

Abstract
M11L, an antiapoptotic protein essential for the virulence of the myxoma poxvirus, is targeted to mitochondria and prevents the loss of mitochondrial membrane potential that accompanies cell death. In this study we show, using a cross-linking approach, that M11L physically associates with the mitochondrial peripheral benzodiazepine receptor (PBR) component of the permeability transition (PT) pore. Close association of M11L and the PBR is also indicated by fluorescence resonance energy transfer (FRET) analysis. Stable expression of M11L prevents the release of mitochondrial cytochrome c induced by staurosporine or protoporphyrin IX (PPIX), a ligand of the PBR. Transiently expressed M11L also prevents mitochondrial membrane potential loss induced by PPIX, or induced by staurosporine in combination with PK11195, another ligand of the PBR. Myxoma virus infection and the associated expression of early proteins, including M11L, protects cells from staurosporine- and Fas-mediated mitochondrial membrane potential loss and this effect is augmented by the presence of PBR. We conclude that M11L regulates the mitochondrial permeability transition pore complex, most likely by direct modulation of the PBR.
Keywords: apoptosis, mitochondria, PK11195, protoporphyrin IX, Poxviridae
Introduction
Apoptotic cell death is central to many aspects of the survival of multicellular organisms (1). For example, initiation of a cellular suicide program after infection serves as an effective means of curtailing viral propagation and provides an early host defense against infection. However, numerous viruses have developed effective strategies to circumvent apoptosis in order to extend the life of the infected cell to their advantage (2, 3). One such virus, with diverse antiapoptotic strategies, is myxoma virus (4).
Myxoma virus is a poxvirus of rabbits and the causative agent of the lethal disease, myxomatosis. This virus encodes M11L, a key virulence factor that inhibits apoptosis of infected cells, particularly monocytes (5, 6). The dramatic contribution of M11L to viral pathogenesis was recognized during characterization of an M11L knockout virus, which cannot produce M11L protein. Infection of rabbits with the M11L knockout virus resulted in highly attenuated disease symptoms and full recovery. In contrast, wild-type virus infection resulted in rapid and complete mortality (7). M11L is also antiapoptotic when expressed autonomously from other viral proteins, is targeted to the cytoplasmic surface of the outer mitochondrial membrane and prevents loss of mitochondrial membrane potential (ΔΨm)* in response to staurosporine-induced apoptosis (6). M11L is therefore functionally implicated in modulating the mitochondrial cell death control point.
Mitochondria play a key role in many apoptotic cascades and apoptotic cell death is frequently accompanied by two marked mitochondrial changes that can occur independently, but frequently occur concurrently (8–11). One of these changes is the release into the cytosol of proteins that are normally retained in the intermembrane space between the inner and outer mitochondrial membranes. Proteins involved in this redistribution include several apoptotic effectors and cofactors that transduce apoptotic signals upon entering the cytoplasm. The other notable mitochondrial change is the abrupt loss of the electrochemical potential (ΔΨm) normally maintained across the inner membrane. Either one of these two mitochondrial events could presage commitment to cell death and consequently, the exact mitochondrial perturbation required to ensure the death of a cell remains controversial.
The loss of ΔΨm occurs as a result of the sudden opening of a mitochondrial megachannel referred to as the permeability transition (PT) pore. The PT pore is thought to consist of three core protein subunits: the voltage-dependent anion carrier (VDAC) that resides in the outer mitochondrial membrane, the adenine nucleotide transporter (ANT) in the inner membrane, and cyclophilin D that is associated with the matrix surface of the ANT (12). Pro- and antiapoptotic members of the Bcl-2 family of proteins are reported to physically associate with the ANT and VDAC subunits and to regulate the PT pore (13, 14). However, the molecular mechanisms underlying the effects of the Bcl-2 proteins are presently unclear. Interestingly, several viral apoptotic modulators also regulate the PT pore by direct interaction with its components (15, 16). In particular, vMIA of cytomegalovirus (17) and Vpr encoded by HIV (18, 19), associate with the ANT, while HBx, encoded by hepatitis B virus, binds to VDAC (20). The PT pore incorporates several accessory proteins including creatine kinase, Hexokinase II, and the peripheral benzodiazepine receptor (PBR; references 8, 12, 21, and 22). The role of these proteins in apoptosis modulation remains poorly understood.
The PBR is an 18-kD integral membrane protein which resides in the outer mitochondrial membrane and copurifies with the VDAC and ANT components of the PT pore (23). PBR protein is concentrated in contact sites between the inner and outer mitochondrial membranes (24), as are PT pore assemblies and Bcl–2 (25). Microscopy has revealed that the PBR is localized on the cytoplasmic surface of mitochondria and is present in pore-like clusters of 4–6 molecules. Each cluster is predicted to be associated with one VDAC molecule (26).
The PBR was first characterized as a benzodiazepine-interacting protein present in certain peripheral tissues (27) and has since been shown to have a wide tissue distribution, being abundant in the adrenals, kidney, heart, and reproductive tract, and also present in nonneuronal brain tissue (21, 22). Within leukocyte subsets, the highest concentrations of the PBR are found in monocytes and polymorphonuclear cells (28, 29). The PBR shows highest affinity for the isoquinoline carboxamide analogue, 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide (PK11195) and the diazepine 4′-chlorodiazepam (Ro 5–4864; references 21 and 22). Porphyrins are the major endogenous ligands of the PBR, with the heme precursor, protoporphyrin IX (PPIX) having the strongest binding interaction (30). The pharmacological properties and tissue distribution of the PBR distinguishes this receptor from the ‘central type’ benzodiazepine receptor that is present on the surface of cells of the central nervous system and preferentially binds the theraputic diazepines (21).
Numerous physiological functions have been proposed for the PBR, including roles in the regulation of cellular growth and proliferation, heme biosynthesis, cholesterol transport, and steroidogenesis (21, 22) in addition to an ability to potentiate inflammation (31, 32). Two lines of evidence also implicate the PBR in apoptosis modulation. First, elevated levels of PBR expression confer protection against apoptosis. Specifically, in hematopoeitic cells, PBR overexpression correlates with resistance to H2O2- and UV-mediated cell death (28, 33). Interestingly, enforced expression of PBR also protects neonatal mice from the otherwise lethal effects of a neurovirulent, proapoptotic strain of Sindbis virus (34). Second, PBR ligands can induce apoptosis, presumably by binding to the PBR and interfering with the function of the PT pore. In particular, exogenous addition of PPIX (35) or PK11195 (36–39) to certain cell types, or addition of PPIX or PK11195 in combination with proapoptotic agents to other cell types (40–46) has been shown to potentiate ΔΨm loss and cell death. Also, PPIX mediates apoptotic death of cells in culture and within brain tumors during photodynamic therapy (47, 48). Notably, Bcl-2 cannot prevent the proapoptotic effects of PK11195 (37, 43–45) or PPIX (35). Therefore, Bcl-2 most likely does not prevent apoptosis by physical interaction with the PBR subunit of the PT pore.
Despite being implicated in apoptotic events, clear evidence to show involvement of the PBR in apoptosis modulation has remained elusive. In this study we show that M11L, an anti-apoptotic, mitochondria-localized protein, physically associates with the PBR subunit of the PT pore and counteracts the proapoptotic effects of PBR ligands. In this way, M11L forestalls transduction of cell death signals at the mitochondrial control point. This is the first report of an apoptotic modulator binding directly to the PBR, thereby identifying a direct role for this PT pore subunit in apoptosis.
Materials and Methods
Cells and Culture Conditions.
The Cos-7 monkey kidney, HeLa human cervical carcinoma, THP-1 human monocyte, and Jurkat T lymphocyte (clone E6) cell lines were obtained from American Type Culture Collection. Cos-7 cells were cultured in DMEM and the other cell lines were cultured in RPMI medium. All media were supplemented with 10% FBS, 200 U/ml penicillin, and 200 μg/ml streptomycin. THP-1 medium also contained 0.1 mM 2-mercaptoethanol. BGMK monkey kidney cells (7) were grown in DMEM supplemented with 10% newborn calf serum, 200 U/ml penicillin, and 200 μg/ml streptomycin. The development of Rat2puro and Rat2M11L cell lines has been described previously (6) and these cells were cultured in DMEM containing 10% FBS, 200 U/ml penicillin, 200 μg/ml streptomycin, and 2.5 μg/ml puromycin. Jurkat-PBR cells (reference 28; supplied by Dr. P. Casellas, Sanofi Research, Montpellier, France) were grown in RPMI medium containing 10% FBS, 200 U/ml penicillin, 200 μg/ml streptomycin, 0.1 mM 2-mercaptoethanol, and 0.6 mg/ml G418.
Cross-linking Assays.
Cos-7 cells (5 × 105) were infected at a multiplicity of infection (MOI) of 10 with a recombinant vaccinia virus that overexpresses M11L (VVM11L; reference 49) or control vaccinia virus (VV601). Cells were then replenished with DMEM lacking serum and containing 35S-labeled cysteine and methionine at a final concentration of 0.1 mCi/ml. 12 h after infection, the cells were harvested and each sample was resuspended in 1 ml cold hypotonic 100 mM potassium phosphate buffer, pH 7.4. Samples were subjected to 35 strokes in a dounce homogenizer and then divided into two aliquots. A DMSO solution of the hydrophobic, noncleavable cross-linker disuccinimidyl suberate (DSS; Pierce Chemical Co.) was added to one aliquot of each sample to a final concentration of 1.25 mM. Samples were incubated for 2 h at 4°C on a nutator and 0.8 ml NP-40/deoxycholate lysis buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 2 mM EDTA, and 1 mM PMSF) was added. Both monomeric and complexed M11L was immunoprecipitated using a polyclonal rabbit anti-M11L antibody (6) and identified by SDS-PAGE and autoradiography. Alternatively, THP-1 or HeLa cells were infected with VV601 or VVM11L, or infection-permissive BGMK cells were infected at an MOI of 10 with either myxoma virus (vMyxlac) or the M11L knockout virus (vMyxM11L−; reference 7). Infected cell samples were subjected to the cross-linking and immunprecipitation procedure described, followed by immunoblot analysis.
Cross-linking and Coimmunoprecipitation Assays.
THP-1 cells (2 × 106) were infected at a MOI of 10 with VVM11L or VV601. 12 h after infection, cells were subjected to hypotonic lysis, dounce homogenization, and cross-linking as described above, except in this case, the hydrophobic, cleavable analogue of DSS, Dithiobis(succinimidylpropionate) (DSP; Pierce Chemical Co.) was used at a final concentration of 0.125 mM. Samples were divided into duplicates and resuspended in 0.8 ml NP-40/deoxycholate lysis buffer. Duplicate samples were then immunoprecipitated with a polyclonal rabbit anti-M11L antibody (6) and cross-linked complexes in one sample from each duplicate were cleaved into their monomeric components by incubation at 65°C in SDS-PAGE loading buffer containing 100 mM dithiothreitol and 0.2% 2-mercaptoethanol. In parallel, the same procedure was conducted on samples immunoprecipitated with a mouse monoclonal anti-PBR antibody that has specificity for human PBR (29) (supplied by Dr. P. Casellas, Sanofi Research). PBR and M11L proteins were detected using SDS-PAGE and immunoblot analysis.
Fluorescence Resonance Energy Transfer Analysis.
A construct to allow the expression of M11L appended to the C terminus of enhanced yellow fluorescent protein (EYFP) was generated by replacing the enhanced green fluorescent protein (EGFP) coding sequence in the plasmid pEGFP-M11L (6) with the EYFP coding sequence obtained from the plasmid pEYFP-C1 (CLONTECH Laboratories, Inc.). EYFP-M11L or EYFP alone were transiently expressed in HeLa cells by transfection of the expression plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Each transfection was conducted with 1 μg of DNA and 1 μl liposome reagent. Transfected cells were cultured in Petri dishes in which the base had been replaced with a glass coverslip (number 1; Fischer) and stained with the PBR-specific fluorescent compound FGIN-1–27-NBD (reference 50; Alexis Biochemicals Corp.) at a dilution of 0.7 μM immediately before visualization.
An acceptor photobleaching approach (51) was used to investigate potential interactions between FGIN-1–27-NBD and EYFP-M11L. Images were collected on a ZEISS Laser Scanning Confocal microscope (LSM 510 META NLO) system mounted on a ZEISS Axiovert 100M microscope using a 40× lens (NA 1.3, Fluar). The Meta detector of the system allowed recording of spectral information of the specimen with 10.7-nm bandwidth resolution. Images were recorded using a pixel resolution of 0.15–0.2 μm and a pixel time of 3.2 μs. The gain and offset values were optimized such that no pixel saturation was observed and the minimum values in the image were near zero. This allowed the full dynamic range of the system to be used. Additionally, images were acquired with a 12-bit pixel depth and an averaging factor of 2 to reduce noise. Image series were recorded using the time-lapse function of the instrument. Three images were recorded before acceptor (EYFP) photobleaching then, two or three more images were recorded after photobleaching in order to calculate the dequenching (fluorescence resonance energy transfer [FRET] efficiency). At least two pairs of EYFP expressing and non-EYFP expressing cells were included in each field analyzed. One EYFP-expressing and one non-EYFP expressing cell was photobleached with the full power of the 514 nm laser line at iterations of between 50 to 150 (mostly 100). The cells that were not photobleached served as an internal control for each image.
The same field-of-view was also imaged with the sequential scanning mode using the Meta detector as a bandpass filter (channel mode). For the NBD signal, a 458 nm-laser line excitation was used and the emission signal was collected between 462 and 537 nm. For EYFP, a 514 nm excitation laser line was used and the emission signal was collected between 526 and 719 nm. An image was acquired before photobleaching using this setting. The effectiveness of photobleaching was monitored by acquiring another image using a higher gain for the EYFP detector channel after photobleaching.
A common difficulty with FRET analysis is bleed through of the donor emission spectrum into the acceptor channel. Using the spectral imaging capability of the Meta detector, serial spectral data ranging from 462.4 to 633.6 nm were recorded with the 458 nm laser line as an excitation source. Reference spectra for EYFP-M11L and FGIN-1–27-NDB in living cells were recorded with the same conditions as with the FRET image acquisitions. Then either these reference spectra or spectra generated from regions of interest were used with the linear-unmixing method (52) to separate the signals. This method permitted the use of the full range of the signal instead of using a narrow bandwidth of the spectra. After linear unmixing, regions of interest were drawn over four cells chosen in each field: a pair that expressed EYPF-M11L and another pair that did not express EYPF-M11L. One cell from each pair was photobleached and the other cell in each pair was not subjected to photobleaching. The average pixel intensity of individual cells was calculated using the LSM Software. The data were exported to Microsoft Excel for further processing.
Theoretically, if FRET occurs, photobleaching of the EYFP-M11L acceptor will induce de-quenching of the donor (FGIN-1–27-NBD) signal. The major limitation of FRET analysis in this experimental system was caused by photobleaching of the FGIN-1–27-NBD compound during normal image acquisition using the 458 nm laser line (by an average amount of 7% per image acquisition under the experimental conditions). To a lesser degree, the FGIN-1–27-NBD signal was also reduced during photobleaching of the acceptor (EYFP) with the 514 nm laser line (∼40% for 100 iterations). The matter was further complicated by a weak excitation of EYFP molecules with the 458 nm laser line used for FRET analysis. To circumvent these problems, we designed the experiments so that internal controls were included in each individual image. Parallel bleaching of another non-EYFP-M11L expressing, FGIN-1–27-NBD-labeled cell with the 514 nm laser line in the same image field served as an intrinsic control for correction of these bleaching problems in the experiments performed. Therefore, the final FRET efficiency was calculated based on the ratio of the average intensity of FGIN-1–27-NBD channel in the photobleached EYFP-M11L–expressing cell versus that of FGIN-1–27-NBD channel in the photobleached non-EYFP-M11L–expressing cell. This approach not only corrected for the photobleaching problems discussed above but it also served as an internal control for the correction of potential fluctuations of laser intensity. Additionally, in order to compensate for intensity variations between cells, the intensity of each cell were expressed as percentage of intensity of the first time point of the same cell. Then, the ratios between FRET and control cells were calculated based on these normalized intensity values.
Briefly, the algorithm used for data processing was: (a) the intensity in the FGIN-1–27-NBD channel at each time point was normalized to the percentage of its intensity of the first time point. (b) The ratios between the averaged pixel intensity of the FGIN-1–27-NBD channel (donor molecule) in the EYFP-M11L-expressing cell and that of the EYFP-M11L nonexpressing cell for each time were calculated. (c) The ratios were plotted for each time lapse point. Other pairs of EYFP and non-EYFP expressing cells in the same image field that were not photobleached served as controls for FRET. The same image processing process was applied to these pairs of cells and the results plotted in the same graph for comparison purposes. We also performed control experiments using cells labeled with the donor (FGIN-1–27-NBD) alone, the acceptor (EYFP-M11L) alone and unlabeled cells. In addition, we analyzed cells that were transfected with the pEYFP-C1 vector and expressed EYFP only and were then stained with FGIN-1–27-NBD. No dequenching phenomena were observed in any of these controls (unpublished data).
Cytochrome c Translocation.
For the determination of cytochrome c translocation into the cytoplasm, Rat2puro or Rat2M11L cells (106) were treated with 5 μM staurosporine (Sigma-Aldrich) for 2, 3, or 4 h, or treated with an equivalent amount of DMSO for 4 h as a solvent control. Cells were harvested by trypsinization and lysed at 4°C in 100 μl digitonin lysis buffer (75 mM NaCl, 1 mM NaH2PO4, 8 mM Na2HPO4, 250 mM sucrose, and 95 μg/ml digitonin). Supernatant fractions were isolated by centrifugation of samples at 15,000 g and 4°C for 15 min and subjected to SDS-PAGE and immunoblot analysis. Cytochrome c present in the supernatant fractions was detected using an anti-cytochrome c antibody (BD Biosciences) and actin levels were determined in the same samples using an anti-actin antibody (Santa Cruz Biotechnology, Inc.). To determine the effects of PPIX on Rat2puro or Rat2M11L cells, the cells were first permeabilized with digitonin lysis buffer, then treated with 20 μM PPIX or DMSO solvent alone at 37°C for 30 min before analyzing the cytosolic levels of cytochrome c or actin as before.
Measurement of ΔΨm in Transiently Transfected HeLa Cells.
HeLa cells (5 × 105) were transfected with the following previously described EGFP constructs: pEGFP-M11L, pEGFP-mt, pEGFP-stop, and pEGFP-Bcl-2 (6). Respectively, these chimeric proteins consist of an NH2-terminal EGFP tag appended with full length M11L, the mitochondrial targeting sequence of M11L alone, a nontargeted, truncated variant of M11L or full-length Bcl-2. HeLa cells were transfected as described above and after 24 h, were split into replicate wells and the cells were subjected to apoptosis induction.
The first treatment involved incubation of cells with staurosporine (Sigma-Aldrich) or staurosporine together with the PBR ligand PK11195 (Sigma-Aldrich) for 4 h. Staurosporine and PK11195 were used at concentrations of 5 and 120 μM, respectively. Replicate cell samples were also individually treated with staurosporine or PK11195 alone. All samples were adjusted to contain the same amount of DMSO solvent and cells treated with PK11195 alone served as controls. The second treatment involved incubation of cells with another PBR ligand, protoporphyrin IX (PPIX; Sigma-Aldrich), at a final concentration of 25 or 30 μM for 2 h. Control cells were incubated with ethanol solvent alone.
During the last half hour of both treatments, the orange fluorescent compound, tetramethyl rhodamine, ethyl ester (TMRE; Molecular Probes), was added to cells at a concentration of 0.2 μM. TMRE is a mitochondrion-specific potentiometric dye that displays increased uptake into mitochondria and increased fluorescence intensity in proportion to increased ΔΨm (53). Cells were harvested by trypsinization, then EGFP and TMRE fluorescence were analyzed in live cells by flow cytometry using a Becton Dickinson FACSCalibur™ instrument equipped with an argon-ion laser with 15 mW of excitation at 488 nm. TMRE fluorescence was collected with a 585/42 nm bandpass filter and green EGFP fluorescence was measured using a 530/30 mn band pass filter. A total of 10,000 cells were analyzed per sample with fluorescence signals at logarithmic scale under conditions that minimized bleed through. The percentage of EGFP-expressing cells that were also TMRE positive was calculated for the gated cell population in both control and treated cells. The reduction in the percentage of dual positive cells after treatment was calculated in each case and the results of three independent experiments (±SD) are shown.
Measurement of ΔΨm in Infected Jurkat Cells.
Jurkat or Jurkat-PBR cells (5 × 105) were mock-infected or infected at a MOI of 20 with either myxoma virus (vMyxlac) or the M11L knockout virus (vMyxM11L−). 10 h after infection, cells were treated with 2 μM staurosporine dissolved in DMSO or an equivalent amount of DMSO only. Alternatively, activating anti-Fas antibody (clone CH11; Upstate Biotechnology) was added directly to the cell culture medium at a concentration of 250 μg/μl. After incubation for 3 h, TMRE was added to cells at a concentration of 0.1 μM and fluorescence was analyzed in live cells by flow cytometry as before using a 585/42 nm bandpass filter. A total of 10,000 cells were analyzed per sample with fluorescence signals at logarithmic scale. The reduction in the percentage of TMRE-positive cells in treated versus control cells was calculated for the gated cell population. The results of three independent experiments (±SD) are shown.
Results
M11L Physically Interacts with the Mitochondrial Permeability Transition Pore.
Previous studies had shown that M11L is antiapoptotic, localizes to mitochondria, and blocks ΔΨm loss in these organelles after apoptosis induction (6). We therefore reasoned that this viral protein is likely to impact the mitochondrial cell death control point, perhaps by direct interaction with protein subunits of the permeability transition pore. To investigate this possibility, M11L was overexpressed in Cos-7 cells by infection with a vaccinia virus expression vector (VVM11L). These cells, together with cells infected with control vaccinia virus (VV601), were radiolabeled with 35S methionine/cysteine for 12 h and lysed by resuspension in hypotonic phosphate buffer and dounce homogenization. Samples were divided into duplicates and the hydrophobic cross-linker, DSS, was added to one sample from each duplicate. In these studies, the use of a hydrophobic cross-linker was found to be essential for demonstrating M11L binding interactions, perhaps because such interactions require the correct conformation of proteins embedded in membranes. Monomeric and complexed M11L was immunoprecipitated and analyzed by SDS-PAGE and autoradiography.
As shown in Fig. 1 A, M11L was only immunoprecipitated from cells infected with the M11L-expressing VVM11L vaccinia virus vector (lanes 1 and 3) and was not detected in cells infected with control vaccinia virus VV601 (lanes 2 and 4). In addition, when proteins were cross-linked, a complex of ∼36 kD was observed (lane 1) and was not observed in the absence of cross-linking (lane 3). The presence of M11L in a 36 kD complex was verified using a similar approach involving infection of THP-1 and HeLa cells with VVM11L or VV601 followed by lysis, cross-linking, immunoprecipitation, SDS-PAGE, and immunoblot analysis. As shown in Fig. 1 B, monomeric and complexed M11L was detected in THP-1 and HeLa cells infected with VVM11L (lanes 1 and 3) but was absent from control VV601-infected cells (lanes 2 and 4). Formation of this cross-linked M11L complex was also observed when experiments were conducted using Cos-7 and HepG2 cells (unpublished data). The same procedure was applied to BGMK cells infected with M11L-expressing myxoma virus (vMyxlac) and the M11L knockout myxoma virus (vMyxM11L−). Monomeric and cross-linked M11L was observed in cells infected with myxoma virus but not the M11L knockout myxoma virus (Fig. 1 C, lanes 1 and 2).
Figure 1.
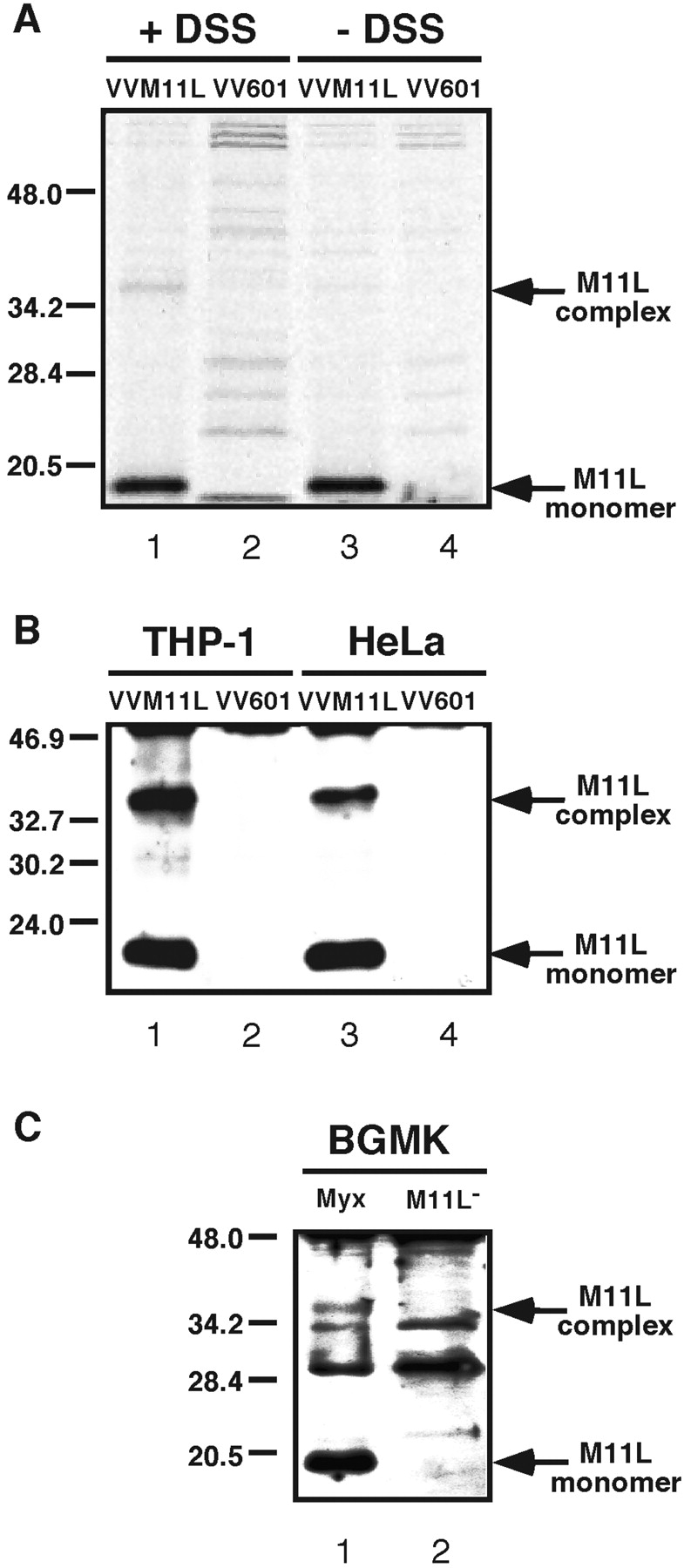
M11L can be cross-linked into a complex (A). Cos-7 cells were infected with the M11L-expressing vaccinia virus VVM11L or the control virus VV601 and labeled with 35S-methionine/cysteine. Proteins present in hypotonic cell lysates were cross-linked with the hydrophobic cross-linker DSS (+DSS) or left uncrosslinked (−DSS), before extraction with an NP-40/deoxycholate buffer, immunoprecipitation with anti-M11L antibody and analysis by SDS-PAGE and autoradiography. (B) THP-1 and HeLa cells infected with VVM11L or VV601 were subjected to hypotonic lysis and DSS cross-linking. M11L was immunoprecipitated as described and detected by SDS-PAGE followed by immunoblot analysis. (C) BGMK cells were infected with the vMyxlac parental myxoma virus (Myx) or the M11L knockout strain vMyxM11L− (M11L−) and again samples were subjected to hypotonic lysis, DSS cross-linking, and immunoprecipitation and analysis of M11L.
Formation of a 36-kD complex suggested that the 18 kD M11L protein associates with a protein of approximately the same molecular weight. The PBR subunit of the PT pore is 18 kD (23), and was therefore a candidate M11L-interacting protein. To investigate whether M11L does interact with the PBR, we conducted cross-linking experiments as described above, using THP-1 monocytes because these cells express a high concentration of the PBR (29). THP-1 monocytes were infected with VVM11L or VV601 and cross-linking was performed with DSP, the cleavable analogue of DSS.
Fig. 2 shows the results following immunoprecipitation of M11L (Fig. 2, A and B) or PBR (Fig. 2, C and D) from lysates of THP-1 cells infected with VVM11L (lanes 1 and 2) or control VV601 (lanes 3 and 4). Samples shown in lanes 1 to 4 were cross-linked before immunoprecipitation and samples in lanes 2 and 4 were subjected to an additional treatment with reducing agents to cleave cross-linked proteins. Control samples included were, in lane 5, M11L immunoprecipitated from VVM11L-infected cells in the absence of cross-linking and, in lane 6, a THP-1 whole cell lysate. M11L was detected in THP-1 cells infected with VVM11L (Fig. 2 A, lanes 1 and 2) but not in cells infected with VV601 (Fig. 2 A, lanes 3 and 4), as expected. The control samples also confirmed that M11L was immunoprecipitated from VVM11L infected cells (Fig. 2 A, lane 5) and was not detected in the THP-1 whole cell lysate (Fig. 2 A, lane 6), as expected. When the same blot was probed with the anti-PBR antibody, PBR was found to be present in samples immunoprecipitated with the anti-M11L antibody (Fig. 2 B, lanes 1 and 2), indicating that PBR coprecipitated with M11L. Monomeric PBR was detected even in the absence of cross-linker cleavage (Fig. 2 B, lane 1), indicating that non–cross-linked protein also coimmunoprecipitated with M11L. A higher level of PBR was identified in samples in which the cross-linker had been cleaved before SDS-PAGE analysis (Fig. 2 B, lane 2). PBR was also detected in the whole cell lysate, as expected (Fig. 2 B, lane 6).
Figure 2.

M11L forms a complex with the PBR. THP-1 cells were infected with VVM11L or VV601. Protein within hypotonic cell lysates was cross-linked with DSP, extracted with an NP-40/deoxycholate buffer, and immunoprecipitated with an anti-M11L (A and B) or anti-PBR (C and D) antibody. Samples subjected to cross-linker cleavage (+) or left uncleaved (−) were analyzed by SDS-PAGE and immunoblotting with either an anti-M11L (A and C) or anti-PBR (B and D) antibody. M11L immunoprecipitated from cells infected with VVM11L in the absence of cross-linking or a THP-1 cell whole cell lysate (WCL) were included as controls.
The reciprocal experiment, in which cross-linked PBR was immunoprecipitated from THP-1 cells infected with VVM11L and VV601 was also conducted. PBR was correctly detected in immunoprecipitates from infected THP-1 cells (Fig. 2 C, lanes 1–4) and in the THP-1 whole cell lysate (Fig. 2 C, lane 6). PBR was not detected in a control M11L immunoprecipitation in the absence of cross-linking (Fig. 2 C, lane 5). When the same blot was analyzed using an anti-M11L antibody, M11L was identified as having coprecipitated with PBR in VVM11L-infected cells (Fig. 2 D, lane 2). As expected, M11L was also detected in a control M11L immunoprecipitate (Fig. 2 D, lane 5) and was absent from samples infected with VV601 and from the THP-1 whole cell lysate (Fig. 2 D, lanes 3, 4, and 6). We conclude from these cross-linking experiments that M11L interacts directly with the PBR.
FRET Analysis of the PBR-M11L Association.
To obtain independent confirmation of a close association between M11L and the PBR, FRET between a EYFP-M11L chimeric protein and the PBR-binding fluorescent compound FGIN-1–27-NBD was assessed. FRET is a process whereby energy is transferred from a molecule (donor) to a neighboring molecule (acceptor) nonradiatively through dipolar–dipolar interactions (54, 55). FRET analysis therefore allows the detection of molecular interactions within a Förster radiance distance (typically less than 10 nm). The technology has been applied extensively for studying molecular interactions within cells (for reviews, see references 56 and 57). When FRET occurs, fluorescence quenching of the donor is observed as a result of donor energy being transferred to the acceptor. We used an acceptor photobleaching technique (51, 57) to investigate potential interactions between FGIN-1–27-NBD (donor) and EYFP-M11L (acceptor). The technique measures the emission of donor molecules before and after selective photobleaching of the acceptor. The increase, or dequenching, of the donor emission is a direct measure of the FRET efficiency.
Fig. 3 shows one representative field of cells out of a total of 38 fields used in FRET analysis. Fig. 3 A shows a HeLa cell monolayer stained with the PBR-specific fluorescent compound FGIN-1–27-NBD and represented in green pseudocolor. Fig. 3 B shows EYFP-M11L expression, represented in red pseudocolor, in the same monolayer. Fig. 3 C shows the unmixed signal using excitation with the 458 nm laser line and Fig. 3 D is the bright field image showing the cell morphology.
Figure 3.
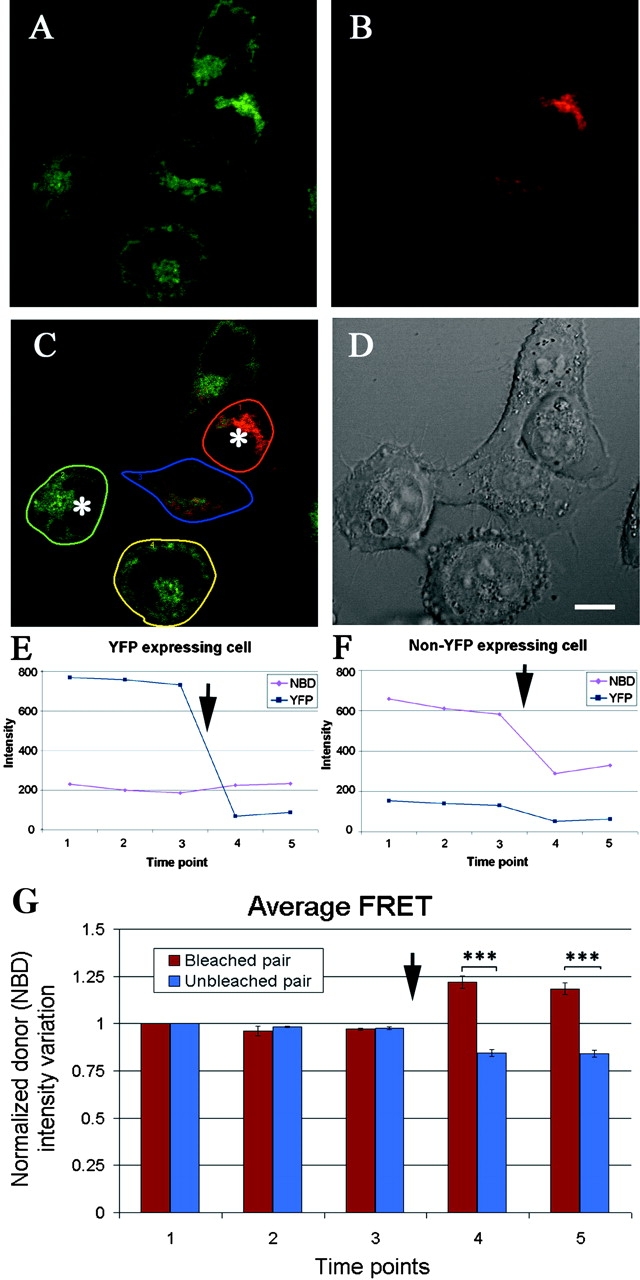
EYFP-M11L and fluorescently labeled PBR undergo FRET (A). Confocal image of a representative HeLa cell monolayer stained with FGIN-1–27-NBD (shown in green pseudocolor) acquired using a 548 nm excitation line and emission filter of 462–537 nm before photobleaching. (B) EYFP-M11L expression in the same monolayer (shown in red pseudocolor) acquired using a 515 nm excitation line and emission filter of 526–719 nm before photobleaching. (C) Unmixed signal obtained using a 458 nm laser line excitation. The cells marked with an asterisk were photobleached. Regions of interest were drawn over the cells to be analyzed. Here, regions designated by red and green are a pair of photobleached cells and regions designated by blue and yellow are an unbleached pair of cells. All cells are stained with FGIN-1–27-NBD and the cells designated by red and blue outlines express EYFP-M11L. (D) DIC image of the field of view showing cell morphology. Scale bar: 10 μm. (E) Intensity of EYFP–expressing cell (red region in C) before and after photobleaching. Notice the dequenching of NBD signal after photobleaching (arrow). (F) Intensity of the non-EYFP expressing cell (green in C) before and after photobleaching (arrow). (G) Averaged normalized intensity changes before and after photobleaching in 38 fields of cells analyzed (standard error, P < 0.0000000233 and 0.0000000186 for time points 4 and 5 respectively). The y axis represents the intensity variation of the FGIN-1-27-NBD (donor) channel. The x axis represents the sequence of time lapse points. Arrow indicates the bleaching point.
FRET was assessed in the cells shown in Fig. 3 C. The cell outlined in red, emitting FGIN-1–27-NBD and EYFP-M11L fluorescence and the cell outlined in green, emitting FGIN-1–27-NBD fluorescence only, were photobleached (asterisk). The cell outlined in blue, emitting FGIN-1–27-NBD and EYFP-M11L fluorescence and the cell outlined in yellow, emitting FGIN-1–27-NBD fluorescence only, were not photobleached. Fig. 3 E shows the intensity of the EYFP and FGIN-1–27-NBD fluorescence channels in the red-outlined cell and Fig. 3 F the readings in both channels for the green-outlined cell. In Fig. 3 E, after photobleaching of the EYFP-M11L acceptor fluorophore (arrow) an increase (dequenching) in the emission of the donor (FGIN-1–27-NBD) fluorophore was observed. In contrast, in a cell not expressing EYFP-M11L (Fig. 3 F), photobleaching resulted in a decrease in the NBD fluorescence. There was also a slight decrease in the signal obtained in the EYFP channel that measured a certain degree of background fluorescence. The averaged dequenching in FGIN-1–27-NBD (donor) fluorescence in 38 pairs of cells is tabulated (Fig. 3 G) and a two-tailed paired t test was used to calculate the mean averaged FRET of the two groups of cells. Dequenching was only observed in EYFP-M11L–expressing cells after photobleaching. This phenomenon was not observed in any of the controls analyzed (unpublished data).
All the pairs of EYFP-M11L–expressing cells analyzed demonstrated a certain degree of FGIN-1–27-NBD dequenching. Given that there are difficulties in measuring the FRET transfer efficiency in living cells due to the complications of varied protein expression between cells and possible incomplete photobleaching of acceptor molecules, we calculated a FRET efficiency in the range of 2.8% to 78% (n = 38) with an average value of 23.9%. No FRET was observed in control experiments conducted using cells that were stained with FGIN-1–27-NBD and expressed EYFP alone (unpublished data).
M11L Acts at the Mitochondrial Cell Death Control Point and Inhibits Cytochrome c Release.
Translocation of cytochrome c into the cytoplasm after induction of apoptosis was evaluated in Rat2puro or Rat2M11L cells treated with staurosporine for 2, 3, or 4 h. Supernatant fractions of digitonin lysates were isolated and subjected to SDS-PAGE and immunoblot analysis using an antibody specific for cytochrome c. As shown in Fig. 4 A, whereas DMSO treatment alone did not induce cytochrome c release from either cell line (Fig. 4 A, lanes 1 and 5), staurosporine treatment of Rat2puro cells triggered a rapid release of cytochrome c into the supernatant fraction (Fig. 4 A, lanes 2–4). In contrast, after staurosporine treatment of Rat2M11L cells, the release of cytochrome c was barely detectable, even after 4 h (Fig. 4 A, lanes 6–8). Equivalent amounts of protein were analyzed as shown by an anti-actin immunoblot (Fig. 4 A, bottom panel).
Figure 4.
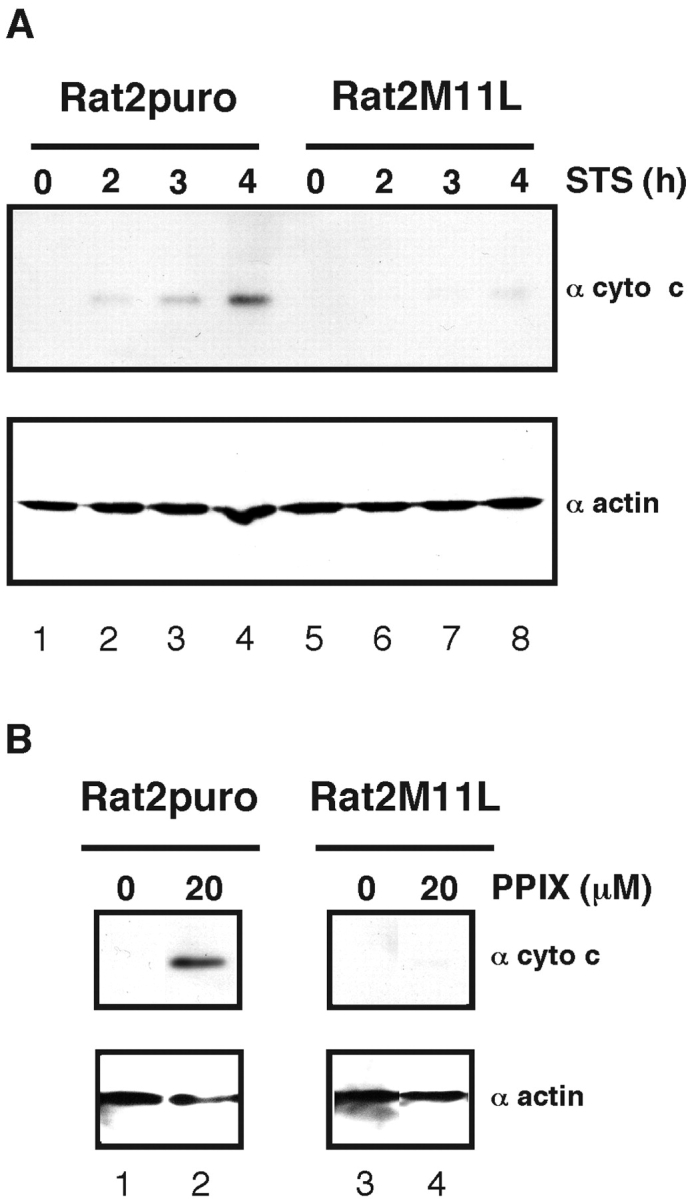
Stable expression of M11L in Rat2 cells provides protection against staurosporine- and PPIX-mediated cytochrome c release. (A) Rat2puro and M11L-expressing Rat2M11L cells were treated with staurosporine (+STS) or DMSO solvent alone (−STS) as control. Cytochrome c redistribution into the cytosolic fraction of cell lysates was detected by SDS-PAGE and immunoblot analysis (top panel). Equivalent amounts of protein were analyzed as shown by the actin immunoblot (bottom panel). (B) Treatment of permeabilized cells with 20 μM PPIX induced greater cytochrome c redistribution in Rat2puro cells than Rat2M11L cells (top panel) when equivalent amounts of the cytosolic fraction were loaded as assessed by actin content (bottom panel).
We wanted to determine whether M11L could prevent cell death mediated by another apoptotic inducer that interacts directly with the PT pore. PPIX is an endogenous ligand of the PBR and is known to induce apoptosis in cell lines (35) or when used as the active agent in photodynamic therapy (47). Permeabilized Rat2puro and Rat2M11L cells were treated with 20 μM PPIX for 30 min at 37°C (Fig. 4 B). DMSO solvent alone did not induce cytochrome c redistribution (Fig. 4 B, top panel, lanes 1 and 3). However, elevated levels of cytochrome c redistribution into the cytosolic fraction were observed in Rat2puro cells in comparison to Rat2M11L cells (Fig. 4 B, top panel, lanes 2 and 4) whereas an anti-actin blot showed comparable protein loading (Fig. 4 B, bottom panel). These results indicate that stable expression of M11L prevents cytochrome c translocation into the cytoplasm in response to apoptotic stimuli. We therefore conclude that M11L prevents apoptotic signals upstream of cytochrome c release and assists in the maintenance of mitochondrial integrity.
M11L Functionally Modulates the Mitochondrial Apoptotic Checkpoint.
To explore the interaction of M11L with the PBR in more detail, we chose to examine the ability of transiently expressed M11L to counteract the proapoptotic effects of PPIX as well as P11195, another PBR ligand that directly targets the PT pore.
The ability of M11L to counteract ΔΨm loss induced by PK11195 combined with staurosporine was assessed in HeLa cells expressing recombinant proteins as EGFP fusion constructs for ease of detection. The following chimeric proteins were expressed with an NH2-terminal EGFP tag: (a) EGFP-stop incorporating a truncated variant of M11L that lacks the mitochondrial targeting sequence, (b) EGFP-M11L which incorporates full length M11L, (c) EGFP-mt which is EGFP targeted to mitochondria by just the mitochondrial targeting sequence of M11L, and (d) EGFP-Bcl-2 which incorporates full-length Bcl-2. ΔΨm in transfected cells was assessed by measuring fluorescence of the potentiometric dye, TMRE. At low concentrations, PK11195 is not proapoptotic but this compound is known to amplify the effects of a number of pro-apoptotic agents (42, 43). Indeed, treatment of cells with PK11195 alone was found not to induce ΔΨm loss or influence the proportion of EGFP-expressing cells that were TMRE positive and these PK11195 treated cells were used to calculate the maximum percentage of cells expressing EGFP constructs that were also TMRE positive. After treatment, the percentage of EGFP-expressing cells that were TMRE positive was also calculated and the reduction in the percentage of dual positive cells was calculated.
The results shown in Fig. 5 A reveal that HeLa cells expressing the negative control protein, EGFP-stop, were not protected from ΔΨm loss after staurosporine treatment in the absence or presence of PK11195. In marked contrast, cells expressing EGFP-M11L were protected from both treatments. EGFP targeted to mitochondria by the 25 amino acid M11L targeting sequence (EGFP-mt) was also not protective. Cells transfected with EGFP-Bcl-2 were protected from staurosporine-mediated apoptosis. However, in keeping with previous studies (43), the protective properties of EGFP-Bcl-2, which are not thought to occur by direct interaction with the PBR, were clearly counteracted by the combined effects of staurosporine and PK11195. From these results, we conclude that M11L is effective in preventing ΔΨm loss in cells following staurosporine treatment, even in the presence of the PBR ligand PK11195. This ability to counteract the combined effects of PK11195 and staurosporine distinguishes M11L functionally from Bcl-2. These findings support the idea that M11L is able to modulate the PT pore and that this might occur via direct interaction with the PBR.
Figure 5.
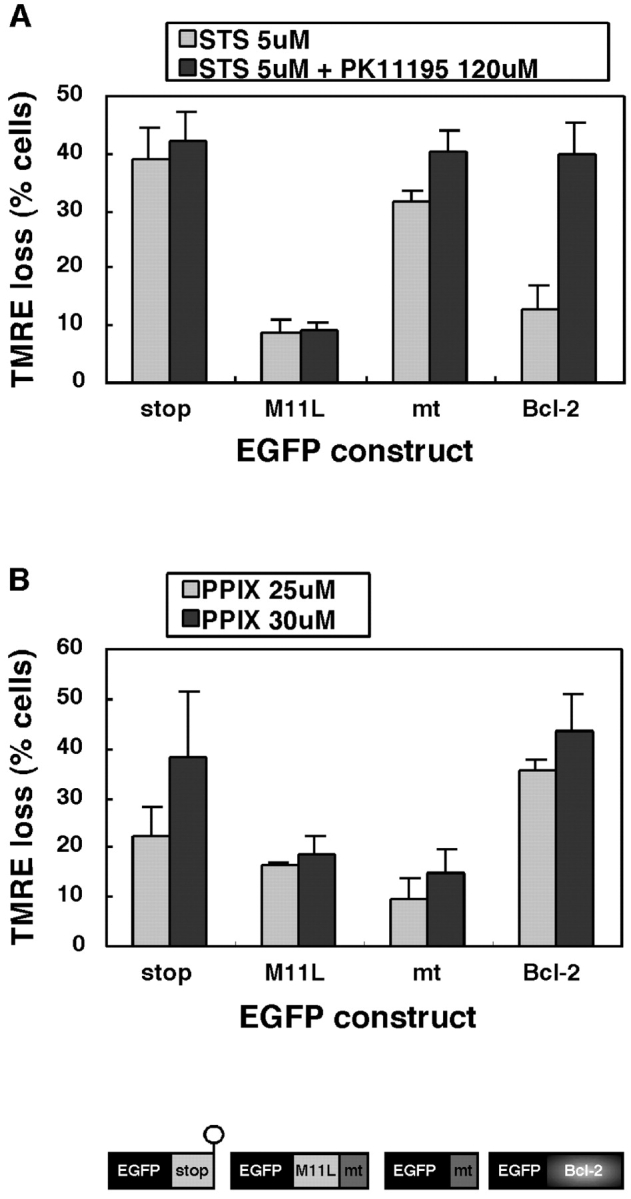
The mitochondrial targeting of M11L is important for inhibition of ΔΨm loss induced by the PBR ligands PK11195 and PPIX. (A) Cells expressing EGFP appended with truncated, nontargeted M11L (stop), full length M11L, the mitochondrial targeting sequence (mt), or Bcl-2 were treated with 5 μM staurosporine (STS) or 5 μM staurosporine in combination with 120 μM PK11195 (STS+PK1195). The reduction in the percentage of TMRE-positive EGFP-expressing cells after treatment is shown. The data represent the average of three independent experiments (±SD). (B) Cells expressing EGFP chimeras were treated with 25 or 30 μM PPIX. The reduction in the percentage of TMRE positive EGFP-expressing cells after treatment is shown. The results represent the average of three independent experiments (±SD).
As with cells that stably express M11L, transiently expressed M11L was also protective against the effects of PPIX. As shown in Fig. 5 B, in contrast to the negative control EGFP-stop, EGFP-M11L was effective in preventing ΔΨm loss mediated by 25 or 30 μM PPIX. EGFP-Bcl-2 was not at all protective against the concentrations of PPIX used, in keeping with other studies (35). Unexpectedly, the EGFP-mt construct, like EGFP-M11L, was able to prevent ΔΨm loss after PPIX treatment, indicating a possible role for the 25 amino acid mitochondrial targeting sequence of M11L in interfering with PPIX binding to the PT pore.
M11L Binding to the PBR Is Biologically Relevant.
To study the biological importance of the physical association between M11L and the PBR, we investigated the ability of myxoma virus infection to protect cells from staurosporine-mediated apoptosis in the context of different PBR expression levels. PBR-deficient Jurkat T lymphocytes or PBR overexpressing Jurkat-PBR cells were infected with the myxoma virus, vMyxlac, to allow the expression of M11L. For control purposes, parallel samples from both cell lines were mock-infected or infected with a M11L-knockout myxoma virus (vMyxM11L−). Cells were treated with 2 μM staurosporine or DMSO solvent as a control and TMRE fluorescence was used to measure ΔΨm loss. As shown in Fig. 6 A, a marked loss of TMRE fluorescence occurred after staurosporine treatment of both Jurkat and Jurkat-PBR cells that had been mock infected, with the presence of PBR only providing a slight protective effect. Myxoma virus infection was found to protect both cell lines from staurosporine-induced ΔΨm loss. However, vMyxlac infection of Jurkat-PBR cells provided a level of protection against ΔΨm loss that was twofold greater than the protection afforded control Jurkat cells. Infection of both cell lines with the M11L-deficient vMyxM11L− did not provide protection against staurosporine-induced ΔΨm loss. Similarly, when apoptosis was induced by Fas stimulation the same trends could be observed (Fig. 6 B).
Figure 6.
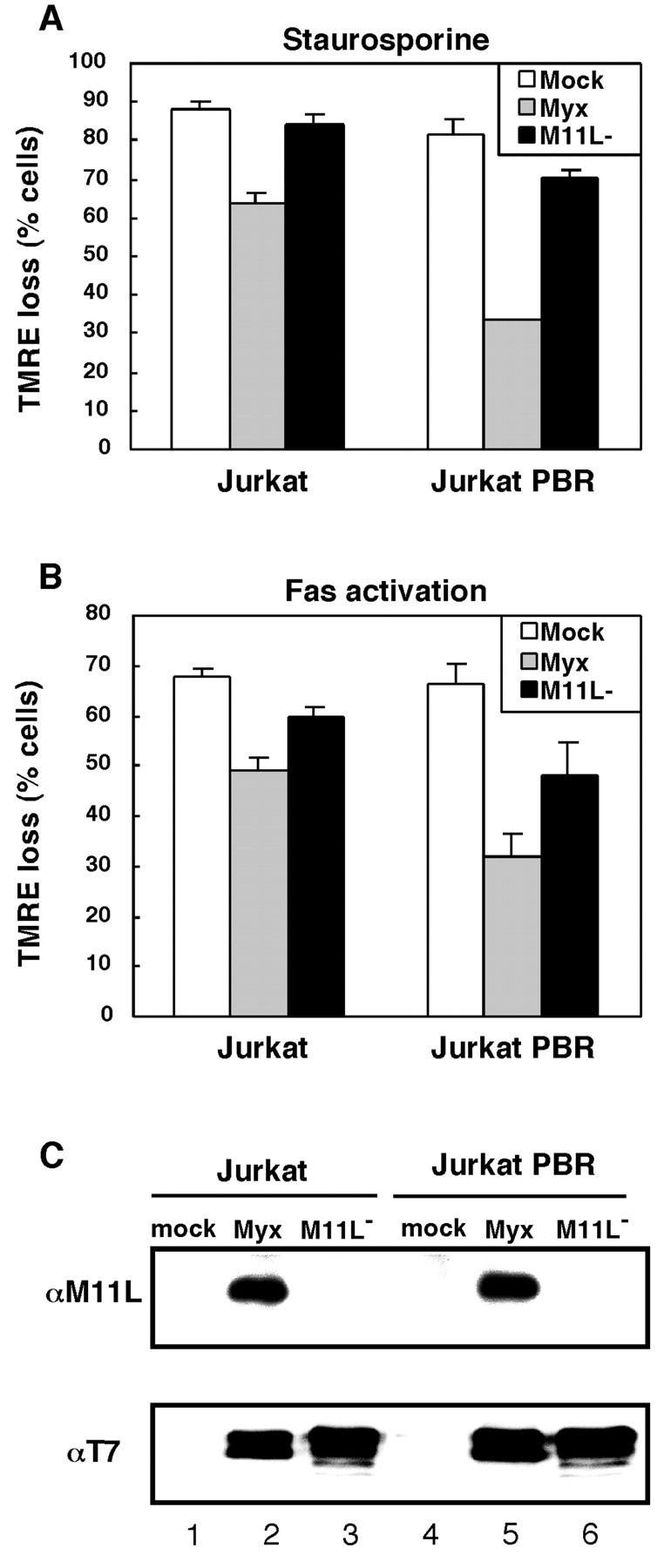
PBR expression augments the protective effects of myxoma virus infection. Jurkat or Jurkat-PBR cells were mock-infected (mock) or infected with myxoma virus (Myx) or the M11L knockout virus (M11L−). The percentage of cells that underwent a loss of TMRE fluorescence as a result of staurosporine treatment (A) or Fas stimulation (B) is shown. Results are the average of three independent experiments (±SD). In parallel, Jurkat or Jurkat-PBR cells that had been mock- or virus-infected were analyzed for the presence of M11L using immunoprecipitation and immunoblotting (C, top panel). Alternatively, virus infection was verified by the production of the early viral protein, M-T7 (C, bottom panel).
For control purposes, virus infection was monitored in Jurkat and Jurkat-PBR cells (Fig. 6 C). Mock-infected as well as vMyxlac- and vMyxM11L−-infected cells were analyzed for the presence of M11L and another viral early protein, M-T7 (58). Immunoblot analysis of immunoprecipitated M11L revealed that, as expected, M11L was not present in mock-infected cells or cells infected with the knockout virus (Fig. 6 C, top panel, lanes 1, 3, 4, and 6), but was only present in vMyxlac-infected cells (Fig. 6 C, lanes 2 and 5). M-T7 was not detected in mock-infected cells (Fig. 6 C, bottom panel, lanes 1 and 4) and levels of M-T7 in lysates of Jurkat and Jurkat-PBR cells infected with vMyxlac and vMyxM11L− viruses were comparable (Fig. 6 C, bottom panel, lanes 2, 3, 5, and 6), indicating that production of early viral proteins was uniform.
Discussion
We have shown previously that M11L is antiapoptotic, localizes to mitochondria, and prevents the loss of mitochondrial inner membrane potential in response to induction of apoptosis (6). M11L is a novel protein and currently has no database homologues outside the poxvirus family (49). However, it is interesting that M11L, like the antiapoptotic Bcl-2 proteins, prevents mitochondrial amplification of apoptotic signals (6). The mechanistic details underlying the functions of Bcl-2 family members are unclear, but it has been proposed that they exert their modulatory effects by direct physical association with the PT pore. In support of this model, Bcl-2 proteins including Bcl-2, Bcl-XL, and Bax have been shown to physically interact with both the ANT and VDAC subunits of the PT pore (12, 14). In addition, another novel viral antiapoptotic protein, vMIA of cytomegalovirus, interacts with the ANT and acts to prevent apoptotic changes downstream of mitochondria (17).
In the current study we have shown, using two independent approaches, that M11L physically associates with the PBR. Specifically, M11L and PBR could be cross-linked into a complex and, in addition, FRET analysis demonstrated a close association between a EYFP-M11L chimera and the PBR-specific fluorescent compound FGIN-1–27-NBD. This represents, to our knowledge, the first evidence that the PBR may physically associate with an antiapoptotic protein able to modulate mitochondrial function. We have also shown that, when expressed in a stable cell line, M11L inhibits cytochrome c release induced by staurosporine and by the PBR ligand, PPIX, therefore indicating that M11L functionally prevents transmission of apoptotic signals that proceed via mitochondria.
Transiently expressed M11L was also characterized with respect to its ability to counteract ΔΨm loss induced by PPIX as well as by the PBR ligand, PK11195, that is known to potentiate the action of several proapoptotic agents. Both of these ligands are able to bind PBR in the outer mitochondrial membrane and in the case of PK11195, this occurs independently of VDAC (59). M11L was found to effectively counteract ΔΨm loss triggered by staurosporine, staurosporine in combination with PK11195, as well as by PPIX.
Transient expression of the nontargeted M11L truncation mutant failed to protect cells from apoptosis. This parallels observations made with Bcl-2. Bcl-2 is targeted to membranes of the endoplasmic reticulum, outer mitochondrial membrane and perinuclear membrane by means of a COOH-terminal signal-anchor sequence (60) and requires membrane association in order to function (61, 62). Bcl-2 constructs specifically targeted by heterologous targeting signals to either mitochondria or the endoplasmic reticulum both possess antiapoptotic activities in discrete systems (63, 64). The current study therefore reinforces the common requirement of membrane association in order for M11L and Bcl-2 to prevent apoptosis.
Differences in M11L and Bcl-2 function were also observed. M11L displayed greater protective properties than Bcl-2. This may reflect a situation in which M11L, a viral protein, intrinsically has more potent antiapoptotic properties than Bcl-2 or may reflect the cell death–sensitizing effects resulting from Bcl-2 overexpression, both these phenomena having been reported previously (65). Also, in keeping with previous studies, Bcl-2 did not protect cells from PK11195 in combination with staurosporine or high concentrations of PPIX (35, 43). In contrast, M11L was protective under these conditions. Bcl-2 has been reported to interact physically with the VDAC and ANT components of the PT pore, but is not thought to bind directly to the PBR. The ability of PK11195 or PPIX to overcome the antiapoptotic effects of Bcl-2 can be rationalized if binding of either ligand to the PBR perturbs the entire pore structure and prevents Bcl-2 from binding the other subunits, an effect that would only be evident when another apoptotic stimulus is applied. The observed differences in the properties of M11L and Bcl-2 supports the idea that M11L directly counteracts the proapoptotic effects of PBR ligands by inhibiting their binding to the PBR or by stabilizing the PBR within the pore structure.
Another interesting observation was that the hydrophobic mitochondrial targeting sequence, mt, might itself possess antiapoptotic activity, particularly against PPIX, although it was not effective against PK11195 and staurosporine. This may relate to the fact that PK11195 and PPIX have different binding sites on the PBR as revealed by binding inhibition studies conducted with an antibody directed against an epitope in the extreme COOH-terminal region of PBR which is orientated toward the cytoplasm (29). Specifically, this antibody was able to block PK11195 but not PPIX binding, indicating that PK11195, but not PPIX, binds the COOH-terminal, cytoplasmic domain of PBR. Our data suggests that the hydrophobic targeting signal of M11L may interact with PBR in a region overlapping the PPIX binding site. In contrast, the full-length M11L protein may be required to prevent binding of PK11195 to the cytoplasmic COOH-terminal region of PBR.
We showed previously that myxoma virus infection of the RL-5 rabbit lymphocyte cell line provides protection against staurosporine-mediated apoptosis (6). Interestingly, we found in this study that infection of Jurkat cells with myxoma virus, but not the M11L knockout virus, protected these cells from ΔΨm loss induced by staurosporine and by Fas activation. This protective effect was accentuated in the presence of elevated levels of PBR. This implies that the physical association of M11L and the PBR is important in infected cells. However, M11L, like Bcl-2, probably has multiple binding interactions that contribute to its antiapoptotic properties.
The ability of M11L to bind to the PBR is intriguing in another context. The PBR is thought to potentiate inflammatory responses, an effect counteracted by the PBR ligand PK11195 (31, 32). We have observed that, during myxoma virus infection, absence of M11L paradoxically results in increased levels of both apoptosis and inflammation (6) implying that, in the infected rabbit, M11L has both antiapoptotic and antiinflammatory roles. We provided a potential explanation for this finding by showing that M11L function is particularly important during infection of monocytes (6). Prevention of monocyte apoptosis during infection has important consequences because, when cells of this lineage undergo apoptosis, they also promote inflammation (6, 66). Within hematopoeitic lineages, PBR is present in highest concentration in monocytes (28), raising the possibility that M11L interaction with the PBR contributes to the observed anti-inflammatory effects of M11L.
In conclusion, we have shown that M11L, an important virulence factor produced by myxoma virus, blocks apoptotic signals that proceed via the mitochondrial cell death control point. M11L binds to and functionally modulates the PBR component of the PT pore. Identification of certain functional similarities between M11L and Bcl-2 also raises the possibility that M11L, like Bcl-2, has multiple functions and binding partners. Investigation of M11L continues to provide valuable insights into how an antiapoptotic protein contributes to virus virulence and also into fundamental aspects of cell death pathways themselves.
Acknowledgments
We wish to thank Dr. Darren Roberts, Dr. Ing Swie Goping, and Kirstin Veugeleurs for critical review of the manuscript. Dr. Pierre Casellas (Sanofi Research) kindly provided the anti-PBR antibody and Jurkat PBR cells.
This work was funded by the Canadian Institutes of Health Research (CIHR) and the National Cancer Institutes of Canada. H. Everett is the recipient of a University of Alberta Dissertation Fellowship. M. Barry is an AHFMR scholar, L.G. Berthiaume is an Alberta Heritage Foundation for Medical Research (AHFMR) Senior Scholar and CIHR Scholar, G. McFadden is a CIHR Senior Scientist and R.C. Bleackley is a Distinguished Scientist of the CIHR, an AHFMR Medical Scientist and a Howard Hughes International Research Scholar. G. McFadden and R.C. Bleackley are both Canada Research Chairs.
Footnotes
Abbreviations used in this paper: ΔΨm, mitochondrial membrane potential; ANT, adenine nucleotide transporter; DSS, disuccinimidyl suberate; DSP, dithiobis(succinimidylpropionate); EGFP, enhanced green fluorescent protein; EYFP, enhanced yellow fluorescent protein; FRET, fluorescence resonance energy transfer; MOI, multiplicity of infection; PBR, peripheral benzodiazepine receptor; PK11195, 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide; PPIX, protoporphyrin IX; PT, permeability transition; STS, staurosporine; TMRE, ethyl ester of tetramethyl rhodamine; VDAC, voltage dependent anion carrier.
References
- 1.Zimmermann, K.C., C. Bonzon, and D.R. Green. 2001. The machinery of programmed cell death. Pharmacol. Ther. 92:57–70. [DOI] [PubMed] [Google Scholar]
- 2.Hay, S.W., and G. Kannourakis. 2002. A time to kill: viral manipulation of the cell death program. J. Gen. Virol. 83:1547–1564. [DOI] [PubMed] [Google Scholar]
- 3.Roulston, A., R.C. Marcellus, and P.E. Branton. 1999. Viruses and apoptosis. Annu. Rev. Microbiol. 53:577–628. [DOI] [PubMed] [Google Scholar]
- 4.Nash, P., J. Barrett, J.-X. Cao, S. Hota-Mitchell, A. Lalani, H. Everett, X.-M. Xu, J. Robichaud, S. Hnatiuk, C. Ainslie, et al. 1999. Immunomodulation by viruses: the myxoma virus story. Immunol. Rev. 168:103–120. [DOI] [PubMed] [Google Scholar]
- 5.Macen, J.L., K.A. Graham, S.F. Lee, M. Schreiber, L.K. Boshkov, and G. McFadden. 1996. Expression of the myxoma virus tumor necrosis factor receptor homologue (T2) and M11L genes is required to prevent virus-induced apoptosis in infected rabbit T lymphocytes. Virology. 218:232–237. [DOI] [PubMed] [Google Scholar]
- 6.Everett, H., M. Barry, S.-F. Lee, X. Sun, K. Graham, J. Stone, R.C. Bleackley, and G. McFadden. 2000. M11L: A novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J. Exp. Med. 191:1487–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Opgenorth, A., K. Graham, N. Nation, D. Strayer, and G. McFadden. 1992. Deletion analysis of two tandemly arranged virulence genes in myxoma virus, M11L and myxoma growth factor. J. Virol. 66:4720–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernardi, P., V. Petronilli, F. Di Lisa, and M. Forte. 2001. A mitochondrial perspective on cell death. Trends Biochem. Sci. 26:112–117. [DOI] [PubMed] [Google Scholar]
- 9.Ferri, K.F., and G. Kroemer. 2001. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3:E255–E263. [DOI] [PubMed] [Google Scholar]
- 10.Wang, X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922–2933. [PubMed] [Google Scholar]
- 11.Parone, P.A., D. James, and J.-C. Martinou. 2002. Mitochondria: regulating the inevitable. Biochimie. 84:105–111. [DOI] [PubMed] [Google Scholar]
- 12.Crompton, M. 1999. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 13.Gross, A., J.M. McDonnell, and S.J. Korsmeyer. 1999. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13:1899–1911. [DOI] [PubMed] [Google Scholar]
- 14.Harris, M.H., and C.B. Thompson. 2000. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 7:1182–1191. [DOI] [PubMed] [Google Scholar]
- 15.Everett, H., and G. McFadden. 2001. Viruses and apoptosis: Meddling with mitochondria. Virology. 288:1–7. [DOI] [PubMed] [Google Scholar]
- 16.Boya, P., B. Roques, and G. Kroemer. 2001. Viral and bacterial proteins regulating apoptosis at the mitochondrial level. EMBO J. 20:4325–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldmacher, V.S., L.M. Bartle, A. Skaletskaya, C.A. Dionne, N.L. Kedersha, C.A. Vater, J.-W. Han, R.J. Lutz, S. Watanabe, E.D. Cahir McFarland, et al. 1999. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA. 96:12536–12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacotot, E., L. Ravagnan, M. Loeffler, K.F. Ferri, H.L.A. Vieira, N. Zamzami, P. Costantini, S. Druillennec, J. Hoebeke, J.P. Briand, et al. 2000. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 191:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacotot, E., K.F. Ferri, C. El Hamel, C. Brenner, S. Druillennec, J. Hoebeke, P. Rustin, D. Metivier, C. Lenoir, M. Geuskens, et al. 2001. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein R and Bcl-2. J. Exp. Med. 193:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahmani, Z., K.-W. Huh, R. Lasher, and A. Siddiqui. 2000. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 74:2840–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casellas, P., S. Galiegue, and A.S. Basile. 2002. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem. Int. 40:475–486. [DOI] [PubMed] [Google Scholar]
- 22.Gavish, M., I. Bachman, R. Shoukrun, Y. Katz, L. Veenman, G. Weisinger, and A. Weizman. 1999. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 51:629–650. [PubMed] [Google Scholar]
- 23.McEnery, M.W., A.M. Snowman, R.R. Trifiletti, and S.H. Snyder. 1992. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA. 89:3170–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Culty, M., H. Li, N. Boujrad, H. Amri, B. Vidic, J.M. Bernassau, J.L. Reversat, and V. Papadopoulos. 1999. In vitro studies on the role of the peripheral-type benzodiazepine receptor in steroidogenesis. J. Steroid Biochem. Mol. Biol. 69:123–130. [DOI] [PubMed] [Google Scholar]
- 25.Kroemer, G., and J.C. Reed. 2000. Mitochondrial control of cell death. Nat. Med. 6:513–519. [DOI] [PubMed] [Google Scholar]
- 26.Papadopoulos, V., N. Boujrad, M.D. Ikonomovic, P. Ferrara, and B. Vidic. 1994. Topography of the Leydig cell mitochondrial peripheral-type benzodiazepine receptor. Mol. Cell. Endocrinol. 104:R5–R9. [DOI] [PubMed] [Google Scholar]
- 27.Braestrup, C., and R.F. Squires. 1977. Specific benzodiazepine receptors in rat brain characterized by high-affinity [3H]diazepam binding. Proc. Natl. Acad. Sci. USA. 74:3805–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carayon, P., M. Portier, D. Dussossoy, A. Bord, G. Petitpretre, X. Canat, G. Le Fur, and P. Casellas. 1996. Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood. 87:3170–3178. [PubMed] [Google Scholar]
- 29.Dussossoy, D., P. Carayon, D. Feraut, S. Belugou, T. Combes, X. Canat, H. Vidal, and P. Casellas. 1996. Development of a monoclonal antibody to immuno-cytochemical analysis of the cellular localization of the peripheral benzodiazepine receptor. Cytometry. 24:39–48. [DOI] [PubMed] [Google Scholar]
- 30.Verma, A., and S.H. Snyder. 1988. Characterization of porphyrin interactions with peripheral type benzodiazepine receptors. Mol. Pharmacol. 34:800–805. [PubMed] [Google Scholar]
- 31.Torres, S.R., T.S. Frode, G.M. Nardi, N. Vita, R. Reeb, P. Ferrara, R.M. Ribeiro-do-Valle, and R.C. Farges. 2000. Anti-inflammatory effects of peripheral benzodiazepine receptor ligands in two mouse models of inflammation. Eur. J. Pharmacol. 408:199–211. [DOI] [PubMed] [Google Scholar]
- 32.Klegeris, A., E.G. McGeer, and P.L. McGeer. 2000. Inhibitory action of 1-(2-chlorophenyl-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide (PK11195) on some mononuclear phagocyte functions. Biochem. Pharmacol. 59:1305–1314. [DOI] [PubMed] [Google Scholar]
- 33.Stoebner, P.-E., P. Carayon, P. Casellas, M. Portier, T. Lavabre-Bertrand, P. Cuq, J.-P. Cano, J. Meynadier, and L. Meunier. 2001. Transient protection by peripheral benzodiazepine receptors during the early events of ultraviolet light-induced apoptosis. Cell Death Differ. 8:747–753. [DOI] [PubMed] [Google Scholar]
- 34.Johnston, C., W. Jiang, T. Chu, and B. Levine. 2001. Identification of genes involved in the host response to neurovirulent alphavirus infection. J. Virol. 75:10431–10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchetti, P., T. Hirsch, N. Zamzami, M. Castedo, D. Decaudin, S.A. Susin, B. Masse, and G. Kroemer. 1996. Mitochondrial permeability transition triggers lymphocyte apoptosis. J. Immunol. 157:4830–4836. [PubMed] [Google Scholar]
- 36.Fischer, R., M. Schmitt, J.G. Bode, and D. Haussinger. 2001. Expression of the peripheral-type benzodiazepine receptor and apoptosis induction in hepatic stellate cells. Gastroenterology. 120:1212–1226. [DOI] [PubMed] [Google Scholar]
- 37.Fennel, D.A., M. Corbo, A. Pallaska, and F.E. Cotter. 2001. Bcl-2 resistant mitochondrial toxicity mediated by the isoquinoline carbozamide PK11195 involves de novo generation of reactive oxygen species. Br. J. Cancer. 84:1397–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maaser, K., M. Hopfner, A. Jansen, G. Weisinger, M. Gavish, A.P. Kozikowski, A. Weizman, P. Carayon, E.O. Reicken, M. Zeitz, and H. Scherubi. 2001. Specific ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in human colorectal cancer cells. Br. J. Cancer. 85:1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chelli, B., A. Falleni, F. Salvetti, V. Gremigni, A. Lucacchini, and C. Martini. 2001. Peripheral-type benzodiazepine receptor ligands: mitochondrial permeability transition induction in rat cardiac tissue. Biochem. Pharmacol. 61:695–705. [DOI] [PubMed] [Google Scholar]
- 40.Pastorino, J.G., G. Simbula, E. Gilfor, J.B. Hoek, and J.L. Farber. 1994. Protoporphyrin IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J. Biol. Chem. 269:31041–31046. [PubMed] [Google Scholar]
- 41.Pastorino, J.G., G. Simbula, K. Yamamoto, P.A. Glascott, R.J. Rothman, and J.L. Farber. 1996. The cytotoxicity of tumor necrosis factor depends on induction of the mitochondrial permeability transition. J. Biol. Chem. 271:29792–29798. [DOI] [PubMed] [Google Scholar]
- 42.Bono, F., I. Lamarche, V. Prabonnaud, G. Le Fur, and J.M. Herbert. 1999. Peripheral benzodiazepine receptor agonists exhibit potent antiapoptotic activities. Biochem. Biophys. Res. Commun. 265:457–461. [DOI] [PubMed] [Google Scholar]
- 43.Hirsch, T., D. Decaudin, S.A. Susin, P. Marchetti, N. Larochette, M. Resche-Rignon, and G. Kroemer. 1998. PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp. Cell Res. 241:426–434. [DOI] [PubMed] [Google Scholar]
- 44.Ravagnan, L., I. Marzo, P. Constantini, S.A. Susin, N. Zamzami, P.X. Petit, F. Hirsch, M. Goulbern, M.-F. Poupon, L. Miccoli, et al. 1999. Londamine triggers apoptosis via a direct, Bcl-2-inhibited effect on the mitochondrial permeability transition pore. Oncogene. 18:2537–2546. [DOI] [PubMed] [Google Scholar]
- 45.Larochette, N., D. Decaudin, E. Jacotot, C. Brenner, I. Marzo, S.A. Susin, N. Zamzami, Z. Xie, J. Reed, and G. Kroemer. 1999. Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp. Cell Res. 249:413–421. [DOI] [PubMed] [Google Scholar]
- 46.Tanimoto, Y., Y. Onishi, Y. Sato, and H. Kizaki. 1999. Benzodiazepine receptor agonists modulate thymocyte apoptosis through reduction of the mitochondrial transmembrane transmembrane potential. Jpn. J. Pharmacol. 79:177–183. [DOI] [PubMed] [Google Scholar]
- 47.Lilge, L., M. Portnoy, and B.C. Wilson. 2000. Apoptosis induced in vivo by photodynamic therapy in normal brain and intracranial tumor tissue. Br. J. Cancer. 83:1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noodt, B.B., K. Berg, T. Stokke, Q. Peng, and J.M. Nesland. 1996. Apoptosis and necrosis induced with light and 5-aminolaevulinic acid-derived protoporphyrin IX. Br. J. Cancer. 74:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graham, K.A., A. Opgenorth, C. Upton, and G. McFadden. 1992. Myxoma virus M11L ORF encodes a protein for which cell surface localization is critical for manifestation of viral virulence. Virology. 191:112–124. [DOI] [PubMed] [Google Scholar]
- 50.Kozikowski, A.P., M. Kotoula, D. Ma, N. Boujrad, W. Tuckmantel, and V. Papadopoulos. 1997. Synthesis and biology of a 7-Nitro-2,1,3-benzoxadiazol-4-yl derivative of 2-Phenylindole-3-acetamide: A fluorescent probe for the peripheral-type benzodiazepine receptor. J. Med. Chem. 40:2435–2439. [DOI] [PubMed] [Google Scholar]
- 51.Bastiaens, P.I., I.V. Majoul, P.J. Verveer, H.D. Soling, and T.M. Jovin. 1996. Imaging the intracellular trafficking and state of the AB5 quaternary structure of cholera toxin. EMBO J. 15:4246–4253. [PMC free article] [PubMed] [Google Scholar]
- 52.Dickinson, M.E., G. Bearman, S. Tilie, R. Lansford, and S.E. Fraser. 2001. Multi-spectral imaging and linear unmixing add a whole new dimension to laser scanning fluorescence microscopy. Biotechniques. 31:1272–1278. [DOI] [PubMed] [Google Scholar]
- 53.Farkas, D.L., M. Wei, P. Febbroriello, J.H. Carson, and L.M. Loew. 1989. Simultaneous imaging of cell and mitochondrial membrane potential. Biophys. J. 56:1053–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stryer, L. 1978. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 47:819–846. [DOI] [PubMed] [Google Scholar]
- 55.Förster, T. 1948. Intermolecular energy migration and fluorescence. Ann. Physiol. 2:55–75. [Google Scholar]
- 56.Bastiaens, P.I., and A. Squire. 1999. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol. 9:48–52. [DOI] [PubMed] [Google Scholar]
- 57.Xia, Z., and Y. Liu. 2001. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 81:2395–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mossman, K., C. Upton, and G. McFadden. 1995. The myxoma virus soluble interferon-γ receptor homolog, M-T7, inhibits interferon-γ in a species specific manner. J. Biol. Chem. 270:3031–3038. [DOI] [PubMed] [Google Scholar]
- 59.Joseph-Liauzun, E., R. Farges, P. Delmas, P. Ferrara, and G. Loison. 1997. The Mr 18,000 subunit of the peripheral-type benzodiazepine receptor exhibits both benzodiazepine and isoquinoline carboxamide binding sites in the absence of the voltage-dependent anion channel or of the adenine nucleotide carrier. J. Biol. Chem. 272:28102–28106. [DOI] [PubMed] [Google Scholar]
- 60.Nguyen, M., D.G. Millar, V.W. Youg, S.J. Korsmeyer, and G.C. Shore. 1993. Targeting of Bcl-2 to the mitochondrial outer membrane by a COOH-terminal signal anchor sequence. J. Biol. Chem. 268:25265–25268. [PubMed] [Google Scholar]
- 61.Nguyen, M., P.E. Branton, P.A. Walton, Z.N. Oltvai, S.J. Korsmeyer, and G.C. Shore. 1994. Role of the membrane anchor domain of Bcl-2 in suppression of apoptosis caused by E1B-defective adenovirus. J. Biol. Chem. 269:16521–16524. [PubMed] [Google Scholar]
- 62.Tanaka, S., K. Saito, and J.C. Reed. 1993. Structure-function analysis of the bcl-2 oncoprotein. J. Biol. Chem. 268:10920–10926. [PubMed] [Google Scholar]
- 63.Zhu, W., A. Cowie, G.W. Wasfy, L.Z. Penn, B. Leber, and D.W. Andrews. 1996. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- 64.Froesch, B.A., C. Aime-Sempe, B. Leber, D. Andrews, and J.C. Reed. 1999. Inhibition of p53 transcriptional activity by Bcl-2 requires its membrane-anchoring domain. J. Biol. Chem. 274:6469–6475. [DOI] [PubMed] [Google Scholar]
- 65.Bellows, D.S., B.N. Chau, P. Lee, Y. Lazebnik, W.H. Burns, and J.M. Hardwick. 2000. Antiapoptotic herpesvirus Bcl-2 homologs escape caspase-mediated conversion to proapoptotic proteins. J. Virol. 74:5024–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Savill, J. 1997. Apoptosis in the resolution of inflammation. J. Leukoc. Biol. 61:375–380. [DOI] [PubMed] [Google Scholar]