

Abstract
The translocation (8;21), generating the AML1-ETO fusion protein, is one of the most frequent chromosomal abnormalities associated with acute myelogenous leukemia (AML). To elucidate its role in oncogenesis, bone marrow (BM) cells were infected with a retroviral vector carrying AML1-ETO and transplanted into mice. In contrast to previous transgenic mouse models, we show that AML1-ETO directly stimulates granulopoiesis, suppresses erythropoiesis, and impairs the maturation of myeloid, B, and T lymphoid cells in vivo. To determine the significance of earlier findings that expression of the tumor suppressor ICSBP is often downregulated in AML myeloblasts, AML1-ETO was introduced into BM cells derived from mice lacking the interferon regulatory factor ICSBP. Our findings demonstrate that AML1-ETO synergizes with an ICSBP deficiency to induce myeloblastic transformation in the BM, reminiscent of AML.
Keywords: leukemia, translocation (genetics), IFN regulatory factor, B cell differentiation, myeloid differentiation
Introduction
Acute myelogenous leukemia (AML)* accounts for >80% of all adult acute leukemias. A distinct subset of these leukemias, making up ∼10% of all AML cases, is characterized by a (8;21)(q22;q22) translocation and, morphologically, by the presence of myeloblasts showing maturation in the neutrophil lineage (1). The t(8;21) juxtaposes the RUNX1 gene on chromosome 21 with the CBFT1 gene on chromosome 8, generating the AML1-ETO fusion transcription factor (2–4). This fusion protein consists of the NH2-terminal portion of the AML1 protein fused in frame to the nearly full-length ETO protein. The fact that the t(8;21) is the sole cytogenetic abnormality in the majority of these cases suggests that AML1-ETO plays a critical role both in the distinctive phenotype and in the establishment of the leukemia clone. However, the mechanism by which AML1-ETO initiates or contributes to AML transformation is not clearly established.
AML1, the amino-terminal partner of the fusion protein, is the DNA-binding subunit of the core-binding factor (CBF) transcription factor complex. It forms heterodimeric complexes with CBFβ and activates transcription from enhancer core motifs (TGT/cGGY), which are present in a number of genes relevant to myeloid and lymphoid development (for a review, see references 5–7). The Runt homology domain (RHD) (8) at the amino terminus of AML1 is included in the fusion protein and has been shown to be required for heterodimerization with CBFβ and for DNA binding (9). In contrast, the transactivation domain, mapped to the COOH terminus of AML1 and shown to interact with the multifunctional transcriptional coactivators p300 and CBP (10), is absent in the fusion protein. In its place is the COOH-terminal partner, ETO, which interacts with the nuclear receptor corepressor/mSin3/histone deacetylase complex (11–14). The RHD of AML1 thus targets the fusion protein, with its associated repressor complex, to AML1 (CBF)-regulated genes, leading to the repression of genes containing CBF-binding sites in their promoter/enhancer region (12, 15, 16). Recent work has shown that the RHD of AML1 also interacts directly with other transcription factors important in lymphoid and myeloid differentiation (17–22), presenting the possibility that the ETO-corepressor complex of AML1-ETO can be recruited to enhancer elements of a broad spectrum of genes.
To establish an effective therapy regimen for t(8;21) AML, it is important to establish an in vivo system in which the critical events and target genes of AML1-ETO–induced transformation can be identified and tested. Initial attempts to mimic the t(8;21) in mice by a knock-in strategy (23, 24) resulted in embryonic lethality, similar to that observed in AML1 knock-out mice, which originally demonstrated the importance of the CBF transcription complex in the establishment of definitive hematopoiesis (25–27). Although these mice confirmed that AML1-ETO is a dominant inhibitor of normal AML1 function, they are not useful models to study leukemia induction. To overcome the embryonic mortality, several groups have established conditional AML1-ETO transgenic mouse strains, in which expression can be induced in myeloid progenitors of adult animals (28–31). Although no overt phenotypic alterations were observed in vivo in these models, AML1-ETO expression was shown to enhance the self-renewal capacity of early multipotent cells in vitro and to prime the cells for leukemic transformation (29–31), demonstrating the importance of secondary genetic lesions for AML1-ETO transformation.
As the hematopoietic stem cell (HSC) is the primary transformed cell in AMLs (32–34), we sought to establish a mouse model for t(8;21) AML by using retroviral vectors which infect this compartment. For these experiments we used FMEV retroviral vectors, which have been optimized to yield high expression levels in the HSC (for a review, see references 35 and 36). Importantly, coexpression of eGFP enabled us to readily detect differences between AML1-ETO transduced and nontransduced cells. Our findings conclusively demonstrate that AML1-ETO directly impairs maturation of multiple lymphohematopoietic lineages and stimulates granulopoiesis in vivo, but is not sufficient to induce leukemia.
Several recent studies have implicated the IFN regulatory factor ICSBP (also known as IRF8) as a tumor suppressor of myeloid neoplasias. The first support of this hypothesis came from studies of mice deficient for ICSBP, which developed a myeloproliferative disease, closely resembling chronic myelogenous leukemia (CML) (37). The ICSBP deficiency results in an expansion of the early myeloid progenitor pool, probably due to an increased sensitivity to proliferation stimuli and a decreased response to apoptotic stimuli (38, 39). The relevance of this observation in human leukemia was confirmed by the demonstration that ICSBP is downregulated in 79 and 66% of patients with CML and AML, respectively (40, 41). A more recent study suggests that ICSBP may be a target gene of deregulated tyrosine kinases that induce myeloproliferation, as constitutive expression of ICSBP inhibited the myeloid transformation capacity of BCR-ABL, a chimeric tyrosine kinase generated by the 9;22 translocation that is a hallmark of CML (42). To determine if ICSBP deficiency cooperates with AML1-ETO–induced impairment of differentiation to induce a acute myeloid disorder, AML1-ETO was introduced into bone marrow (BM) cells derived from ICSBP−/− mice. Our results demonstrate that an ICSBP deficiency synergizes with expression of AML1-ETO in the induction of myeloid tumorigenesis.
Materials and Methods
Retroviral Vectors and Generation of Infectious Pseudotyped Viral Particles.
The full-length AML1-ETO cDNA was excised from plasmid pUHD-AML1-ETO (provided by D. Tenen, Harvard Institutes of Medicine, Boston, MA) by XbaI digestion, blunt-ended, and inserted into the plasmid pSF91-I-eGFP-PRE (R780) at the unique NotI site. SF91-EGFP-PRE is a FMEV type vector (43) that differs from the previously described SF91 retroviral vector (44) by the insertion of an internal ribosome entry site (IRES)-eGFP cassette and the posttranscriptional regulatory element (PRE) of the woodchuck hepatitis virus (45). To generate retroviral pseudotypes, retroviral vector plasmid DNA was contransfected with a plasmid expressing the ecotropic env (46) into Phoenix-GP packaging cells (provided by G. Nolan, Stanford University, Palo Alto, CA), as described previously (47).
Retroviral Infection of BM Cells and Transplantations.
C57BL/6J- Ly5.1-Pep3b mice were used as recipients for all BM transplantations. C57BL/6J-Ly5.2 or ICSBP−/−-C57BL/6J-Ly5.2 mice between 12 to 16 wk of age were used as a source of BM cells. In this genetic background and when maintained as homozygotes, ICSBP−/− mice did not develop acute hematological disorders in over a 2-y observation period. Mice had slightly increased spleen weights (0.35 vs. 0.11 g) and increased number of myeloid progenitors and precursors in BM and spleen, but normal blood counts. BM cells were harvested from tibiae and femura of male donor mice 4 d after intraperitoneal administration of 5-fluorouracil (150 mg/kg; Lederle) and infected as described previously (48). 12 h after the last infection, 106 cells were transplanted by tail vein injection into lethally irradiated (9.5 Gy) Ly5.2+ female recipients. To ensure normal erythropoiesis in engrafted mice receiving ICSBP−/− BM, the transplanted cells were supplemented with one-tenth amounts of nontransduced ICSBP+/+ BM cells. Serial transplantation was performed with transduced ICSBP+/+ and ICSBP−/− BM to ensure that the vector-expressing cells were of HSC origin. All animal experiments were approved by the Hamburg Office of Health (permit no. 04/2000).
Western Blot Analysis.
Nuclear protein from Kasumi-1 cells (49) or Phoenix cells transfected with the retroviral vector plasmid FMEV/AE or FMEV/GFP were prepared as described previously (50). 5 mg cell lysates were immunoprecipitated with the polyclonal anti-ETO antibody (Oncogene Research Products) and one-third of the precipitate was size-separated by 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Western blot analysis were performed as described previously (50). A 1:200 dilution of the polyclonal anti-AML1 antibody (N-20, Santa Cruz Biotechnology, Inc.) was used to detect protein expression. After incubation with the first antibody, the bound antibody was detected with the appropriate secondary antibody conjugated with HRP and visualized using the ECL (Amersham Pharmacia Biotech).
Flow Cytometry.
For lineage marker analysis, single-cell suspensions (106 cells) were prepared from hematopoietic tissues after lysis of erythroid cells with PharmMLyse™ (BD PharMingen) and incubated at 4°C for 30 min in PBS containing 2% BSA with PE- or APC-conjugated mAbs (BD PharMingen). Nonspecific binding of mAbs was prevented by preincubation with Fc Block™ (BD PharMingen). Cells were washed twice with PBS containing 2% BSA and applied for analysis on a FACScalibur™ (BD Biosciences) FACS®. A MoFlow™ Cell Sorter (Cytomation Bioinstruments) was used to sort transduced cells before colony assays.
In Vitro Colony-forming Assays.
BM cells from either FMEV/AE or FMEV/GFP mice were isolated from femura and tibiae and sorted for GFPpos expression. Single-cell suspensions (5 × 104 cells per milliliter and 105 cells per milliliter) were plated in triplicates in methylcellulose (MethCult™ GF M3434, StemCell Technologies) and incubated at 37°C. Individual colonies were scored 9 d after plating. Cytospins were made from representative colonies and stained with Giemsa solution to confirm or determine cell morphology within a colony.
Histologic and Immunohistologic Methods.
Splenic and hepatic tissue specimens were fixed with 4% formalin containing 1% acetic acid. For histologic inspection of BM, sterna were dissected and fixed in a formalin containing solution specifically developed for fixation of iliac crest trephine biopsies (Protaqs Calfix, Quaratett) and appropriately decalcified (Protaqs Calless, Quaratett) before paraffin embedding. Deparaffinated sections were stained according to standard laboratory protocols with H&E, periodic acid Schiff solution, or a modified Wright-Giemsa stain (all obtained from Sigma-Aldrich). For myeloperoxidase immunostaining of granulocytic cells an indirect immunoperoxidase staining technique was applied. In brief, for antigen retrieval, deparaffinated sections were treated with a commercial “Target Unmasking Fluid” (TUF, Dako) at 98°C for 20 min in a microwave oven. Incubation with 1:15,000 diluted polyclonal rabbit anti–human myeloperoxidase (Dako) antibody was done overnight at 4°C. Specifically bound primary antibodies were detected using a highly sensitive peroxidase- and polymer-conjugated anti–rabbit Ig detection system (Envision, Dako).
Results
Transduction and Transplantation of BM Cells with eGFP and AML1-ETO–expressing Vectors.
The AML1-ETO cDNA was introduced into a FMEV retroviral vector, which coexpresses the eGFP behind an IRES (Fig. 1 A). Both virus titers and eGFP expression levels were reduced in infected cells expressing the FMEV/AE construct as compared with the control vector FMEV/GFP (Fig. 1 B), which may be partly due to AML1-ETO transcriptional repression of the vector via a pivotal CBF-binding site in the retroviral LTR (51). Western blot analysis of protein extracted from FMEV/ AE-infected cells confirmed the presence of the AML1-ETO protein with the same molecular weight as that found in Kasumi-1 cells, a cell line established from a t(8;21) AML patient (49) (Fig. 1 C). Exact quantification of AML1-ETO protein levels produced by infected cells was precluded by the immunoprecipitation step, but they appear to be somewhat lower as observed in Kasumi-1 cells.
Figure 1.

Coexpression of eGFP with AML1-ETO allows detection of both transduced and nontransduced cells. (A) AML1-ETO cDNA was inserted into the FMEV/GFP vector, in which the translation of eGFP is initiated from an IRES, to generate FMEV/AE. Transcription is initiated from the SFFVp LTR. The PRE from woodchuck hepaptitis virus increases the expression levels. (B) eGFP fluorescence was measured by flow cytometry in BM cells obtained from mice transplanted with infected BM cells. Reduced levels of expression from FMEV/AE may be attributable, in part, to transcriptional downregulation by binding of AML1-ETO to the CBF-binding site in the LTR. (C) Western blot analysis of protein extracted from FMEV/AE- and FMEV/GFP-infected human cells was performed to confirm AML1-ETO expression. Protein extracted from Kasumi-1 cells served as a control.
BM cells were infected with comparable titers of FMEV/AE or FMEV/GFP and then transplanted into lethally irradiated donors. At the time of transplantation, 10–15% of the BM cells expressed the viral vector, as measured by eGFP expression. After 3–6 mo, three animals of each group (first cohort) were killed and the BM was used to transplant five new mice each (second cohort).
Higher Proportion of AML1-ETO Transduced Cells in BM as in Blood.
For both FMEV/AE and FMEV/GFP transduced mice, a total of 14 and 9 mice, respectively, were killed between 2 and 6 mo after BM transplantation. At least 4–5 animals from each cohort were analyzed. Significantly, no difference in leukocyte and differential counts in the blood were observed between experimental and control animals. The percentage of BM cells carrying the vector varied considerably between individual mice for both FMEV/AE and FMEV/GFP (Table I), ranging from 6.1 to 73.6% and 3.8 to 51.5%, respectively. Although overall higher levels of transduction were observed in BM receiving FMEV/AE, this was not statistically significant. Thus, AML1-ETO expression neither imparts a selective advantage nor disadvantage to repopulating cells.
Table I.
Percentage of Transduced Cells in BM and Blood of Transplanted Mice
| Percentage of GFPpos cells
|
Ratio GFPpos(blood/BM) | |||||
|---|---|---|---|---|---|---|
| Transduced vector | Mouse no. | Cohort | Daysa | Blood | BM | |
| FMEV-GFP | 9 | 1 | 166 | 23.7 | 19.2 | 1.23 |
| 11 | 1 | 166 | 8.4 | 7.9 | 1.06 | |
| 13 | 1 | 133 | 48.2 | 30.5 | 1.58 | |
| 23 | 1 | 242 | 57.8 | 51.5 | 1.12 | |
| 91 | 2 | 69 | 44.0 | 21.6 | 2.04 | |
| 92 | 2 | 137 | 51.2 | 48.8 | 1.05 | |
| 94 | 2 | 69 | 13.9 | 5.6 | 2.48 | |
| 108 | 2 | 131 | 8.8 | 3.8 | 2.32 | |
| 112 | 2 | 118 | 23.1 | 9.8 | 2.36 | |
| x = 1.69 ± 0.61 | ||||||
| FMEV-AE | 3 | 1 | 194 | 8.8 | 34.2 | 0.26 |
| 4 | 1 | 194 | 6.3 | 28.5 | 0.22 | |
| 4.1 | 1 | 127 | 9.7 | 38.5 | 0.25 | |
| 30 | 1 | 218 | 2.1 | 6.1 | 0.34 | |
| 30.1 | 1 | 188 | 2.8 | 7.4 | 0.38 | |
| 86 | 2 | 69 | 16.4 | 40.7 | 0.40 | |
| 87 | 2 | 69 | 26.0 | 73.6 | 0.35 | |
| 89 | 2 | 137 | 3.6 | 17.8 | 0.20 | |
| 96 | 2 | 126 | 3.3 | 8.5 | 0.39 | |
| 97 | 2 | 106 | 8.0 | 12.8 | 0.63 | |
| 98 | 2 | 78 | 11.8 | 26.0 | 0.45 | |
| 102 | 2 | 118 | 7.2 | 19.4 | 0.37 | |
| 103 | 2 | 78 | 5.4 | 10.9 | 0.50 | |
| 106 | 2 | 131 | 10.2 | 31.1 | 0.33 | |
| x = 0.36 ± 0.11b | ||||||
Number of days after transplantation.
P < 0.001 as compared to GFP control mice.
However, a striking difference was observed when the level of transduced cells in BM and blood was compared. Whereas mice receiving FMEV/GFP–infected BM showed similar or slightly higher levels of transduced (GFPpos) cells in the blood as compared with the BM, the number of transduced (AEpos) cells in the blood was reduced by a mean factor of 2.7 relative to the BM in FMEV/AE mice (Table I). This was observed in all mice analyzed, regardless of whether they received BM directly after retroviral transduction (first cohort; n = 5) or BM from primary transplants (second cohort; n = 9). In addition, no significant change in the ratio was observed in mice killed at different time points (ranging from 69 to 218 d after transplantation). This reduction in transduced cells in the blood must thus be a direct consequence of AML1-ETO expression.
The distribution of myeloid, as well as B and T lymphoid cells in the transduced (AEpos; GFPpos) or nontransduced (AEneg; GFPneg) populations in the blood was determined by FACS® analysis to ascertain if a particular lineage was underrepresented in the AEpos transduced blood cells (Fig. 2, A and B). Greatly reduced numbers of transduced lymphoid cells, in particularly those of the T cell lineage, was observed in the AEpos population. This distortion in lineage ratio could not be attributed to a bias generated by retroviral transduction, as the lineage ratio of GFPpos transduced cells was comparable to the nontransduced populations. Significantly, in addition to a decrease in both B and T lymphoid cells (∼13 and 3% of the expected number, respectively), a 48% decrease in the expected number of myeloid cells was also observed in the AEpos transduced population in the blood. Again, no significant difference was observed between the lineage distribution in blood between the first cohort (n = 4) and the second cohort (n = 8). Notably, no difference in spleen weights were observed between AE and GFP mice and the spleen architecture appeared normal by standard histological techniques. A reduction in the proportion of AEpos transduced cells located in the spleen as compared with the BM was also observed, however due to great variation between individual mice in both control and experimental cohorts, this was not quantified.
Figure 2.
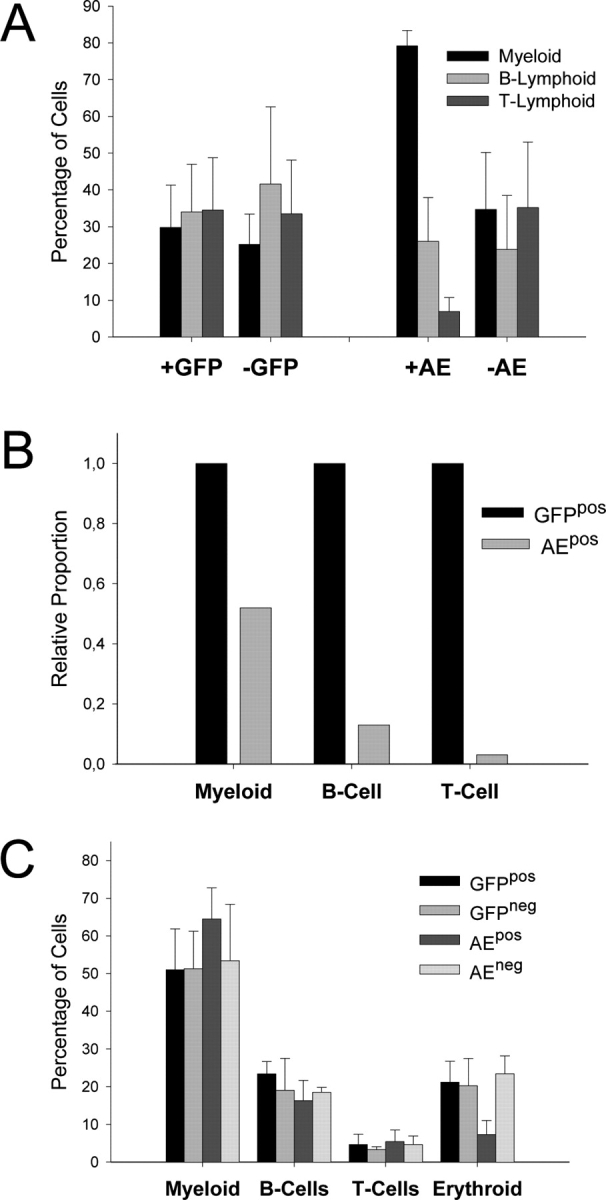
Number and lineage distribution of AEpos and GFPpos cells in blood and BM. (A) Lineage distribution within the transduced (pos) and nontransduced (neg) populations of blood from AE and GFP mice (n = 12 and n = 7, respectively) was determined by measuring lineage-specific marker expression against GFP expression. Expression of CD11b (MAC-1), CD45R (B220), and a CD4/CD8 cocktail was used to determine the percentage of myeloid, B and T lymphoid cells, respectively, within a given population. Shown are mean values with SDs. (B) The relative decrease in cell numbers in the AEpos blood population for each particular lineage as compared with the expected value is depicted. The expected value (set at 1.0 for each lineage) is based on the lineage distribution of GFPpos cells in the blood. (C) Lineage distribution of transduced and nontransduced cell populations in BM of GFP and AE mice (n = 7 and n = 10, respectively) was determined as described above. Expression of TER119 was used to define cells of the erythroid lineage.
AML1-ETO Expression Perturbs Maturation of B Cell Lymphopoiesis in BM.
The observation that AEpos transduced cells are underrepresented in the blood as compared with the BM suggested that either lineage differentiation was blocked or that lineage-committed cells were unable to egress the BM due to inefficient maturation. To address this issue, the lineage distribution and the maturation status of cells in the BM was determined by FACS® analysis. As the lymphoid lineage was greatly reduced in AEpos population in the blood, we first assessed their numbers and cell surface markers. As depicted in Fig. 2 C, no significant difference in the proportion of B cells was observed in the AEpos fraction as compared with either the AEneg fraction in the same group of mice, or the GFPpos fraction in control mice. However, a striking difference in the expression level of the B220 surface marker expression was observed in AEpos cells from mice of both the first and second cohorts. A representative analysis is shown in Fig. 3 A. Whereas the untransduced AEneg cell population showed relative equal levels of a B220med and B220hi cells, the AEpos cells were almost exclusively B220med. Equal proportions of B220hi and B220med were present in the GFPpos transduced population from control mice, ruling out a bias due to the retrovirus per se (Fig. 3 A). This difference in expression levels between AEpos versus AEneg lymphoid cells was also observed with CD45, which is a product of the same gene encoding B220, but due to alternative splicing does not contain the B cell specific epitope present in B220. Notably, the levels of CD45 expression on other cell types was not reduced (unpublished data).
Figure 3.
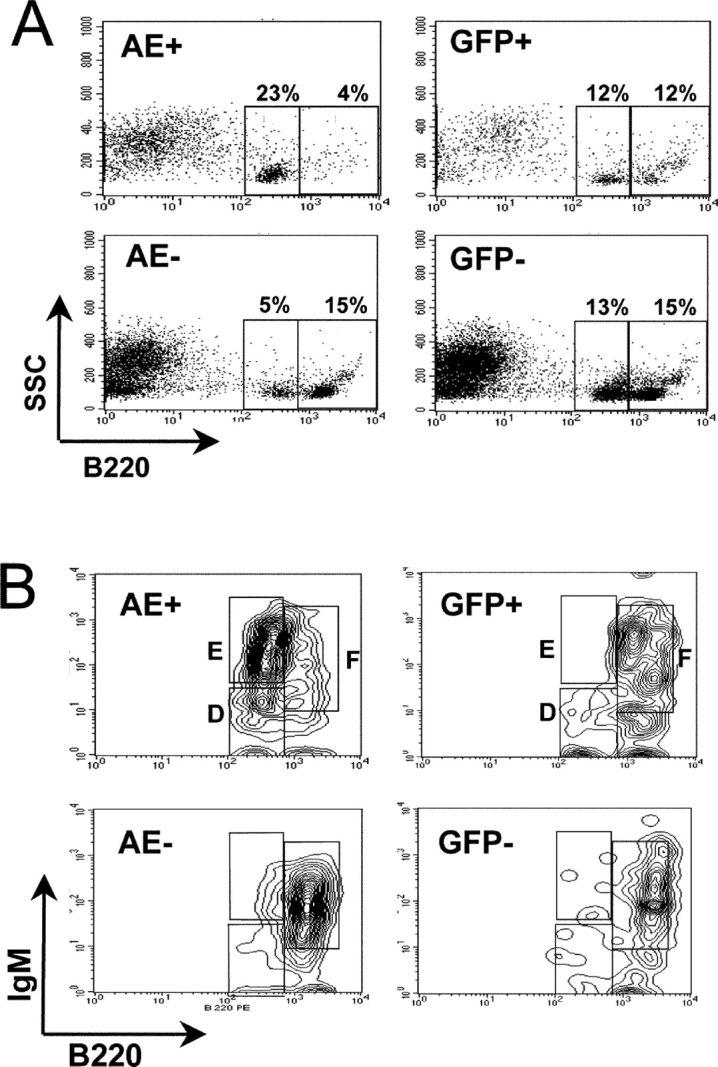
B cells expressing AML1-ETO in the BM are impaired in maturation at the immature B cell stage. (A) Flow cytometric analysis of the cell surface marker CD45RA (B220) on BM cells from representative mice receiving transplants of FMEV/AE- or FMEV/GFP-infected cells. GFP expression was used to gate nontransduced (AEneg or GFPneg) or transduced (AEpos or GFPpos) cells. The ratio of B220med to B220hi cells (circled populations) for AEpos cells (7.9 ± 3.7; n = 7) was significantly higher (P < 0.001) than AEneg (1.1 ± 0.9; n = 7) and control GFPpos cells (1.1 ± 1.0; n = 6). Furthermore, this increase was observed in the AEpos fraction of animals from both the first and second cohorts (n = 4 and 3, respectively). (B) Histograms displaying the different B cell maturation stages based on IgM and B220 expression levels. BM cells from transplanted mice were gated for GFPpos/AEpos or GFPneg/AEneg expression. Fraction D, E, and F, corresponding to preB cells, immature and mature B cells, respectively, as defined by Hardy et al. (reference 52) are indicated.
B220med expression is a characteristic of late pro-B, pre-B, and immature B cells (52), thus cells were stained with antibodies against IgM and CD43, two cell surface markers that can resolve these differentiation stages. The majority (74%; mean value of three mice) of the B220pos AEpos cell fraction were IgMhi CD43neg B220med and thus can be classified as immature B cells (Fraction E; reference 52); in contrast, these immature B cells made up only 10% (mean value of four mice) of the B220pos fraction of AEneg or GFPpos controls (Fig. 3 B). Indeed, the majority (65%) of cells in the control transduced (GFPpos) or untransduced populations were mature B cells (B220hi CD43neg IgMmed-hi; Fraction F), which made up only a small proportion of the AEpos population.
In summary, B cell maturation of AEpos transduced cells is impaired at the final stages of maturation. This impairment appears not to be absolute, however, as an average of 47.1% of the B220 positive cells in the AEpos fraction in the blood expressed high levels of B220 (n = 6). Although this is significantly lower than in the AEneg population of the same mice (87%) or the GFPpos fraction of control mice (90.7%; n = 4), end maturation does occur. No obvious correlation between AML1-ETO levels (as measured by GFP levels) and maturation levels were discernible, but this possibility cannot be entirely ruled out. Finally, histological examination of BM did not reveal a significant increase in apoptotic or necrotic cell death, thus we postulate that the accumulation of AEpos immature B cells contribute to the increased cellularity observed in the BM (see below).
AML1-ETO Mice Exhibit a Reduction in Erythropoiesis Coupled with an Increase in Granulopoiesis Shifted to Immature Cell Types.
Although the proportion of lymphoid cells in BM did not vary significantly between the different transduced and nontransduced populations, a striking decrease in the proportion of erythroid cells was seen in the AEpos population (Fig. 2 C). In contrast to GFPpos or AEneg BM cells, which contained a mean of 21.2% and 23.4% erythroid cells, respectively, this lineage made up only 7.3% of the AEpos population (P < 0.001 for both controls). A relative increase in granulopoiesis accompanied the erythrocytic hypoplasia in the AEpos transduced fraction in BM, with an ∼10% increase over the mean proportion of myeloid cells in the nontransduced AEneg and control transduced GFPpos populations (Fig. 2 C). Microscopial inspection of BM sections of mice containing >25% AEpos transduced cells confirmed the medullary granulocytosis and decrease in erythropoiesis (Fig. 4, A and B). Granulocytic hypercellularity was observed in the BM in 4 out of 5 FMEV/AE transduced mice examined, which was confirmed by immunohistochemical demonstration of myeloperoxidase (unpublished data). Thus, both granulopoiesis and, to a lesser degree, lymphopoiesis is stimulated, whereas erythropoiesis is significantly suppressed.
Figure 4.
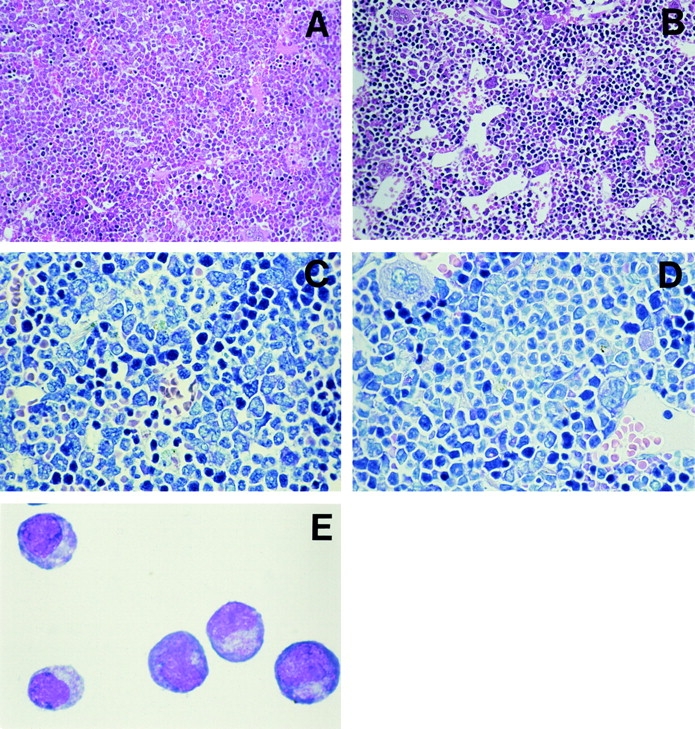
Morphological findings in mice transduced with AML1-ETO. (A) Sternal BM of a mouse with 41% AEpos cells from the second cohort showing considerable hypercellularity associated with left shifted granulocytosis and reduced numbers of erythroid cells (note the compressed and obscured sinusoidal vascular system). (B) In comparison, the marrow of an control animal is of normal cellularity with regular granulocytic and erythrocytic compartments and a distended, well-defined sinusoidal vascular system. Higher magnification reveals increased numbers of blast-like and immature granulocytic cells in the BM of a the FMEV/AE transduced mouse (C) as compared with control mouse (D). (E) Typical myeloblast morphology of cells picked from an immature colony and cytocentrifuged onto a glass slide. (A and B) H&E, magnification: ×180. (C and D) Modified Giemsa stain, magnification: ×450. (E) Wright-Giemsa stain, original magnification: ×500.
Closer examination of BM sections and cytospins from FMEV/AE–infected mice also revealed a shift to immature granulopoiesis, with an increased proportion of blast forms (promyelocytes and myeloblasts) (Fig. 4, C and D). Differential counts of 500–1,000 nucleated cells revealed an average of 10.1% blasts (n = 3), as compared with 3.1% (n = 3) blasts in GFP-infected controls. As only 20–41% AEpos cells are present in the BM, the relative increase in blast cells in the transduced cells must be higher. A reduction in mature myeloid cells was confirmed by analysis of Gr-1 expression levels by flow cytometry, where the transduced and nontransduced population could be examined independently. The level of Gr-1 (Ly-6G) expression is directly correlated with granulocyte differentiation and maturation (53). As representatively shown in Fig. 5, Gr-1 expression levels were consistently reduced by a mean factor of 6.8 on AEpos cells as compared with AEneg cells in both the first and second cohorts (n = 2 and 3, respectively), as determined by mean fluorescence intensities after staining. In contrast, no significant difference in Gr-1 expression levels was observed between GFPpos and GFPneg cells (n = 6) in control animals.
Figure 5.
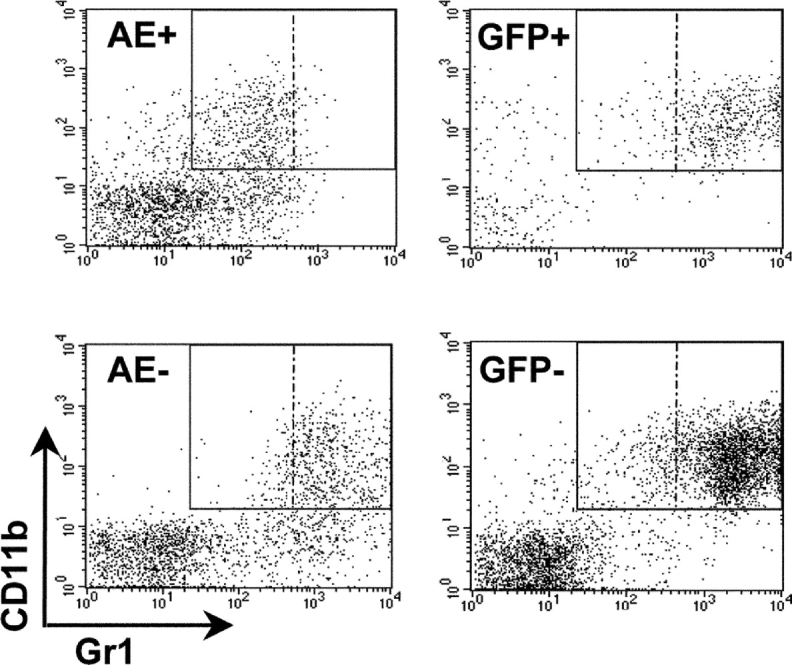
A shift to more immature forms in the granulocytic compartment is confirmed by expression levels of the GR-1 antigen. Representative FACS® profile of BM cells from transplanted mice as described in Fig. 3. The mean fluorescence of GR-1 expression was reduced by a mean factor of 6.8 ± 4.4 in AEpos cells (n = 5) as compared with the nontransduced AEneg cells, in contrast to the GR-1 mean fluorescence of GFPpos and GFPneg, which did not vary (ratio of 1.1 ± 0.1; n = 6).
AML1-ETO BM Cells Contain a High Percentage of In Vitro Clonogenic Blasts.
To assess the observed changes in myeloerythroid differentiation, in vitro colony assays were performed to determine if increased numbers of myeloid progenitors and/or decrease levels of erythroid progenitors could be confirmed. BM cells from either FMEV/AE or FMEV/GFP mice were sorted for GFPpos expression and plated in methylcellulose under conditions that detect both myeloid and erythroid progenitors. As a second control, unsorted cells from FMEV/GFP were also assayed. At day 9, a fourfold increase in total colony numbers was observed in plates containing AEpos BM cells as compared with control GFPpos cells sorted and assayed in parallel (Table II). The majority of the AEpos colonies (92%) displayed a unique but consistent morphology, characterized by a tightly compacted center surrounded by a few dispersed cells. Representative colonies were picked and the cell morphology analyzed microscopically. These colonies contained predominately myeloblasts and promyelocyes (>75%), with some macrophages and occasional myelocytes and basophilic granulocytes (Fig. 4 E). In >50 colonies of this morphology that were screened, only rare metamyelocytes and segmented and banded neutrophils were observed. Thus, the AEpos BM population contains a large number of clonogenic myeloid progenitors that are impaired in differentiation. Colony assays were only performed with BM cells from mice from the second cohort (between 2.5 and 4.5 mo after transplantation) and not the first cohort, so we cannot exclude that secondary transplantation has resulted in the selection of this aberrant population.
Table II.
Clonogenic Progenitor Assay Reveals a Differentiation Block in Myeloid Progenitors
| BMa
|
No. of colonies per 105 cellsb
|
Mean distribution of mature colonies (%)
|
|||||
|---|---|---|---|---|---|---|---|
| Immature | Mature | CFU-Mix | CFU-GM | CFU-G | CFU-M | BFU-E | |
| AEpos | 196 ± 60 | 17 ± 7 | 6.1 | 19.3 | 7.2 | 42.0 | 24.2 |
| GFPpos | 0 | 49 ± 22 | 2.3 | 14.9 | 20.15 | 29.4 | 33.8 |
| Unsortedc | 0 | 50 ± 28 | 8.2 | 15.2 | 17.7 | 29.4 | 31.2 |
BM was isolated from femurs and tibiae and then either sorted for eGFP expression or assayed directly.
The mean value and SD from three independent mice for each group performed in triplicate at two different cell concentrations are presented.
Unsorted cells from FMEV-GFP–infected transplants.
Reduced levels of mature day 9 colonies were also observed in AEpos as compared with GFPpos controls (Table II), however, it is difficult to assess the significance of this, as the large numbers of immature colonies may have inhibited their growth. Variations in the distribution of these mature colonies with regard to colony-forming type was also seen, with a decrease in CFU-G (P = 0.07) and a relative increase in CFU-M (P = 0.01). No relative difference in the proportion of BFU-E and CFU-GM colonies were observed, suggesting that AML1-ETO either effects the proliferation of these lineages at later stages of differentiation or that both cell types are reduced at similar levels.
AML1-ETO Inhibits Myeloid and Lymphoid Differentiation of ICSBP-deficient BM Cells and Imparts a Selective Repopulating Ability In Vivo.
The results presented above demonstrate that expression of AML1-ETO impairs myeloblast differentiation. However, the BM environment is nonpermissive for the growth and expansion of these differentiation impaired cells. Thus, we sought to determine if the deletion of the gene encoding ICSBP, which is often downregulated in AML myeloblasts (41), would collaborate with AML1-ETO expression to induce an acute myeloid disorder. BM cells of ICSBP−/− mice were infected with FMEV/AE and FMEV/GFP and used to transplant lethally irradiated ICSBP+/+ mice. A second cohort of mice was established after 90 d as described above.
Strikingly, the AEpos ICSBP−/− population had a selective growth advantage in vivo over the nontransduced cells, which was not observed in the GFPpos ICSBP−/− population. Although both groups of the first cohort received 15% transduced cells, mice receiving FMEV/AE transduced cells had an average of 36.7 ± 22.2% transduced cells after 5–7 mo (n = 12), in contrast to an average of 4.9 ± 7.7% in mice receiving cells transduced with FMEV/GFP (n = 11; P < 0.001 in a unpaired Student's t test). The rapid loss of GFPpos ICSBP−/− BM cells, as compared with the relative stable levels of GFPpos ICSBP+/+ BM cells in the first and second cohorts, probably reflects a competitive disadvantage of the ICSBP−/− cells during repopulation as compared with ICSBP+/+ BM cells that were added during the transplantation to ensure sufficient levels of erythropoiesis in the regenerating BM, which is deficient in ICSBP−/− mice (37). Nevertheless, the AEpos transduced ICSBP−/− cells could compete with wild-type cells for repopulation.
Mice receiving either FMEV/AE or FMEV/GFP ICSBP−/− BM with >6.5% transduction frequencies were killed and screened for hematopoietic abnormalities in the blood, spleen, and BM between 2 and 6 mo after transplantation. Similar to the ICSBP+/+ mice, blood leukocyte levels and differentiation counts were normal in both FMEV/AE and FMEV/GFP ICSBP−/− transduced mice. In addition, all FMEV/AE mice of both the first and second cohorts (n = 3 and 6, respectively) showed the same block in lymphohematopoietic differentiation as described above for ICSBP+/+-BM transplanted animals, as evidenced by the number of transduced cells in the blood versus BM (0.33 ± 0.18 versus 1.45 ± 0.47 for GFP controls), as well as an increase in B220med over B220hi expression levels in BM cells (5.3 ± 2.2 for the AEpos population, in contrast to 1.1 ± 1.1 and 1.3 ± 0.7 for AEneg and GFPpos populations, respectively) and a 5.4 reduction in the mean fluorescence of Gr-1 levels in the AEpos versus AEneg BM populations.
ICSBP Deficiency Synergizes with AML1-ETO to Transform Myeloblasts in the BM and to Induce Granulocytic Sarcoma-like Neoplasias.
In striking contrast to the ICSBP+/+ BM recipients, mice receiving ICSBP−/− BM showed markedly increased granulopoiesis with enhanced blast cell formation in the BM, as revealed by histological and immunohistochemical staining and confirmed by FACS® analysis (Fig. 6, A and B, and unpublished data). Differential counts of cytospins of FMEV/AE transduced BM cells revealed an average of 25.3 ± 5.4% blasts (n = 4), as compared with 5 ± 1.2% (n = 3) in control FMEV/GFP transduced ICSBP−/− BM mice. In addition to their increased numbers, blasts displayed an abnormal morphology (e.g., enhanced basophilic cytoplasm and enlarged nuclei with pathological chromatin structures) (Fig. 6 C). In further support of a neoplastic transformation, immature myeloid cells were observed invading periosseous tissue of the bone (e.g., sternum and femur) via the nutrient foramen to form granulocytic sarcoma-like tumors in one out of three mice from the first cohort and three out of five mice from the second cohort (Fig. 6 D). Significantly, granulocytic sarcomas are observed at a relatively high incidence in t(8;21) AML (54, 55), and can be the first manifestation of AML (56, 57). Similar lesions were not observed in control mice receiving FMEV/GFP infected ICSBP−/− BM, in mice receiving FMEV/AE ICSBP+/+ BM, nor in nontransplanted ICSBP−/− mice maintained in parallel in our facilities. Despite the unrestricted local proliferation of BM cells observed in these AE ICSBP−/− mice, the spleen, liver, and blood remained devoid of infiltrating blasts. Their absence in the spleen was striking, as lymphohematopoietic neoplasias often arise simultaneously in the splenic red pulp and BM of mice (unpublished data). One explanation is offered by the observation that BM provides the more favored microenvironment for granulopoiesis after transplantation and thus may offer the necessary milieu for the transformation potential of AML1-ETO.
Figure 6.
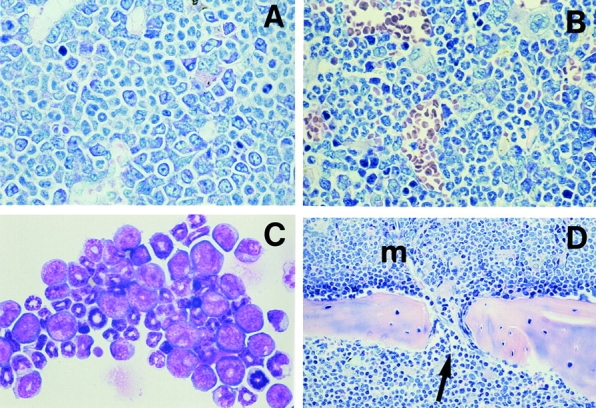
Morphological findings in ICSBP−/− mice transduced with AML1-ETO. (A and B) The sternal BM of an AML-ETO1 transduced ICSBP−/− mouse with 40% AEpos cells (A) is characterized by left-shifted granulocytosis with many blasts and immature granulocytic cells and the absence of erythrocytes, while a nontransduced ICSBP−/− control animal (B) reveals a moderate and mature granulocytosis, and a moderately decreased erythrocytosis which is constitutive for the ICSBP null mutation. (C) Greatly increased numbers of myeloblast, promyelocytes, and myelocytes are present in a cytocentrifuged cell preparation of BM cells from an AML1-ETO transduced ICSBP−/− mouse with 55% AEpos cells as compared with the more mature granulocytic differentiation of nontransduced ICSBP−/− marrow cells. (D) Predominately immature granulocytic BM cells invading via a nutrient foramen of the sternum (arrow) into the periosseous connective tissue (bottom section of the microphotograph). (A and B) Modified Giemsa stain, magnification: ×450. (C) Wright-Giemsa stain, magnification: ×500. (D) Modified Giemsa stain, magnification: ×210.
Approximately 12 mo after transplantation, three mice from each of the first cohorts receiving FMEV/AE-infected ICSBP−/− or ICSBP+/+ BM were killed and analyzed. Mice receiving AE-transduced wild-type BM showed the same phenotype as mice analyzed six months earlier, with no signs of disease progression. In contrast, in one of the three mice receiving AE-transduced ICSBP−/−, transformed myeloid blast cells were no longer restricted to the bone, but had infiltrated the liver, kidneys, and spleen in large numbers, forming granulocytic sarcomas exhibiting myeloperoxidase immunopositivity (unpublished data). The relatively long latency and clonality of this tumor, as assessed by the retroviral integration site, supports the hypothesis that additional mutation(s) is/are required for the transformed myeloblasts, generated by the synergistic action of AML1-ETO expression and ICSBP deficiency, to progress to a full-blown AML.
Discussion
Several groups have attempted to demonstrate the oncogenic potential of AML1-ETO expression in inducible transgenic mouse models, but no overt alterations in vivo have been observed or reported previously (28–31). In contrast, by introducing AML1-ETO by retroviral transduction into murine adult HSC, the origin of t(8;21) (33) and the target cell of AML transformation (32, 34), we could demonstrate that normal hematopoiesis is disrupted at several levels by AML1-ETO expression in vivo. Similar results were also recently reported by de Guzman et al. using a retroviral approach (58). Although it is not entirely clear why transgenic animals models failed to manifest these hematopoietic lesions, several factors must be considered. Clearly, in addition to expression levels, both the temporal and spatial patterns of expression are sure to be decisive factors in the ensuing phenotype. In our system, expression levels are relatively constant in all hematopoietic cell compartments, including the HSC (35), thus ensuring that expression is achieved in the appropriate target cells. Another contributing factor to the success of a retroviral approach, may be due to the perturbation of the HSC pool during the BM cell transplantation. The activation of this compartment may trigger the necessary cellular milieu for the transforming action of AML1-ETO. Although Higuichi et al. also transplanted BM from their knock-in mice into syngeneic recipients (31), subtle changes in BM cells expressing AML1-ETO may have gone undetected due to their inability to track these cells with a marker such as GFP. Finally, the potential importance of slight variations in the genetic background in these different mouse models cannot be ignored.
The most striking observation in our mouse model of AML1-ETO was the inhibition of maturation of both lymphoid and myeloid cells. This was initially most evident in the lymphoid lineage, where the majority (>85%) of AML1-ETO cells remained at immature stages of differentiation, as revealed by strikingly low levels in the blood. We could demonstrate that the impaired B cell maturation in cells expressing AML1-ETO occurred at a late stage, as evidenced by the accumulation of B220med IgMhi phenotype in the AML1-ETO–expressing BM. Maturation was not completely blocked, however, as mature B220hi B cells in the AEpos fraction were found in the blood, albeit at low levels. Preliminary results also suggest that the T cell development is also blocked at a late stage of maturation, as double positive (DP) CD4+/CD8+ thymocytes are present at normal levels in the AML1-ETO transduced population as compared with controls in the thymus (unpublished data), but very few AML1-ETO T cells are found in the blood. This hypothesis is supported by recent studies that have demonstrated the importance of the AML1/CBFβ complex in the transition and maturation of DP CD4+/CD8+ to single positive thymocytes (59, 60). Significantly, the knock-in transgenic model of Higuichi et al., also reported the unexpected absence of AML1-ETO transcripts in mature T cells (31). More important, although AML1-ETO transcripts are detectable in HSC with lymphohematopoietic potential in AML patients, they were not found in mature T cells and only detectable in B cells at a frequency one-tenth of that observed in monocytes (33). We postulate that mature B and T cells preferentially arise from HSCs not carrying t(8;21), due to the impaired maturation of these lineages in cells expressing AML1-ETO.
In addition to the maturation block in lymphopoiesis, a pronounced increase in granulopoiesis accompanied by a decrease in erythropoiesis was another consistent phenotype observed in AML1-ETO transduced BM. The latter phenotype must be a direct consequence of AML1-ETO expression and not a displacement effect due to granulocytosis, as a 70% reduction in erythroid cell proportions was specifically observed in AEpos cells and not in AEneg nontransduced cells. In addition to increased levels of granulopoiesis, a distinct left shift, characterized by the accumulation of more immature forms including blasts, was documented both by FACS® and immunohistochemical analysis. The differentiation defect in the myeloid lineage was most strikingly demonstrated in in vitro colony assays, where up to 92% of the clonogenic colonies contained primarily myeloblast or promyelocytic cells. Notably, and consistent with the t(8;21) AML phenotype (1), granulocytic differentiation was impaired but not completely blocked in our mouse model, as evidenced by the development of normal granulocytic colonies in vitro from AEpos-sorted cells, as well as GR-1hi–expressing cells in the blood and spleen in the AEpos fraction. Although it has been previously postulated that AML1-ETO exerts its effect by blocking granulocytic differentiation (18, 61–64), the use of leukemic cell lines, which have multiple mutations and limited differentiation potential, limited the interpretation of these earlier results. Interestingly, a left shift in granulopoiesis was not previously observed in transgenic AML1-ETO mouse models, but in vitro culturing of cells from these mice did reveal defects in myeloid maturation (23, 29). Thus, in vitro culture conditions seem to be more permissive for the manifestation of this defect. Importantly, a recent study in which human CD34+ enriched cells were transduced with a retroviral vector expressing AML1-ETO also noted a pronounced bias toward immature myeloid cells after in vitro culture (65).
What is the molecular basis of the maturation block in both the lymphoid and myeloid lineages and disruption of normal erythropoiesis? Although several myeloid genes are regulated by CBF (6, 7), accumulating evidence suggest that AML1-ETO may exert its effect by protein interaction with other transcription factors, such as PU.1 and C/EBPα (18, 19, 64), both of which play a pivotal role in myeloid differentiation, as demonstrated in knock-out mice (66–68). This idea is supported by the recently observed downregulation of the gene encoding C/EBPα (an autoregulatory gene) in 30% of AML patients with a t(8;21) karotype (21). With regard to the lymphoid compartment, previous studies have shown that components of the antigen receptor in both B and T cells are regulated by CBF (5, 69). Thus, the downregulation of one or more of these genes by AML1-ETO transcriptional repression may interfere with lymphoid maturation by reducing receptor levels. Alternatively, genes encoding proteins that act downstream of antigen stimulation, e.g. the gene encoding the B cell lymphocytic kinase blk that is also regulated by CBF (70), may be important targets. Recent findings have also shown that AML1 can interact with the B cell transcription factor Pax5 (70). Potential target genes of AML1-ETO transformation in erythroid cells have recently been identified (71, 72). Our mouse model provides a reproducible system to further define the target genes of AML-ETO transformation.
Despite the profound effects on differentiation and maturation observed in BM cells expressing AML1-ETO cells, the mice remained aleukemic, underlining the importance of secondary mutations in leukemia induction. The recent finding that the gene encoding the tumor suppressor ICSBP is downregulated in the hematopoietic compartment in a large percentage of AML patients (41) prompted us to determine if the differentiation impairment of AML1-ETO–transduced myeloid cells would collaborate with ICSBP deficiency to induce leukemia. AML1-ETO expression in the ICSBP−/− background resulted in the accumulation of morphologically altered myeloblasts (20–30%) in the BM and the generation of granulocytic sarcoma-like tumors, both pathognomonic features of t(8;21) AML (1, 54, 55). Thus, the ICSBP defect collaborates with AML1-ETO to expand and transform a myeloblast population in vivo. To our knowledge, this is the first identification of a tumor suppressor that cooperates with AML1-ETO in myeloid transformation. Although the exact mechanism by which ICSBP regulates growth control is not known, genes regulating apoptosis (e.g., Bcl-x) (39) and growth signaling (e.g., Dab2) (73) have been implicated as targets of ICSBP regulation. It has recently been proposed that ICSBP expression is downregulated by the CML oncogene BCR-ABL, thus it will be of interest to determine if ICSBP expression is a downstream target of other tyrosine kinases, e.g., Flt3 and c-kit, which have been found to be mutated in patients with t(8;21) AML (74, 75).
In summary, our results clearly demonstrate that AML1-ETO expression can disrupt the proliferation and maturation controls of several hematopoietic lineages in vivo. Furthermore, we have identified ICSBP deficiency as a secondary mutation that cooperates with AML1-ETO to induce localized neoplasias, characterized by the accumulation of >20% myeloblasts in the BM and the generation of granulocytic sarcoma-like tumors. Noteworthy, these myeloblastic transformants remain confined to the bone (except for one important exception), demonstrating the need for subsequent mutations to induce the expansion and dissemination of these cells into other organs, including the blood. Currently, we are using retroviral insertional mutagenesis in the ICSBP−/− model to define collaboration oncogenes of AML1-ETO–induced AML.
Acknowledgments
We gratefully acknowledge the technical support of Karin Heigl, Susanne Roscher, and Marion Ziegler and the help of Andreas Rimek with cell sorting. We also thank the “HOX group” in the laboratory of Wolfram Ostertag for stimulating discussions.
This work was supported from grants from the Deutsche Forschungsgemeinschaft, the Deutsche José Carreras Leukämie Stiftung, and the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (BioRegio Hamburg/ViSioN7). The Heinrich-Pette-Institut is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit.
Footnotes
Abbreviations used in this paper: AML, acute myelogenous leukemia; BM, bone marrow; CBF, core-binding factor; CML, chronic myelogenous leukemia; HSC, hematopoietic stem cell; IRES, internal ribosome entry site; PRE, posttranscriptional regulatory element; RHD, Runt homology domain.
References
- 1.Brunning, R., E. Matutes, N. Harris, G. Flandrin, J. Vardiman, J. Bennett, and D. Head. 2001. Acute myeloid leukemias. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. E. Jaffe, N. Harris, H. Stein, and J. Vardiman, editors. IARC Press, Lyon. 77–80.
- 2.Erikson, P., J. Gao, K.-S. Chang, T. Look, E. Whisenant, S. Raimondi, R. Lasher, J. Trujillo, J. Rowley, and H. Drabkin. 1992. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, AML-ETO, with similarity to Drosophila segmentation gene, runt. Blood. 80:1825–1831. [PubMed] [Google Scholar]
- 3.Miyoshi, H., K. Shimizu, T. Kozu, N. Maseki, Y. Kaneko, and M. Ohki. 1991. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc. Natl. Acad. Sci. USA. 88:10431–10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyoshi, H., T. Kozu, K. Shimizu, K. Enomoto, N. Maseki, Y. Kaneko, N. Kamada, and M. Ohki. 1993. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 12:2715–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leiden, J. 1993. Transcriptional regulation of T cell receptor genes. Annu. Rev. Immunol. 11:539–570. [DOI] [PubMed] [Google Scholar]
- 6.Tenen, D., T. Hromas, J. Licht, and D.-E. Zhang. 1997. Transcription factors, normal myeloid development, and leukemia. Blood. 90:489–519. [PubMed] [Google Scholar]
- 7.Licht, J. 2001. AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene. 20:5660–5679. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa, E., M. Maruyama, H. Kagoshima, M. Inuzuka, J. Lu, M. Satake, K. Shigesada, and Y. Ito. 1993. PEBP2/PEA2 represents a new family of transcription factor homologous to the products of the Drosophila runt and the human AML1 gene. Proc. Natl. Acad. Sci. USA. 90:6859–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyers, S., J. Downing, and S. Hiebert. 1993. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for DNA binding and protein-protein interactions. Mol. Cell. Biol. 13:6336–6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitabayashi, I., A. Tokoyama, K. Shimazu, and M. Ohki. 1998. Interaction and functional cooperation of the leukemia-associated factors AML1 and p300 in myeloid cell differention. EMBO J. 17:2994–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amann, J., J. Nip, D. Strom, B. Lutterbach, H. Harada, N. Lenny, J. Downing, S. Meyers, and S. Hiebert. 2001. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol. Cell. Biol. 21:6470–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lutterbach, B., J. Westendorf, B. Linggi, A. Patten, M. Moniwa, J. Davie, K. Huynh, V. Bardwell, R. Lavinsky, M. Rosenfeld, et al. 1998. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol. Cell. Biol. 18:7176–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang, J., T. Hoshino, R. Redner, S. Kajigaya, and J. Liu. 1998. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3A/HDAC1 complex. Proc. Natl. Acad. Sci. USA. 95:10860–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelmetti, V., J. Zhang, M. Fanelli, S. Minucci, P. Pelicci, and M. Lazar. 1998. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol. Cell. Biol. 18:7185–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank, R., J. Zhang, S. Meyers, S. Hiebert, and S. Nimer. 1995. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML 1B. Oncogene. 11:2667–2674. [PubMed] [Google Scholar]
- 16.Meyers, S., N. Lenny, and S. Hiebert. 1995. The t(8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Mol. Cell. Biol. 15:1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang, D., C. Hetherington, S. Meyers, K. Rhoades, C. Larson, H.-M. Chen, S. Hiebert, and D. Tenen. 1996. CAAT enhancer-binding protein (C/EBP) and AML1(CBFα2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol. Cell. Biol. 16:1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westendorf, J., C. Yamamoto, N. Lenny, J. Downing, M. Selsted, and S. Hiebert. 1998. The t(8;21) fusion product, AML-1-ETO, associates with C/EBP-α, inhibits C/EBP-α-dependent transcription, and blocks granulocytic differentiation. Mol. Cell. Biol. 18:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petrovick, M., S. Hiebert, A. Friedman, C. Hetherington, D. Tenen, and D.-E. Zhang. 1998. Multiple functional domains of AML1: PU.1 and C/EBPα synergize with different regions of AML1. Mol. Cell. Biol. 18:3915–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao, S., R. Frank, J. Zhang, Y. Miyazaki, and S. Nimer. 1999. Functional and physical interactions between AML1 proteins and an ETS protein, MEF: implications for the pathogenesis of t(8;21)-positive leukemias. Mol. Cell. Biol. 19:3635–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pabst, T., B. Mueller, N. Harakawa, C. Schoch, T. Haferlach, G. Behre, W. Hiddemann, D.-E. Zhang, and D. Tenen. 2001. AML1-ETO downregulates the granulocytic differentiation factor C/EBP-α in t(8;21) myeloid leukemia. Nat. Med. 7:1–8. [DOI] [PubMed] [Google Scholar]
- 22.Giese, K., C. Kingsley, J. Kirschner, and R. Grosschedl. 1995. Assembly and function of a TCR alpha enhancer complex is dependent on LEF-1-induced DNA bending and multiple protein-protein interactions. Genes Dev. 9:995–1008. [DOI] [PubMed] [Google Scholar]
- 23.Okuda, T., Z. Cai, S. Yang, N. Lenny, C.-J. Lyu, J. van Duersen, H. Harada, and J. Downing. 1998. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood. 91:3134–3143. [PubMed] [Google Scholar]
- 24.Yergeau, D., C. Hetherington, Q. Wang, P. Zhang, A. Sharpe, M. Binder, M. Marín-Padilla, D. Tenen, N. Speck, and D.-E. Zhang. 1997. Embryonic lethality and impairment of haematopoiesis in mice heterozygous for an AML1-ETO fusion gene. Nat. Genet. 15:303–306. [DOI] [PubMed] [Google Scholar]
- 25.Wang, Q., T. Stacy, M. Binder, M. Marín-Padilla, A. Sharpe, and N. Speck. 1996. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc. Natl. Acad. Sci. USA. 93:3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang, Q., T. Stacy, J. Miller, A. Lewis, T.-L. Gu, X. Huang, J. Bushweller, J.-C. Bories, F. Alt, G. Ryan, et al. 1996. The CBFβ subunit is essential for CBFα2 (AML1) function in vivo. Cell. 87:697–708. [DOI] [PubMed] [Google Scholar]
- 27.Okuda, T., J. van Deursen, S. Hiebert, G. Grosveld, and J. Downing. 1996. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 84:321–330. [DOI] [PubMed] [Google Scholar]
- 28.Buchholz, F., Y. Refaeli, A. Trump, and J. Bishop. 2000. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep. 1:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhoades, K., C. Hetherington, N. Harakawa, D. Yergeau, L. Zhou, L.-Q. Liu, M.-T. Little, D. Tenen, and D.-E. Zhang. 2000. Analysis of the role of AML1-ETO in leukemogenesis, using an inducible transgenic mouse model. Blood. 96:2108–2115. [PubMed] [Google Scholar]
- 30.Yuan, Y., L. Zhou, T. Miyamato, H. Iwasaki, N. Harakawa, C. Hetherington, S. Burel, E. Lagasse, I. Weissman, K. Akashi, and D.-E. Zhang. 2001. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc. Natl. Acad. Sci. USA. 98:10398–10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higuichi, M., D.O. Brien, P. Kumaravelu, N. Lenny, E.-J. Yeah, and J. Downing. 2002. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell. 1:63–74. [DOI] [PubMed] [Google Scholar]
- 32.Bonnet, D., and J. Dick. 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3:730–737. [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto, T., I. Weissman, and K. Akashi. 2000. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc. Natl. Acad. Sci. USA. 97:7521–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raskind, W., and P.J. Fialkow. 1987. The use of cell markers in the study of human hematopoietic neoplasia. Adv. Cancer Res. 49:127–167. [DOI] [PubMed] [Google Scholar]
- 35.Baum, C., W. Ostertag, C. Stocking, and D. von Laer. 2001. Retroviral vector design for cancer gene therapy. 2nd ed. Gene Therapy of Cancer. E. Lattime and S. Gerson, editors. Academic Press, San Diego. 3–29.
- 36.Stocking, C., and C. Baum. 1997. Gene transfer in hematopoietic cells. Bailliére's Clinical Hematology. A. Whetton, editor. Baillière Tindall, London. 445–466.
- 37.Holtschke, T., J. Löhler, Y. Kanno, T. Fehr, N. Giese, F. Rosenbauer, J. Lou, K.-P. Knobeloch, L. Gabriele, J. Waring, et al. 1996. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 87:307–317. [DOI] [PubMed] [Google Scholar]
- 38.Scheller, M., J. Foerster, C. Heyworth, J. Waring, J. Lohler, G. Gilmore, R. Shadduck, T. Dexter, and I. Horak. 1999. Altered development and cytokine responses of myeloid progenitors in the absence of transcription factor, interferon consensus sequence binding protein. Blood. 94:3764–3771. [PubMed] [Google Scholar]
- 39.Gabriele, L., J. Phung, J. Fukumoto, D. Segal, I.-M. Wang, P. Giannakakou, N. Giese, K. Ozato, and H. Morse, III. 1999. Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein. J. Exp. Med. 190:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt, M., A. Hochhaus, A. Nitsche, R. Hehlmann, and A. Neubauer. 2001. Expression of nuclear transcription factor interferon consensus sequence binding protein in chronic myeloid leukemia correlates with pretreatment risk features and cytogenetic response to interferon-α. Blood. 97:3648–3650. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt, M., S. Nagel, J. Proba, C. Thiede, M. Ritter, J. Waring, F. Rosenbauer, D. Huhn, B. Wittig, I. Horak, and A. Neubauer. 1998. Lack of interferon consensus binding protein (ICSBP) transcripts in human myeloid leukemia. Blood. 91:22–29. [PubMed] [Google Scholar]
- 42.Hao, S., and R. Ren. 2000. Expression of interferon consensus sequence binding protein (ICSBP) is downregulated in bcr-abl-induced murine chronic myelogenous leukemia-like disease, and forced coexpression of ICSBP inhibits bcr-abl-induced myeloproliferative disorder. Mol. Cell. Biol. 20:1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baum, C., S. Hegewisch-Becker, H.-G. Eckert, C. Stocking, and W. Ostertag. 1995. Novel retroviral vectors for efficient expression of the multidrug resistance (mdr-1) gene in early hematopoietic cells. J. Virol. 69:7541–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hildinger, M., K. Abel, W. Ostertag, and C. Baum. 1999. Design of 5′untranslated sequences in retroviral vectors developed for medical use. J. Virol. 73:4083–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schambach, A., H. Wodrich, M. Hildinger, J. Bohne, H. Krausslich, and C. Baum. 2000. Context dependence of different modules for posttranscriptional enhancement of gene expression from retroviral vectors. Mol. Ther. 2:435–445. [DOI] [PubMed] [Google Scholar]
- 46.Morita, S., T. Kojima, and T. Kitamura. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063–1066. [DOI] [PubMed] [Google Scholar]
- 47.Beyer, W., M. Westphal, W. Ostertag, and D. von Laer. 2002. Oncoretrovirus and lentivirus vectors pseudotyped with lymphocytic choriomeningitis virus glycoprotein: generation, concentration, and broad host range. J. Virol. 76:1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wahlers, A., M. Schwieger, Z. Li, D. Meier-Tackmann, C. Lindemann, H.-G. Eckert, D. von Laer, and C. Baum. 2001. Influence of multiplicity of infection and protein stability on retroviral vector-mediated gene expression in hematopoietic cells. Gene Ther. 8:477–486. [DOI] [PubMed] [Google Scholar]
- 49.Asou, H., S. Tahiro, K. Hamamoto, A. Otsuji, K. Kita, and N. Kamada. 1991. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood. 77:2031–2036. [PubMed] [Google Scholar]
- 50.Meyer, J., M. Jücker, W. Ostertag, and C. Stocking. 1998. Carboxyl-truncated STAT5β is generated by a nucleus-associated serine protease in early hematopoietic progenitors. Blood. 91:1901–1908. [PubMed] [Google Scholar]
- 51.Baum, C., K. Itoh, J. Meyer, C. Laker, Y. Ito, and W. Ostertag. 1997. The potent enhancer activity of the polycythemic strain of spleen focus-forming virus in hematopoietic cells is governed by a binding site for Sp1 in the upstream control region and by a unique enhancer core motif, creating an exclusive target for PEBP/CBF. J. Virol. 71:6323–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hardy, R., C. Carmack, S. Shinton, J. Kemp, and K. Hayakawa. 1991. Resolution and characterization of pro-B and pre-B-cell stages in normal mouse bone marrow. J. Exp. Med. 173:1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hestdal, K., F. Ruscetti, J. Ihle, S. Jacobsen, C. Dubois, W. Kopp, D. Longo, and J. Keller. 1991. Characterization and regulation of RB6-8C5 antigen expression on murine bone marrow cells. J. Immunol. 147:22–28. [PubMed] [Google Scholar]
- 54.Tallman, M., D. Hakimian, J. Shaw, G. Lissner, E. Russell, and D. Variakojis. 1993. Granulocytic sarcoma is associated with the 8;21 translocation in acute myeloid leukemia. J. Clin. Oncol. 11:690–697. [DOI] [PubMed] [Google Scholar]
- 55.Abe, R., H. Umezu, T. Uchida, S. Kariyone, N. Maseki, Y. Kaneko, and M. Sakurai. 1986. Myeloblastoma with an 8;21 chromosome translocation in acute myeloblastic leukemia. Cancer. 58:1260–1264. [DOI] [PubMed] [Google Scholar]
- 56.Meis, J., J. Butler, B. Osborne, and J. Manning. 1986. Granulocytic sarcoma in non-leukemic patients. Cancer. 58:2697. [DOI] [PubMed] [Google Scholar]
- 57.Neiman, R., M. Barcos, C. Berard, H. Bonner, R. Mann, R. Rydell, and J. Bennett. 1981. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer. 48:1426–1437. [DOI] [PubMed] [Google Scholar]
- 58.de Guzman, C., A. Warren, Z. Zhang, L. Gartland, P. Erikson, H. Drabkin, S. Hiebert, and C. Klug. 2002. Hematopoietic stem cell expansion and distinct myeloid developmental abnormalities in a murine model of the AML1-ETO translocation. Mol. Cell. Biol. 22:5506–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayashi, K., W. Natsume, T. Watanabe, N. Abe, N. Iwai, H. Okada, Y. Ito, M. Asano, Y. Iwakura, S. Habu, et al. 2000. Diminution of the AML1 transcription factor function causes differential effects on the fates of CD4 and CD8 single-positive T cells. J. Immunol. 165:6816–6824. [DOI] [PubMed] [Google Scholar]
- 60.Hayashi, K., N. Abe, T. Watanabe, M. Obinata, M. Ito, T. Sato, S. Habu, and M. Satake. 2001. Overexpression of AML1 transcription factor drives thymocytes into the CD8 single-positive lineage. J. Immunol. 167:4957–4965. [DOI] [PubMed] [Google Scholar]
- 61.Sakakura, C., Y. Yamaguchi-Iwai, M. Satake, S.-C. Bae, A. Takahashi, E. Ogawa, A. Hagiwara, T. Takahashi, A. Murakami, K. Makino, et al. 1994. Growth inhibition and induction of differentiation of t(8;21) acute myeloid leukemia cells by the DNA-binding domain of PEBP2 and the AML1/MTG8(ETO)-specific antisense oligonucleotide. Proc. Natl. Acad. Sci. USA. 91:11723–11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahn, M., G. Huang, S. Bae, J. Wee, W. Kim, and Y. Ito. 1998. Negative regulation of granulocyte differentiation in the myeloid precursor cell line 32Dcl3 by ear-2, a mammalian homolog of Drosophila seven-up, and a chimeric leukemogenic gene, AML1/ETO. Proc. Natl. Acad. Sci. USA. 95:1812–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitabayashi, I., K. Ida, F. Morohoshi, A. Yokoyama, N. Mitsuhashi, K. Shimizu, N. Nomura, Y. Hayashi, and M. Ohki. 1998. The AML1-MTG8 leukemic fusion protein forms a complex with a novel member of the MTG8(ETO/CDR) family, MTGR1. Mol. Cell. Biol. 18:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burel, S., N. Harakawa, L. Zhou, T. Pabst, D. Tenen, and D. Zhang. 2001. Dichotomy of AML1-ETO functions: apoptosis versus block of differentiation. Mol. Cell. Biol. 21:5577–5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mulloy, J., J. Cammenga, K. MacKenzie, F. Berguido, A. Moore, and S. Nimer. 2002. The AML1-ETO fusion protein promotes the expansion of human hematopoietic stem cells. Blood. 99:15–23. [DOI] [PubMed] [Google Scholar]
- 66.McKercher, S., B. Torbett, K. Anderson, G. Henkel, D. Vestal, H. Baribault, M. Klemsz, A. Freeney, G. Wu, C. Paige, and R. Maki. 1996. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- 67.Scott, E., M. Simon, J. Anastasi, and H. Singh. 1994. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 265:1573–1577. [DOI] [PubMed] [Google Scholar]
- 68.Zhang, D., P. Zhang, N. Wang, C. Hetherington, G. Darlington, and D. Tenen. 1997. Absence of G-CSF signaling and neutrophil development in C/EBP α deficient mice. Proc. Natl. Acad. Sci. USA. 94:569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Erman, B., M. Cortes, B. Nikolajczyk, N. Speck, and R. Sen. 1998. ETS-core binding factor: a common composite motif in antigen receptor gene enhancers. Mol. Cell. Biol. 18:1322–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Libermann, T., Z. Pan, Y. Akbarali, C. Hetherington, J. Boltax, D. Yergeau, and D.-E. Zhang. 1999. AML1 (CBFa2) cooperates with B cell-specific activating protein (BSAP/Pax5) in activation of the B cell-specific BLK gene promoter. J. Biol. Chem. 274:24671–24676. [DOI] [PubMed] [Google Scholar]
- 71.Harada, H., Y. Harada, D. O'Brien, D. Rice, C. Naeve, and J. Downing. 1999. HERF1, a novel hematopoiesis-specific RING finger protein, is required for terminal differentiation of erythroid cells. Mol. Cell. Biol. 19:3808-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harada, Y., H. Harada, J. Downing, and A. Kimura. 2001. A hematopoietic-specific transmembrane protein, Art-1, is possibly regulated by AML1. Biochem. Biophys. Res. Commun. 284:714–722. [DOI] [PubMed] [Google Scholar]
- 73.Rosenbauer, F., A. Kallies, M. Scheller, K.-P. Knobeloch, C. Rock, M. Schwieger, C. Stocking, and I. Horak. 2002. Disabled-2 is transcriptionally regulated by interferon consensus sequence binding protein and augments macrophage spreading and adhesion. EMBO J. 21:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beghini, A., P. Peterlongo, C. Ripamonti, L. Larizza, R. Cairoli, E. Morra, and C. Mecucci. 2000. C-kit mutations in core binding factor leukemias. Blood. 95:726–727. [PubMed] [Google Scholar]
- 75.Yokota, S., H. Kiyoi, M. Nakao, T. Iwai, T. Misawa, T. Okujda, Y. Sonodo, T. Abe, K. Kahsima, Y. Matsuo, and T. Naoe. 1997. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodyspastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia. 11:1605–1609. [DOI] [PubMed] [Google Scholar]