

Abstract
Autoreactive CD4+ T lymphocytes are critical to the induction of autoimmune disease, but because of the degenerate nature of T cell receptor (TCR) activation such receptors also respond to other ligands. Interaction of autoreactive T cells with other non–self-ligands has been shown to activate and expand self-reactive cells and induce autoimmunity. To understand the effect on the autoreactivity of naive cross-reactive T cells of activation with a potent nonself ligand, we have generated a TCR transgenic mouse which expresses a TCR with a broad cross-reactivity to a number of ligands including self-antigen. The activation of naive transgenic recombination activating gene (Rag)2 − / − T cells with a potent non–self-ligand did not result in a enhancement of reactivity to self, but made these T cells nonresponsive to the self-ligand and anti-CD3, although they retained a degree of responsiveness to the non–self-ligand. These desensitized cells had many characteristics of anergic T cells. Interleukin (IL)-2 production was selectively reduced compared with interferon (IFN)-γ. p21ras activity was reduced and p38 mitogen-activated protein kinase (MAPK) was relatively spared, consistent with known biochemical characteristics of anergy. Surprisingly, calcium fluxes were also affected and the anergic phenotype could not be reversed by exogenous IL-2. Therefore, activation with a hyperstimulating non–self-ligand changes functional specificity of an autoreactive T cell without altering the TCR. This mechanism may preserve the useful reactivity of peripheral T cells to foreign antigen while eliminating responses to self.
Keywords: T lymphocyte anergy, autoimmunity, T cell cross-reactivity, superagonist, selective peripheral tolerance
Introduction
It is increasingly appreciated that the peripheral T cell repertoire of normal healthy humans and rodents is highly cross-reactive and contains cells with the potential to respond to a wide range of self-peptides presented by self-MHC molecules (1, 2). This arises both because T lymphocyte survival in the thymus relies on activation by self-peptides for positive selection (3), and because TCRs are inherently cross-reactive, so the complete elimination of all TCRs with the potential to respond to self would also significantly compromise responses to pathogens (4). In many models of organ-specific autoimmune diseases, the activation of these self-reactive T cells is sufficient to initiate a delayed-type hypersensitivity (DTH)-like reaction directed against the self-antigen in the target organ and resulting in an autoimmune disease (5, 6). Understanding how cells which have the potential to cross react with autoantigens are regulated in the periphery, and how they become activated, is thus crucial to understanding the pathology of these conditions.
Two mechanisms have been proposed to explain autoreactive T cell activation, bystander activation, and molecular mimicry (7, 8). The first proposes the activation of autoreactive cells in the proinflammatory milieu engendered by infection of the target tissue, and the second the cross-reactive activation of autoreactive T cells by foreign peptide mimics. In both these cases, it is held that because memory cells have a lower activation threshold than naive T cells (9), they may be more liable to cause disease upon reactivation with a foreign antigen. To investigate the question of the potential of cross-reactive peptides of different potencies (hierarchies) to influence the activation of an autoreactive TCR, we have developed a TCR transgenic system using a receptor (the 1B6 TCR) with a hierarchy of responses to different peptide ligands, including the autoantigenic myelin proteolipid (PLP) peptide 139–151 (HSLGKWLGHPDKF; W144).
The 1B6 TCR was derived from a T cell clone which was generated by immunization of a female SJL mouse with the altered peptide Q144 (HSLGKQLGHPDKF) in which glutamine (Q) has replaced the primary TCR contact tryptophan (W) in the autoantigenic peptide (10). The 1B6 TCR is highly cross-reactive and is able to recognize a number of other ligands with substitutions at position 144, including the self-ligand W144, and the altered self-ligands Y144 (HSLGKYLGHPDKF) and L144 (HSLGKLLGHPDKF) which stimulate cells much more strongly than other ligands (see Table I). This finding raised the possibility that activation with L144 would change the response of naive 1B6 cells to W144, making them more responsive to the autoantigen. In contrast, we show in this paper that the activation of naive 1B6 cells with the hyperstimulatory ligand L144, compared with activation with Q144, leads to a reduction in all responses and particularly a loss of response to activation with anti-CD3 and the autoantigen W144. We found significant differences in the biochemical profiles of the 1B6 T cells activated with the hyperstimulatory ligand L144, in that these T cells showed many of the biochemical hallmarks of anergic T cells.
Table I.
Hierarchy of Response of the Q1.1B6 T Cell Clone to Various Ligands
| Cytokine (pg/ml)
|
|||||||
|---|---|---|---|---|---|---|---|
| Ligand | Sequence | Relative MHC binding |
Proliferative response |
IFN-γ | TNF-α | IL-2 | IL-4 |
| L144 | HSLGKLLGHPDKF | 0.8 | ++++ | 9,040 | 420 | 1,350 | 390 |
| *Y144 | HSLGKYLGHPDKF | 0.6 | +++ | N.D. | N.D. | N.D. | N.D. |
| Q144 | HSLGKQLGHPDKF | 0.6 | ++ | 9,052 | <50 | <50 | 200 |
| W144 | HSLGKWLGHPDKF | 1.0 | + | 420 | <50 | <50 | <50 |
| Anti-TCR antibody | – | – | ++ | 8,350 | <50 | <50 | 60 |
Data summarized from Nicholson et al. 1998 (reference 10) from a representative experiment, except *Y144 compiled from data obtained from 1B6 T cell hybridoma and 1B6 transgenic T cells. The Q1.1B6 T cell clone was derived from an SJL mouse immunized with Q144. L144 is a superagonist ligand. Anti-TCR antibody activation is similar to activation with the cognate ligand Q144. N.D., not determined.
Materials and Methods
Animals.
4–6-wk-old female SJL mice were purchased from the The Jackson Laboratory and housed under specific pathogen-free conditions. To generate the 1B6 TCR transgenic (Tg) mouse, the TCRs AV1S, Ja10 and BV10S1A2, Db1, and Jb2.1 were cloned into shuttle vectors provided by Dr. M. Davis (Stanford University, Palo Alto, CA) and the constructs coinjected into the pronuclei of fertilized FVB oocytes. Seven founder lines were generated, of which two have been maintained and crossed onto the SJL genetic background. In these SJL transgenic mice, T cell thymic selection was skewed into the CD4 single-positive compartment (57 vs. 32% in littermate controls) with a reduced selection into the CD8 single-positive compartment (3 vs. 11%). This skewing is also seen in peripheral lymph nodes (CD4+: 74 vs. 68%; CD8+: 10 vs. 26%). The hierarchy of response to L144, Y144, Q144, and W144 (see Fig. 1) was comparable to the pattern seen with the 1B6 T cell clone (see Table I; reference 10). All mice were maintained in accordance with the guidelines of the Committee on Animals of Harvard Medical School, Boston.
Figure 1.

Phenotype of naive ex-vivo transgenic 1B6 T cells. The 1B6 TCR transgenic T cells retain the same hierarchy of response to peptide ligands as the Q1.1B6 T cell clone. LNCs from a TCR transgenic mouse were stimulated with different peptides and proliferation was assessed by measuring the incorporation of [3H]thymidine added after 48 h of culture.
Generation of the 1.16 Anti-1B6 TCR Antibody.
Balb/c mice were immunized subcutaneously with a mixture of Rag2 − / − 1B6 transgenic lymph node and spleen cells emulsified in complete Freund's adjuvant. They were boosted intravenously with Rag2 − / − 1B6 transgenic lymphocytes five times over the course of one year and the spleen from the mouse with the highest anti-1B6 titre, as assessed by flow cytometry, was fused with myeloma cells (SP2.0 cells; American Type Culture Collection CRL8006) 4 d after the final boost, using polyethylene glycol 1450 and selection in HAT medium. Supernatants from all viable fusants were screened using a 1B6 T cell hybridoma and a control (9F8) T cell hybridoma. Antibodies that were positive by FACS® on 1B6 but not 9F8 cells were cloned at least twice by limiting dilution. Further screening was performed for specific growth inhibition of 1B6 T cell hybridoma and activation of 1B6 transgenic T cells. Anti-1B6 antibodies stained 1B6 T cells, but not T cell clones expressing Vβ10 or Vα1 paired with other TCR chains. The fusion generated three clonotypic antibodies and one anti-Vβ10 antibody. One clonotypic antibody (1.16; an IgG1,κ isotype) was selected for further expansion and used in analysis.
Antigens.
Peptide antigens with COOH-terminal amides were synthesized by Quality Controlled Biochemicals. Most peptides were >90% pure, as determined by HPLC, and were not purified further. The peptides used in these experiments were variants of the native PLP 139–151 peptide W144 (HSLGKWLGHPDKF), Q144 (HSLGKQLGHPDKF), Y144 (HSLGKYLGHPDKF) and L144 (HSLGKLLGHPDKF), and the control peptide Neuraminidase 101–120 (EALVRQGLAKVAYVYKPNNT). The relative binding of the PLP analogue peptides to I-As compared with W144 is 0.6 (Q144), 0.6 (Y144), and 0.8 (L144).
Antibodies.
FACS® staining for CD3, TCR, and CD4 was performed with fluorochrome conjugated antibodies (BD Biosciences). Analysis of cell death was performed using 7-AAD and annexin-PE according to the manufacturer's instructions (BD Biosciences). Immunoprecipitations and Western blotting were done with the following antibodies: biotinylated anti-CD3ɛ, anti–c-Jun NH2-terminal kinase (JNK; BD Biosciences), anti-phosphotyrosine (4G10; Upstate Biotechnology), anti-phospho p44,42 mitogen-activated protein kinase (MAPK),* anti-phospho p38 MAPK, anti-phospho JNK, anti-p44,42 MAPK, anti-p38 MAPK (Cell Signaling Technology), and anti-ZAP-70 (Santa Cruz Biotechnology, Inc.). All secondary reagents were from Jackson ImmunoResearch Laboratories.
Generation of T Cell Lines.
CD4+ lymph node cells (LNCs) of 1B6 TCR Tg Rag2 − / − mice were sorted on a FACS VantageSE™ (Becton Dickinson), yielding purities of >99%. Sorted CD4+ cells were cultured in 24-well plates (106 cells/well) with irradiated SJL splenocytes (107 cells/well) and the indicated concentrations of peptides in DMEM supplemented with 10% heat-inactivated fetal bovine serum (BioWhittaker, Inc.), 0.1 mM nonessential amino acids, sodium pyruvate (1 mM), l-glutamine (2 mM), MEM essential vitamin mixture (1×), penicillin (100 U/ml), streptomycin (100 U/ml), arginine (0.15 mM), asparagine (0.1 mM), folic acid (0.1 mg/ml), and 2-mercaptoethanol (5 × 10−5 M; Sigma-Aldrich). From day 2 onwards, T cells were expanded by feeding with culture medium supplemented with 0.6% T cell growth factor (T-Stim; Collaborative Biomedical Research) and 0.06% recombinant IL-2. Cells were fed every 2–3 d and restimulated every 14 d by the appropriate antigen and irradiated SJL spleen cells.
In Vitro Proliferation Assays and Cytokine Determinations.
T cells (5 × 104/well) were cultured in triplicate in 96-well round-bottomed plates (Falcon) together with (a) irradiated SJL spleen cells (5 × 105/well) and the indicated concentrations of peptide or anti-CD3 mAb, or (b) PMA plus ionomycin. After 48 h, [3H]thymidine (1 μCi/well) was added for the last 16 h before harvesting the cells. The incorporation of [3H]thymidine was determined in a Beckman scintillation counter (Model LS 5000; Beckman Coulter). Supernatants were collected after 40 h, diluted 1:2, and cytokine concentrations were measured by specific capture ELISA according to the manufacturer's instructions (BD Biosciences).
Cell Stimulation for Biochemistry, Immunoprecipitation, and Immunoblot Analysis.
DAS cells (fibroblasts transfected with I-As and expressing B7.1; a gift from Dr. S. Miller, Northwestern University, Evanston, IL) were taken as APCs for biochemical experiments. They were pulsed with 100 μg/ml L144 peptide for 2 h at 37°C. All cells were kept in DMEM without supplements on ice before activation. Two different modes of stimulation were chosen: (a) T cells (2 × 107/sample) were spun together for 30 s at 4,000 rpm with 107 peptide-pulsed DAS cells and transferred immediately to 37°C or (b) biotinylated anti-CD3ɛ (10 μg/ml) and avidin (25 μg/ml) were added to the T cell suspension (2 × 107/sample) and transferred immediately to 37°C. The cells were lysed at the indicated time points in lysis buffer (1% Brij96, 50 mM Tris, pH 7.6, 150 mM NaCl, 200 μM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 mM NaF, 1 mM Na3VO4, and 10 mM Na4P2O7). Immunoprecipitations of the lysates were done for 2 h at 4°C with the indicated mAbs and 50 μl of Streptavidin-Agarose (Upstate Biotechnology) for biotinylated anti-CD3ɛ, or Protein A-Sepharose (Amersham Biotech) for anti–ZAP-70. For the Western blots of kinases, aliquots of the cell lysates (corresponding to 2 × 106 T cells) were used. Proteins were electrophoresed in SDS-polyacrylamide gels, transferred to Immobilon-P membranes (Millipore), and visualized after Western blotting by the ECL detection system (Amersham Biosciences).
Measurement Of Calcium Flux.
T cells (5 × 106 cells/ml) were incubated for 1 h at 37°C with 4 μg/ml Indo-1-AM (Molecular Probes) in HBSS/5% FCS. Dye-loaded cells were washed twice and kept at RT for 30 min before use. For APC/peptide stimulation, 106 T cells were spun together for 30 s at 4,000 rpm with 106 peptide-pulsed I-As and B7 expressing fibroblast (DAS) cells, gently resuspended, and immediately analyzed. For mAb stimulation, biotinylated anti-CD3ɛ (10 μg/ml) and avidin (25 μg/ml) were added to the T cell suspension (1.5 × 106 cells/sample). Calcium fluxes were read by determining the ratio of 410 nm/485 nm over time on a FACS VantageSE™ (Becton Dickinson).
Determination of p21ras Activity.
The activity of p21ras was determined by immunoprecipitating the active, GTP-bound form of p21ras with Raf1-Ras binding domain agarose beads and subsequent Western blotting with anti-p21ras of the immunoprecipitated proteins according to the manufacturer's instructions (Upstate Biotechnology).
Results
Generation and Characterization of the 1B6 TCR Transgenic Mouse.
To investigate the effects of activating T cells with different cross-reactive ligands we developed a TCR transgenic system using a previously well characterized T cell clone (10; see Materials and Methods for details). The Q1.1B6 T cell clone was generated from a female SJL mouse (H-2s) immunized with the peptide Q144 (HSLGKQLGHPDKF), which is an altered peptide ligand of the native encephalitogenic autoantigenic peptide W144 (PLP 139–151; HSLGKWLGHPDKF). The TCR uses Vα1 and Vβ10. Detailed analysis of the clone, with peptides with single amino acid substitutions at position 144, allowed the characterization of a panel of APLs with equivalent binding affinity to MHC class II, but a broad hierarchy in stimulatory potential for the 1B6 TCR (10). The clone responded to both Q144, the self-antigen W144, and the altered peptide Y144. For this clone the peptide L144 was identified as a superagonist ligand (10). It achieved half-maximal proliferation at much lower antigen concentrations than Q144. At higher concentrations it induced the secretion of proinflammatory cytokines not induced by the agonist peptide Q144 (the phenotype of the Q1.1B6 clone is summarized in Table I). This wide dose response gives us the potential to investigate the phenotype of naive transgenic T cells exposed to varying strengths of stimulation with cross-reactive ligands and determine the effects of this cross-reactive activation and expansion on responses to an autoantigen at the level of a single clonotype.
Using the TCR from the Q1.1B6 T cell clone, we generated a TCR transgenic mouse (the 1B6 mouse) which was bred onto the SJL genetic background. The peripheral CD4+ T cells of the 1B6 transgenic mouse recapitulate the hierarchy of responses seen in the Q1.1B6 T cell clone, in that the self-peptide W144 is a weaker ligand and L144 is a superagonist ligand compared with the agonist Q144 peptide (Fig. 1). As there is no commercially available anti-TCR α or β chain monoclonal antibody for this receptor, we also developed a clonotypic monoclonal antibody for the 1B6 TCR. This allowed us to investigate the behavior of this clone in vivo. This antibody was specific for the 1B6 receptor by the criteria that it stained and activated 1B6 T cells, but did not stain T cell clones in which Vα1 or Vβ10 were paired with other TCR chains. With these tools we examined functional T cell responses of naive Rag2 −/− 1B6 T cells following activation with cross-reactive ligands.
Loss of Responsiveness to Self-Ligands After Activation with Nonself.
We found that 1B6 T cells could be successfully activated and maintained in culture with different ligands, including Q144 and L144 over a wide range of peptide concentrations. However, over time the phenotype of the cells changed, in a manner which suggested a change in the functional specificity of the T cell response. When the functional responses of these cells were tested after 2 rounds of activation in vitro, (∼30 d), we found that cells which had been restimulated with 50 μg/ml of Q144 or lower concentrations (0.005 μg/ml) of L144 responded briskly to activation with all the ligands L144, Q144, or W144 (Fig. 2 A). We describe these as responsive cells. In contrast cells which had been restimulated with 50 μg/ml L144 had a global impairment in their ability to proliferate. Their response to L144 was reduced, while their response to the autoantigen W144 and activation with anti-CD3 was completely abrogated. We describe these cells as desensitized cells (Fig. 2 A). We found that activation in the presence of IL-2 did not restore the responsiveness of the desensitized cells (unpublished data). We reasoned that if these cells represented a population of transgenic 1B6 cells with nonspecific defects in nuclear transcription, then they should also be unresponsive to activation with PMA/ionomycin. However, as shown in Fig. 2 B, desensitized cells retained responsiveness to activation with PMA/ionomycin which bypasses the proximal signaling machinery, arguing that rather than developing a generalized defect of activation, they had a proximal deficit in signaling.
Figure 2.

Culture with the superagonist peptide L144 affects the functional response of transgenic 1B6 T cells. Lymph node CD4+ T cells from 1B6 TCR transgenic Rag2 − / − mice were stimulated three times in vitro with 50 μg/ml Q144, 50 μg/ml L144, or 0.005 μg/ml L144. 2 wk after the third stimulation they were incubated with SJL/J splenocytes as APCs and the indicated concentrations of (A) L144, Q144, W144 peptides, or anti-CD3 and APCs, or (B) PMA and ionomycin (c1 = 100 ng/ml PMA, 2 μg/ml ionomycin; subsequent concentrations diluted 1:5) shown compared with activation by anti-CD3 (c1 = 10 μg/ml, subsequent concentrations diluted 1:5). Proliferation was assessed by pulsing the cultures with [3H]thymidine after 48 h.
When we compared the secretion of cytokines, the production of IFN-γ was similar between all the groups (Fig. 3). This suggests that the signaling pathways necessary for the induction of IFN-γ are equivalent between the different groups of cells. In contrast, the secretion of IL-2 is selectively blunted in the desensitized cells activated with L144 and APCs (Fig. 3), although the desensitized cells do retain the potential to secrete IL-2 when they are activated with PMA/ionomycin (Fig. 3). When we analyzed the secretion of the Th2 cytokines IL-4 and IL-10 in these experiments there were no clear differences in the patterns of secretion of these cytokines between groups (unpublished data). Because superagonist peptide treatment in other TCR transgenic systems has been associated with apoptosis (11) and with receptor and coreceptor down-regulation (12), we investigated if cell death and/or changes in the expression of the TCR was the mechanism responsible for the phenotype of the desensitized 1B6 cells.
Figure 3.
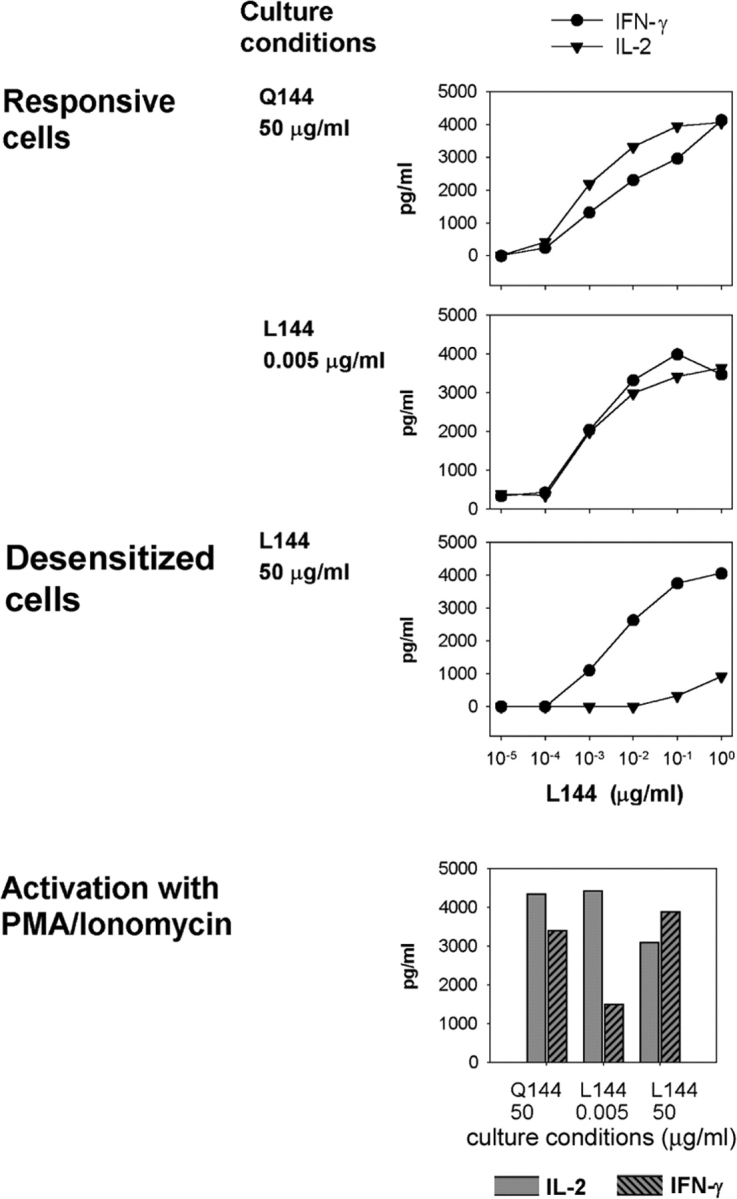
Selective loss of IL-2 but not IFN-γ production from desensitized 1B6 T cells. CD4+ 1B6 Rag2 − / − T cells were cultured as described in Fig. 1 and activated with L144 peptide and APC or with PMA/ionomycin. Supernatants were harvested after 40 h and analyzed for IL-2 and IFN-γ concentration by ELISA. Activation with antigen, but not PMA/ionomycin fails to induce IL-2 secretion from the desensitized cells.
Activation-induced Cell Death and Receptor Down-Regulation Is Normal in Superagonist-activated Transgenic 1B6 T Cells.
Despite complete unresponsiveness to stimulation with anti-CD3, cells cultured with L144 at 50 μg/ml were healthy and proliferated vigorously on restimulation. We wondered if activation of naive 1B6 T cells with L144 led to massive apoptosis and the outgrowth of a minor subpopulation of unresponsive T cells. To quantitate differences in cell death on activation with different ligands, we analyzed activation-induced cell death (AICD) directly by staining annexin V and 7-AAD (Fig. 4 A). There was no evidence of dramatically increased apoptosis, as there was no significant increase in the frequency of annexin-positive CD4+ cells in the L144 (50 μg/ml) activated cells, or in the frequency of the annexin positive plus 7-AAD–positive cells in this group, compared with the other groups. After the first restimulation, viable rested T cells were present under all activation conditions and, in fact, cells activated with higher concentrations of L144 appeared to have expanded better than cells activated with low concentrations of L144 or with Q144 (unpublished data). To determine whether the ability of 1B6 cells to survive activation with the superagonist was the consequence of receptor or coreceptor down regulation, we examined the expression of TCR and CD4 by flow cytometry immediately before restimulation. As shown in Fig. 4 B there were no differences in the expression of these molecules on 1B6 T cells activated with different ligands.
Figure 4.

AICD and receptor/coreceptor expression are comparable. (A) Naive CD4+ 1B6 Rag2 − / − T cells were cultured with different antigens for 48 h and then assessed for AICD by annexin and 7-AAD staining. The fraction of CD4+ cells undergoing AICD was similar in all antigen-treated groups. (B) CD4+ 1B6 Rag2 − / − T cell lines were assessed for expression of antigen receptor and CD4 before they were used in the assays shown in Figs. 1 and 2. The level of receptor expression was equivalent in the different populations.
Taken together, this phenotype of normal TCR and CD4 expression, with normal IFN-γ secretion but deficient IL-2 secretion, is characteristic of anergic T cells generated by suboptimal stimuli (e.g., by activation with fixed APCs, plate bound anti-CD3, or weak agonists), but has not been described after activation with a superagonist ligand. However, although in vitro the 1B6 cells activated with L144 expanded and were healthy, it was possible that in vivo they would not be viable but would be deleted, as has been described for another TCR transgenic cell activated with a superagonist ligand (11). To investigate this possibility we studied transgenic 1B6 T cells which had been transferred into naive SJL recipients using the clonotypic antibody 1.16.
Immunization with Superagonist L144 Generates Desensitized Cells In Vivo.
To determine whether immunizing SJL mice with L144 peptide compared with Q144 peptide in CFA would have different effects on the functional phenotype of transgenic 1B6 T cells in vivo, we transferred LNCs from naive 1B6 transgenic mice or littermate controls into normal female SJL mice. 10 d after immunization the recipient mice were killed and their LNCs were analyzed by flow cytometry and by proliferation assay (Fig. 5). Flow cytometry showed that transgenic 1B6 cells were detectable above background in Q144 and L144 immunized mice and that there was an ∼3-4-fold greater expansion of 1.16-positive 1B6 cells in the lymph nodes of mice immunized with L144 compared with mice immunized with Q144 (Fig. 5 A). This demonstrated that activation in vivo with L144 did not eliminate 1B6 T cells from the peripheral repertoire. Because we had immunized with L144 and Q144 we could not formally distinguish the response of 1B6 cells from those of endogenous L144 or Q144 reactive cells by performing a proliferation assay with these antigens, although there is evidence that the transfer of antigen-specific transgenic T cells can suppress the endogenous T cell response (13). Therefore, to investigate the functional phenotype of the 1B6 cells we determined how well they responded both to L144 and Q144 and also to cross-reactive peptides in vitro after immunization in vivo. Because of the well-characterized hierarchy of response of the 1B6 TCR, we could activate with several different peptides specific for the 1B6 receptor, or with the clonotypic antibody 1.16. In agreement with the in vitro data, cells from mice immunized with Q144 responded to all the ligands tested and to the clonotypic antibody 1.16 (Fig. 5 B). In contrast, mice immunized with L144, or mice that received littermate nontransgenic LNCs, did not respond well to Y144, or to activation with anti-1B6 1.16 antibody and their responses to immunization with L144 or Q144 were lower than the responses detected in mice which received 1B6 cells and were immunized (Fig. 5 B). This shows that in vivo immunization with L144 can induce desensitized cells within 10 d. All groups showed some response to W144. We have previously described enhanced cross-reactive responses to W144 after activation (14), which is due to an expanded endogenous pool of W144-reactive cells that is found in all I-As mice (15). Taken together, the in vitro and in vivo data show that activation with hyperstimulatory ligands expands the transgenic T cells but these expanded cells are hyporesponsive to self-ligand (W144). Based on our in vitro data, the unresponsiveness appeared to be due to a proximal signaling defect rather than TCR or CD4 down-regulation. To determine the mechanism of this proximal signaling defect we analyzed the biochemical phenotype of the desensitized 1B6 T cells.
Figure 5.
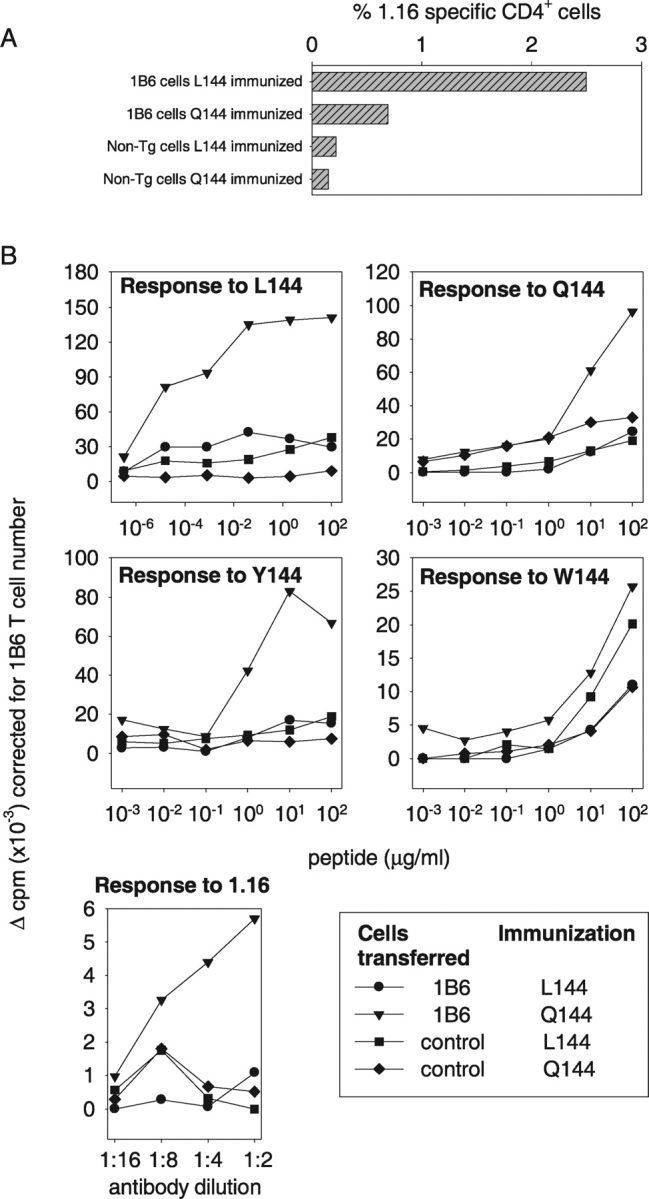
1B6 T cells are hyporesponsive but not deleted in vivo after immunization with L144. Naive female SJL mice were reconstituted with transgenic 1B6 T cells or LNCs from littermate controls and immunized with L144 (50 μg/ml) or Q144 (50 μg/ml). 10 d after immunization, LNCs were assessed for the number of surviving 1B6-clonotype–positive CD4+ cells by flow cytometry (A) and for their functional response by proliferation (B). 1B6 T cells expanded well in mice immunized with L144, but were hyporesponsive compared with cells obtained from mice immunized with Q144 when they were tested in a proliferation assay ex vivo.
p21ras, p44,42 MAPK, and JNK Activation Are Blocked in Desensitized 1B6 T Cells.
If activation with a superagonist induces T cell anergy, then desensitized cells will have the same biochemical characteristics as cells which have been anergized conventionally. One of the fundamental blocks in signal transduction upon TCR occupancy in anergic cells lies at the level of p21ras (16, 17). This small GTPase is inactive when bound to GDP, but becomes activated when GDP is exchanged for GTP. To analyze the activation status of p21ras in desensitized and responsive cells, the GTP-bound form of p21ras was immunoprecipitated with Raf-1 RBD beads and the amount of active p21ras in the immunoprecipitate was analyzed by Western blot. The responsive cells show an activation-induced increase in GTP-bound p21ras, which is already apparent 1 min after stimulation, further increased at 3 min, and is still pronounced at 12 min (Fig. 6 A). In comparison, the desensitized cells show only a weak and retarded increase in the amount of active p21ras, which was not apparent after 1 min but could be seen at later time points.
Figure 6.

Impaired activity of p21ras, p44,42 MAPK, and JNK in the desensitized cells. (A) T cells of the responsive (cultured with L144 0.005 μg/ml) and desensitized (cultured with L144 50 μg/ml) cell lines were stimulated by cross-linking anti-CD3. At the indicated time points the cells were lysed and the active, GTP-bound form of p21ras was immunoprecipitated with Raf-1 RBD agarose beads. The immunoprecipitated proteins were analyzed by SDS-PAGE and Western blot for p21ras. As a control for equal content of total p21ras protein in the different samples before immunoprecipitation, an aliquot of the cell lysate (corresponding to 2 × 106 cells) was separately analyzed by Western blot for p21ras (bottom half of the panel). (B) T cells from the responsive and desensitized cell lines were stimulated by cross-linking anti-CD3 as described for panel A. At the indicated time points the cells were lysed and aliquots of the cell lysates (corresponding to 2 × 106 cells) were analyzed by SDS-PAGE and Western blot with phosphorylation-state–specific mAbs for the phosphorylated forms of p44,42 MAPK, JNK, and p38 MAPK. To control for equal loading, parallel Western Blots with mAbs for each kinase are shown below the respective blots for the phospho-form.
The inability to activate p21ras is expected to lead to a defect in activation of two of the three major mitogen-activated protein kinase (MAPK) pathways, p44,42 MAPK and JNK, while in conventionally anergized cells, phosphorylation of the third major MAPK, p38 MAPK, is usually preserved. The activity of these MAPKs correlates with their phosphorylation on threonines and tyrosines by upstream kinases. We therefore determined the inducibility of the three MAPK pathways in the desensitized T cells by using phosphorylation state–specific antibodies. Upon cross-linking of CD3 the activation of p44,42 MAPK and JNK is severely reduced in the desensitized cells compared with the control responsive cells (Fig. 6 B). When we analyzed the third MAPK pathway in desensitized cells, we found that the phosphorylation of p38 MAPK, although slightly diminished, was relatively spared compared to that of p44,42 MAPK and JNK, suggesting that this phenotype is the result of a specific and selective inhibition of T cell activation. To assess this further we analyzed the status of calcium flux in desensitized cells.
The Sustained Calcium Flux Is Impaired in the Desensitized Cells.
Signaling via calcium influx is a major pathway that is stimulated after TCR occupancy and is spared in most models of anergy, but has been shown to be exclusively affected in some models of anergic human T cells (18). Proximal phosphorylation events lead to the activation of phospholipase Cγ1 (PLCγ1) and the subsequent production of inositol-1,4,5-trisphosphate (IP3), the release of calcium from intracellular stores, and the calcium-triggered influx of extracellular calcium (19). To study the signal transduction via calcium in the desensitized cells, we labeled these cells and the responsive cells with the calcium binding dye Indo-1-AM, stimulated them with anti-CD3 or with APCs and peptide, and analyzed the resulting increase in calcium concentration by flow cytometry (20). Bypassing upstream signaling events by stimulation with the calcium ionophore ionomycin induced equivalent calcium fluxes in both cell types; however, activation through the antigen receptor led to patterns of calcium flux which were strikingly different in the two cell types (Fig. 7). Whereas the initial Ca2+ peak was comparable in the two cell populations, the sustained plateau in free cytosolic calcium concentration ([Ca2+]i), that is apparent in the responsive T cell population, is completely absent in the desensitized cells. This pattern is seen upon anti-CD3 stimulation and with all the tested concentrations of the peptide L144 presented by APCs. The amount of fluxing relative to stimulation with ionomycin is reduced by an average of 55% in the desensitized cells under all activating conditions (Fig. 7). The phenotype of calcium flux in the desensitized cells cannot be reproduced in normally responsive cells by lowering the antigen concentration of the stimulating peptide, demonstrating again that the desensitized cells are fundamentally different from the normally responsive cells in terms of their calcium flux. Therefore, although bypassing proximal signaling with PMA/ionomycin or activation with the superagonist L144 can activate desensitized cells as well as the responsive cells, they have pronounced impairment of both the calcium signaling pathways as well as downstream MAPK signaling pathways (p44,42 MAPK, JNK, but not p38 MAPK).
Figure 7.

Impairment in sustained calcium influx in the desensitized cells. T cells of the responsive and desensitized cell lines were loaded with the indicator dye Indo-1-AM. Calcium mobilization of the cells was monitored continuously by flow cytometry upon stimulation of the T cells with ionomycin, cross-linking anti-CD3, or with I-As/B7 expressing DAS cells, that had been loaded with the indicated concentrations of L144 peptide (start of stimulation indicated by arrow). Calcium mobilization in antibody and antigen stimulated cells was measured and compared with activation with ionomycin, by comparing the number of gated events above a baseline in the total time after stimulation (converted to arbitrary units and taking flux induced with ionomycin as 1.0).
TCR-induced Phosphorylation of TCRζ, CD3ɛ, and ZAP-70 Is Impaired in the Desensitized Cells.
Stimulation of T cells with suboptimal variant ligands has been associated with various changes in the proximal tyrosine phosphorylation patterns. Normally upon stimulation of a T cell via the TCR by anti-CD3 or cognate peptide plus APC, ITAMs of the TCRζ and TCR ɛ chains become phosphorylated via Src family kinases. The Syk family kinase ZAP-70 binds to phosphorylated ITAM motifs and is itself phosphorylated and activated by Src kinases. ZAP-70/Syk family PTKs are then thought to induce tyrosine phosphorylation of important downstream targets including LAT, SLP-76, and PLCγ1 (21). Anergy induced by activation with fixed APCs has been reported to induce near normal patterns of TCRζ and ZAP-70 tyrosine phosphorylation (22), while anergy induced with APLs has been shown to be associated with changes in the ratio of p21/p23 TCRζ (23). It was therefore important to investigate the possibility of a very proximal impairment or block in the TCR signal transduction within the desensitized cells, and determine whether activation with the superagonist L144 induced normal patterns of tyrosine phophorylation. We stimulated desensitized cells with APCs and peptide, immunoprecipitated the CD3 complex at various time points after activation, and determined the extent of tyrosine phosphorylation of both the CD3ɛ chains and the coimmunoprecipitated TCRζ chains (Fig. 8 A). As expected, both the responsive and desensitized cells showed slight constitutive tyrosine phosphorylation of the TCRζ chain, demonstrated by the detection of the p21 phospho-form of TCRζ before stimulation. This form of TCRζ is associated with nonphosphorylated, inactive ZAP-70 kinase (24). Upon TCR-mediated stimulation a pronounced increase in TCRζp21 is detectable in the responsive cells. Additionally, the appearance of the p23 form of TCRζ signals the presence of fully phosphorylated TCRζ chain ITAMs, a prerequisite for phosphorylation and activation of ZAP-70 (23, 25, 26). In comparison, the desensitized cells show a drastically reduced phosphorylation of TCRζ chains. The p21 phospho-form is briefly enhanced but returns to baseline at 12 min and there is only weak induction of the p23 form. The ratio of p23/p21, which is a critical indicator of the degree of proximal T cell activation (23, 25), is at least twofold higher in the responsive cells than in the desensitized cells (Fig. 8 A). Overall, the induction of TCRζ phosphorylation in the desensitized cells is retarded, weaker, and terminated earlier compared with the responsive cells. Similarly striking is the absence of an increase in phosphorylation of the CD3ɛ chain in the desensitized cells compared with the pronounced induction of phosphorylation in the control cells.
Figure 8.

Impaired tyrosine phosphorylation of TCRζ, CD3ɛ, and ZAP-70 in desensitized cells. (A) T cells of the responsive and desensitized cell lines (2 × 107 cells for each time point) were stimulated with I-As/B7 expressing DAS cells (loaded with 100 μg/ml of L144 peptide) as APCs. At the indicated times, the cells were lysed and the lysates subjected to immunoprecipitation with anti-CD3ɛ mAb. The immunoprecipitated proteins were analyzed by Western blot with an anti-phosphotyrosine–specific mAb. The relative ratio of p23 and p21 were measured as indicated by image analysis using the Scion Image package. We were unable to determine the total loading of CD3ɛ by stripping and reprobing. (B) The responsive and tolerant cells were activated with L144 peptide and I-As/B7 expressing DAS cells. After lysis and immunoprecipitation with anti-ZAP-70 antibody, Western blotting with anti-phosphotyrosine-specific mAb was performed and showed reduced ZAP-70 phosphorylation in the desensitized cells. After stripping the blot was reprobed with anti-ZAP-70 to control for loading.
Finally, we looked for the activation-induced association of phosphorylated ZAP-70 with the CD3 complex/TCRζ chain in both cell lines. When the responsive cells were stimulated with anti-CD3 or L144 plus APC, a phospho-band of ∼70 kD was apparent in the anti-phosphotyrosine Western blots of the CD3ɛ immunoprecipitations (unpublished data), whereas there was no detectable band in the case of the desensitized cells. To confirm that ZAP-70 phosphorylation was absent or diminished in the desensitized cells, we immunoprecipitated ZAP-70 at various time points following stimulation and tested in anti-phosphotyrosine Western blots. Again a pronounced activation-induced phosphorylation of ZAP-70 was notable in the responsive cells. By means of direct immunoprecipitation of ZAP-70 we were able to pick up a slight phosphorylation of the kinase even in the desensitized cells, but the difference between the two cell lines remains striking (Fig. 8 B). Stripping and reprobing the blot with anti-ZAP70 confirmed comparable amounts of unphosphorylated protein at all time points (Fig. 8 B).
Discussion
The 1B6 TCR transgenic mouse provides a novel opportunity to investigate the response of T cells to self-antigen, after encounter with cross-reactive ligands. We show in this paper that when T cells are exposed to superagonist ligands they become desensitized, lose responses to self-peptides and to anti-CD3, but still retain the ability to respond to the superagonist ligand. In this system superagonist-activated cells are viable in vitro and in vivo, and express TCR at the same level as cells activated by weaker stimuli, but surprisingly the phenotype of these desensitized cells is very similar to that reported for cells that have been anergized by activation with suboptimal stimulation. This contrasts with experiments that show that activation in vivo with superagonist peptides can lead to peripheral T cell deletion (11), a point that emphasizes that the results in this system reflect part of a spectrum of possible responses to T cell activation.
It is not certain from these experiments whether the state which is induced is the result of the induction of specific anergy inducing proteins in all cells, or the selection of certain cells from the whole population of T cells, and therefore we cannot come to a definitive conclusion as to whether we are dealing with instruction or selection. As with experiments to address this same question in studies of differentiation to a Th1 or Th2 phenotype, this is a difficult question to answer. The majority of all activated T cells undergo AICD, and selection of one population over another is argued to be a major mechanism for achieving T cell differentiation (27). We have not observed a major “crash” of L144 activated cells compared with Q144 activated cells over the duration of the experiments reported in this paper and if selection of a sub-population of cells which is already anergic is critical for survival after activation with L144 peptide, then this subpopulation must be a large fraction of the CD4 cells that survive AICD in vitro after activation with L144.
It is clear that although globally desensitized, these cells retain a brisk response to L144 at nM concentrations of peptide, but lose responsiveness to the autoantigen W144, a change in functional specificity that could be significant in autoimmune disease. That the mechanism is specific and not a global hyporesponsiveness is argued by the data showing that downstream responses induced by PMA/ionomycin are retained, and by the specific defect in IL-2 but not IFN-γ secretion. In addition, the changes in signaling biochemistry indicate a specific block in activation affecting p21ras, as has been described in other models of anergy.
The desensitization induced by the superagonist peptide has various interesting aspects, which may distinguish it from previously known types of anergy. T cell anergy is generally assumed to result from partial T cell activation (18). Examples of this type of anergy induction are TCR stimulation without costimulation, either by fixed APCs with agonist peptides (28), by live APCs with suboptimal ligands (29), or by cross-linking the TCR with antibody alone (30). In human T cells anergy has also been induced by expanding T cells in the presence of large doses of IL-10 and antigen (31). Two different biochemical mechanisms have been identified in anergic cells: in the classical models of anergy there is defective p21ras activation (16,17), whereas in human T cells, anergy has sometimes been attributed to defective calcium fluxes (18). In addition to these mechanisms, anergy induced by partial agonists has been shown to be associated with incomplete CD3ζ chain and ZAP-70 phosphorylation (23, 25). Several in vivo models of anergy have confirmed these findings (32, 33) or pointed to defects in calcium signaling (34–36) in anergic cells.
When we examined the proximal CD3 associated signaling pathways induced by L144, we found that the superagonist peptide L144 induces a fully agonistic pattern of TCRζ chain tyrosine phosphorylation in naive 1B6 TCR transgenic cells (unpublished data). However, after activation with the hyperstimulating peptide L144, this pattern changes dramatically. Stimulation with the same peptide now produces a TCRζ phosphorylation pattern (Fig. 8 A) which recapitulates the findings originally described in a T cell clone stimulated with an anergy-inducing APL (23). Although Sloan-Lancaster et al. (23) reported sustained phosphorylation of the CD3ζ chain for at least 20 min upon agonist stimulation, in our experimental system the p21 and p23 forms of CD3ζ decrease earlier in both cell lines (Fig. 8). It is difficult to judge if this reflects suboptimal stimulation even in the responsive Q144 cell line as the systems differ with regard to the antigen, the cell type (short term T cell line as opposed to established T cell clones), and the mode of stimulation. Similar to our observation, in newly generated AND TCR transgenic T cell lines, ζ chain phosphorylation upon stimulation with the high-affinity ligand moth cytochrome c (MCC) is already terminated at 15 min (37). In our view it is therefore only safe to deduce that within our system the responsive and the desensitized cells clearly differ in their pattern of proximal tyrosine phosphorylation, most notably in the ratio of p21/p23. The desensitized cells also show defective activation of the p21ras pathway, and they also have abnormal patterns of calcium mobilization, with a reduction in sustained calcium influx after activation. Sustained elevation of intracellular Ca2+, which is required for efficient transcription of the IL-2 gene (19, 38, 39), requires the influx of extracellular calcium through activated calcium channels, and this may be uncoupled in the desensitized cells induced by activation with L144 peptide. The calcium phenotype of the desensitized cells in this regard resembles that of Itk−/− cells (40). Induction of selective unresponsiveness toward lower-affinity ligands has recently been analyzed in AND TCR transgenic cells (37). Compared with a transgenic Th1 cell population, the Th2 cells fail to recruit TCR complex members to lipid rafts, which seems to account for their impaired signaling capacity. It remains to be elucidated if a similar difference in membrane microdomain partitioning is operative in our system of superagonist-induced unresponsiveness.
The novel type of superagonist-induced anergy we describe here is clearly not the consequence of partial activation of the T cells. Furthermore, it seems to involve the p44,42 and JNK MAPK signaling pathways and signaling via calcium, which are simultaneously severely impaired. This biochemical phenotype points to a profound block in activation. This is also reflected in the fact that exogenous IL-2 is not able to rescue the anergic cells. This is in contrast to conventional anergy which can be reversed by culturing the anergic cells in IL-2 containing medium (41). However, this feature of superagonist-induced anergy is not unique, in that IL-2 cannot reverse anergy in a murine model system of partial agonist-induced anergy (42) and in IL-10–induced anergy in human T cells (31). Taken together, it is clear that anergy is not one unique entity, rather, different modes of induction seem to induce different degrees of anergy (18). Superagonist-induced anergy occupies a space at the very refractory end of this spectrum.
This description of desensitization of peripheral T cells is clearly reminiscent of findings described in thymic selection in vivo in a Vα2/Vβ8.1 transgenic mouse (43). The TCR these mice express is specific for a lymphocytic choriomeningitis virus (LCMV) peptide, and due to the Vβ8.1 TCR chain usage also possesses potential reactivity toward Mls-1a and staphylococcal enterotoxin B (SEB). When Mls-1a was introduced into these mice during thymic selection it resulted in CD8+ T cells that lost reactivity selectively to Mls-1a, while cells with normal reactivity toward the LCMV peptide and the superantigen SEB were retained. The authors concluded from these data that thymocytes rendered tolerant of low-affinity self-antigen can develop into T cells which retain responsiveness for high-affinity foreign antigens. Similar divergent changes in reactivity toward weak and strong agonists have been noted in other models of thymic differentiation (44, 45), in which developing thymocytes maintain reactivity to strong agonists, while simultaneously losing reactivity to weak agonists. It was recently reported in a double transgenic model with TCR specificity for MCC and transgenic expression of the antigen, that peripheral T cells in these mice are hypoproliferative toward MCC while retaining their reactivity toward the superantigen SEA (46). The authors could show in this experimental system that the anergic T cells had escaped negative selection in the thymus by expressing endogenous α-chains and that they behaved essentially as low avidity naive cells in the periphery. We could rule out the possibility that our in vitro culture conditions selected for such double α-chain expressing cells by using T cell lines derived from Rag2 −/− mice, in which all T cells express only the 1B6 transgenic αβ TCR.
We speculate that these changes in functional specificity may be useful in the context of a normal immune response in that they preserve useful reactivity to a foreign specificity but reduce reactivity with self. Such a scenario leads to an increase in specificity at the expense of sensitivity, which is clearly a viable trade off in terms of the responses to L144 which occur over such a wide dose response. Our observations fit into the concept of flexible T cell activation thresholds, that can be tuned upon activation of the TCR with stimuli of different strengths (12, 47, 48). This paper presents experimental evidence for such a flexible threshold and changes in the reactivity of a T cell after activation with a cross-reactive ligand. Indeed in these circumstances, the functional specificity of the T cell changes without changing the level of expression or the specificity of the TCR. This may be of particular interest in studies of autoimmune disease where infection is widely regarded as an important precipitating factor for induction of autoimmune disease; our experiments provide an example of how in some circumstances infection may induce peripheral tolerance by reducing responsiveness to autoantigen. Thus, we have described a novel mechanism by which a cross-reactive TCR may retain functional responses to nonself but lose responses to self-ligand without deleting the clonotype from the peripheral repertoire.
Acknowledgments
The authors would like to thank R. Nazareno for maintaining and screening mice, B. Rich and J. Murphy who provided Rag2 − / − mice for breeding, and N. Price who provided technical assistance. We would also like to thank R. McGilp for help with analysis of calcium flux, and Edward Greenfield for performing the fusion that produced the 1.16 antibody.
M. Munder was supported by grant Mu1547/1-1 of the Deutsche Forschungsgemeinschaft. This work was also supported by grants from the National Institutes of Health (K08 AI01557-01) and the National Multiple Sclerosis Society, New York (RG3257) to L.B. Nicholson and from the National Institutes of Health (R01NS30843, R01NS35685, P01AI39671-01A1, PO1NS38037-01A1), and the National Multiple Sclerosis Society (NMSS), New York (RG2571, RG2320) to V.K. Kuchroo. J.M. Slavik is a fellow of the NMSS.
M. Munder's present address is Medizinische Klinik und Poliklinik V, Ruprecht-Karls-Universität Heidelberg, Hospitalstrasse 3, 69115 Heidelberg, Germany.
Footnotes
Abbreviations used in this paper: AICD, activation-induced cell death; LNC, lymph node cell; MAPK, mitogen-activated protein kinase; MCC, moth cytochrome c.
References
- 1.Kersh, G.J., and P.M. Allen. 1996. Essential flexibility in the T-cell antigen recognition of antigen. Nature. 380:495–498. [DOI] [PubMed] [Google Scholar]
- 2.Mason, D. 1998. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today. 19:395–404. [DOI] [PubMed] [Google Scholar]
- 3.Bevan, M.J. 1997. In thymic selection, peptide diversity gives and takes away. Immunity. 7:175–178. [DOI] [PubMed] [Google Scholar]
- 4.Goldrath, A.W., and M.J. Bevan. 1999. Selecting and maintaining a diverse T-cell repertoire. Nature. 402:255–262. [DOI] [PubMed] [Google Scholar]
- 5.Zamvil, S.S., and L. Steinman. 1990. The T lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol. 8:579–621. [DOI] [PubMed] [Google Scholar]
- 6.Kuchroo, V.K., N. Sarvetnick, D.A. Hafler, and L.B. Nicholson. 2002. Cytokine and Autoimmune Diseases. Humana Press, Totowa, New Jersey.
- 7.Vanderlugt, C.J., and S.D. Miller. 2002. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2:85–95. [DOI] [PubMed] [Google Scholar]
- 8.Oldstone, M.B.A. 1998. Molecular mimicry and immune-mediated diseases. FASEB J. 12:1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.London, C.A., M.P. Lodge, and A.K. Abbas. 2000. Functional responses and costimulator dependence of memory CD4+ T cells. J. Immunol. 164:265–272. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson, L.B., H.-P. Waldner, A. Carrizosa, A. Sette, M. Collins, and V.K. Kuchroo. 1998. Heteroclitic proliferative responses and changes in cytokine profile induced by altered peptides: implications for autoimmunity. Proc. Natl. Acad. Sci. USA. 95:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderton, S.M., C.G. Radu, P.A. Lowrey, E.S. Ward, and D.C. Wraith. 2001. Negative selection during the peripheral immune response to antigen. J. Exp. Med. 193:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholson, L.B., A.C. Anderson, and V.K. Kuchroo. 2000. Tuning T cell activation threshold and effector function with cross-reactive peptide ligands. Int. Immunol. 12:205–213. [DOI] [PubMed] [Google Scholar]
- 13.Kedl, R.M., K.A. Rees, D.A. Hildeman, B. Schaefer, T. Mitchell, J. Kappler, and P. Marrack. 2000. T cells compete for access to antigen-bearing antigen-presenting cells. J. Exp. Med. 192:1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson, A.C., H.-P. Waldner, V. Turchin, C. Jabs, M. Prabhu Das, V.K. Kuchroo, and L.B. Nicholson. 2000. Autoantigen responsive T cell clones demonstrate unfocused TCR cross-reactivity towards multiple related ligands: implications for autoimmunity. Cell. Immunol. 202:88–96. [DOI] [PubMed] [Google Scholar]
- 15.Anderson, A.C., L.B. Nicholson, K.L. Legge, V. Turchin, H. Zaghouani, and V.K. Kuchroo. 2000. High frequency of autoreactive myelin proteolipid protein-specific T cells in the periphery of naive mice: Mechanisms of selection of the self-reactive repertoire. J. Exp. Med. 191:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, W., C.D. Whaley, A. Mondino, and D.L. Mueller. 1996. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science. 271:1272–1276. [DOI] [PubMed] [Google Scholar]
- 17.Fields, P.E., T.F. Gajewski, and F.W. Fitch. 1996. Blocked Ras activation in anergic CD4+ T cells. Science. 271:1276–1278. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz, R.H. 1996. Models of T cell anergy: is there a common molecular mechanism. J. Exp. Med. 184:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guse, A.H. 1998. Ca2+ signaling in T-lymphocytes. Crit. Rev. Immunol. 18:419–448. [DOI] [PubMed] [Google Scholar]
- 20.June, C.H., and P.S. Rabinovitch. 1994. Intracellular ionized calcium. Methods Cell Biol. 41:149–174. [PubMed] [Google Scholar]
- 21.Wange, R.L., and L.E. Samelson. 1996. Complex complexes: signaling at the TCR. Immunity. 5:197–205. [DOI] [PubMed] [Google Scholar]
- 22.Madrenas, J., R.H. Schwartz, and R.N. Germain. 1996. Interleukin 2 production, not the pattern of early T-cell antigen receptor-dependent tyrosine phosphorylation, controls anergy induction by both agonists and partial agonists. Proc. Natl. Acad. Sci. USA. 93:9736–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sloan-Lancaster, J., A.S. Shaw, J.B. Rothbard, and P.M. Allen. 1994. Partial T cell signaling: altered phospho-zeta and lack of Zap70 recruitment in APL-induced T cell anergy. Cell. 79:913–922. [DOI] [PubMed] [Google Scholar]
- 24.van Oers, N.S., N. Killeen, and A. Weiss. 1994. ZAP-70 is constitutively associated with tyrosine-phosphorylated TCR zeta in murine thymocytes and lymph node T cells. Immunity. 1:675–685. [DOI] [PubMed] [Google Scholar]
- 25.Madrenas, J., R.L. Wange, J.L. Wang, N. Isakov, L.E. Samelson, and R.N. Germain. 1995. Zeta phosphorylation without ZAP-70 activation induced by TCR antagonists or partial agonists. Science. 267:515–518. [DOI] [PubMed] [Google Scholar]
- 26.Kersh, E.N., A.S. Shaw, and P.M. Allen. 1998. Fidelity of T cell activation through multistep T cell receptor zeta phosphorylation. Science. 281:572–575. [DOI] [PubMed] [Google Scholar]
- 27.Coffman, R.L., and S.L. Reiner. 1999. Immunology - instruction, selection, or tampering with the odds? Science. 284:1283–1285. [DOI] [PubMed] [Google Scholar]
- 28.Jenkins, M.K., and R.H. Schwartz. 1987. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 165:302–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sloan-Lancaster, J., B.D. Evavold, and P.M. Allen. 1993. Induction of T-cell anergy by altered T-cell-receptor ligand on live antigen-presenting cells. Nature. 363:156–159. [DOI] [PubMed] [Google Scholar]
- 30.Jenkins, M.K., C. Chen, G. Jung, D.L. Mueller, and R.H. Schwartz. 1990. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J. Immunol. 144:16–22. [PubMed] [Google Scholar]
- 31.Groux, H., M. Bigler, J.E. de Vries, and M.G. Roncarolo. 1996. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J. Exp. Med. 184:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Migita, K., K. Eguchi, Y. Kawabe, T. Tsukada, Y. Ichinose, S. Nagataki, and A. Ochi. 1995. Defective TCR-mediated signaling in anergic T cells. J. Immunol. 155:5083–5087. [PubMed] [Google Scholar]
- 33.McKay, D.B., H.Y. Irie, G. Hollander, J.L. Ferrara, T.B. Strom, Y. Li, and S.J. Burakoff. 1999. Antigen-induced unresponsiveness results in altered T cell signaling. J. Immunol. 163:6455–6461. [PubMed] [Google Scholar]
- 34.Kimura, M., M. Yamashita, M. Kubo, M. Iwashima, C. Shimizu, K. Tokoyoda, J. Chiba, M. Taniguchi, M. Katsumata, and T. Nakayama. 2000. Impaired Ca/calcineurin pathway in in vivo anergized CD4 T cells. Int. Immunol. 12:817–824. [DOI] [PubMed] [Google Scholar]
- 35.Blackman, M.A., T.H. Finkel, J. Kappler, J. Cambier, and P. Marrack. 1991. Altered antigen receptor signaling in anergic T cells from self-tolerant T-cell receptor beta-chain transgenic mice. Proc. Natl. Acad. Sci. USA. 88:6682–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Utting, O., S.J. Teh, and H.S. Teh. 2000. A population of in vivo anergized T cells with a lower activation threshold for the induction of CD25 exhibit differential requirements in mobilization of intracellular calcium and mitogen-activated protein kinase activation. J. Immunol. 164:2881–2889. [DOI] [PubMed] [Google Scholar]
- 37.Balamuth, F., D. Leitenberg, J. Unternaehrer, I. Mellman, and K. Bottomly. 2001. Distinct patterns of membrane microdomain partitioning in Th1 and th2 cells. Immunity. 15:729–738. [DOI] [PubMed] [Google Scholar]
- 38.Scharenberg, A.M., and J.P. Kinet. 1998. PtdIns-3,4,5-P3: a regulatory nexus between tyrosine kinases and sustained calcium signals. Cell. 94:5–8. [DOI] [PubMed] [Google Scholar]
- 39.Dolmetsch, R.E., R.S. Lewis, C.C. Goodnow, and J.I. Healy. 1997. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 386:855–858. [DOI] [PubMed] [Google Scholar]
- 40.Liu, K.Q., S.C. Bunnell, C.B. Gurniak, and L.J. Berg. 1998. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J. Exp. Med. 187:1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beverly, B., S.M. Kang, M.J. Lenardo, and R.H. Schwartz. 1992. Reversal of in vitro T cell clonal anergy by IL-2 stimulation. Int. Immunol. 4:661–671. [DOI] [PubMed] [Google Scholar]
- 42.Ryan, K.R., and B.D. Evavold. 1998. Persistence of peptide-induced CD4+ T cell anergy in vitro. J. Exp. Med. 187:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawai, K., and P.S. Ohashi. 1995. Immunological function of a defined T-cell population tolerized to low-affinity self antigens. Nature. 374:68–69. [DOI] [PubMed] [Google Scholar]
- 44.Davey, G.M., S.L. Schober, B.T. Endrizzi, A.K. Dutcher, S.C. Jameson, and K.A. Hogquist. 1998. Preselection thymocytes are more sensitive to T cell receptor stimulation than mature T cells. J. Exp. Med. 188:1867–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lucas, B., I. Stefanova, K. Yasutomo, N. Dautigny, and R.N. Germain. 1999. Divergent changes in the sensitivity of maturing T cells to structurally related ligands underlies formation of a useful T cell repertoire. Immunity. 10:367–376. [DOI] [PubMed] [Google Scholar]
- 46.Girgis, L., M.M. Davis, and B.F. de St Groth. 1999. The avidity spectrum of T cell receptor interactions accounts for T cell anergy in a double transgenic model. J. Exp. Med. 189:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grossman, Z., and W.E. Paul. 1992. Adaptive cellular interactions in the immune system: The tunable activation threshold and the significance of subthreshold responses. Proc. Natl. Acad. Sci. USA. 89:10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grossman, Z., and A. Singer. 1996. Tuning of activation thresholds explains flexibility in the selection and development of T cells in the thymus. Proc. Natl. Acad. Sci. USA. 93:14747–14752. [DOI] [PMC free article] [PubMed] [Google Scholar]