of pathological neovascularization (Fig. 5). In our model of pathological retinal neovascularization, CTL-mediated cellular immunity is thought to play a critical role in the defense against the ectopic proliferation of endothelial cells. This is also supported by the findings that CD8-positive T lymphocytes from rats undergoing pathological neovascularization resulted in the FasL-mediated apoptosis of endothelial cells in vitro (Fig. 5). The role of immunotherapy for retinal neovascular diseases is currently under investigation.
Acknowledgments
This work was funded by Eyetech Pharmaceuticals. Drs. Adamis and Shima are employees and shareholders of Eyetech Pharmaceuticals. The anti-VEGF165 aptamer used in these experiments is a product of Eyetech Pharmaceuticals and is currently in clinical trial.
S. Ishida, T. Usui, and K. Yamashiro contributed equally to this work.
References
- 1.Stone, J., A. Itin, T. Alon, J. Pe'er, H. Gnessin, T. Chan-Ling, and E. Keshet. 1995. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 15:4738–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stone, J., T. Chan-Ling, J. Pe'er, A. Itin, H. Gnessin, and E. Keshet. 1996. Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 37:290–299. [PubMed] [Google Scholar]
- 3.Ozaki, H., M.S. Seo, K. Ozaki, H. Yamada, E. Yamada, N. Okamoto, F. Hofmann, J.M. Wood, and P.A. Campochiaro. 2000. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am. J. Pathol. 156:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joussen, A.M., T. Murata, A. Tsujikawa, B. Kirchhof, S.E. Bursell, and A.P. Adamis. 2001. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am. J. Pathol. 158:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stalmans, I., Y.S. Ng, R. Rohan, M. Fruttiger, A. Bouche, A. Yuce, H. Fujisawa, B. Hermans, M. Shani, S. Jansen, et al. 2002. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J. Clin. Invest. 109:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barleon, B., S. Sozzani, D. Zhou, H.A. Weich, A. Mantovani, and D. Marme. 1996. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 87:3336–3343. [PubMed] [Google Scholar]
- 7.Melder, R.J., G.C. Koenig, B.P. Witwer, N. Safabakhsh, L.L. Munn, and R.K. Jain. 1996. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat. Med. 2:992–997. [DOI] [PubMed] [Google Scholar]
- 8.Miyamoto, K., S. Khosrof, S.E. Bursell, Y. Moromizato, L.P. Aiello, Y. Ogura, and A.P. Adamis. 2000. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am. J. Pathol. 156:1733–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim, I., S.O. Moon, S.H. Kim, H.J. Kim, Y.S. Koh, and G.Y. Koh. 2001. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J. Biol. Chem. 276:7614–7620. [DOI] [PubMed] [Google Scholar]
- 10.Gille, H., J. Kowalski, B. Li, J. LeCouter, B. Moffat, T.F. Zioncheck, N. Pelletier, and N. Ferrara. 2001. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J. Biol. Chem. 276:3222–3230. [DOI] [PubMed] [Google Scholar]
- 11.Keyt, B.A., L.T. Berleau, H.V. Nguyen, H. Chen, H. Heinsohn, R. Vandlen, and N. Ferrara. 1996. The carboxyl-terminal domain (111-165) of vascular endothelial growth factor is critical for its mitogenic potency. J. Biol. Chem. 271:7788–7795. [DOI] [PubMed] [Google Scholar]
- 12.Whitaker, G.B., B.J. Limberg, and J.S. Rosenbaum. 2001. Vascular endothelial growth factor receptor-2 and neuropilin-1 form a receptor complex that is responsible for the differential signaling potency of VEGF(165) and VEGF(121). J. Biol. Chem. 276:25520–25531. [DOI] [PubMed] [Google Scholar]
- 13.Naug, H.L., J. Browning, G.A. Gole, and G. Gobe. 2000. Vitreal macrophages express vascular endothelial growth factor in oxygen-induced retinopathy. Clin. Exp. Ophthalmol. 28:48–52. [DOI] [PubMed] [Google Scholar]
- 14.Wigginton, J.M., E. Gruys, L. Geiselhart, J. Subleski, K.L. Komschlies, J.W. Park, T.A. Wiltrout, K. Nagashima, T.C. Back, and R.H. Wiltrout. 2001. IFN-gamma and Fas/FasL are required for the antitumor and antiangiogenic effects of IL-12/pulse IL-2 therapy. J. Clin. Invest. 108:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freeman, M.R., F.X. Schneck, M.L. Gagnon, C. Corless, S. Soker, K. Niknejad, G.E. Peoples, and M. Klagsbrun. 1995. Peripheral blood T lymphocytes and lymphocytes infiltrating human cancers express vascular endothelial growth factor: a potential role for T cells in angiogenesis. Cancer Res. 55:4140–4145. [PubMed] [Google Scholar]