

Abstract
Endothelial cells (ECs) are believed to be an important component in the protection from lipopolysaccharide (LPS)-induced endotoxic shock. However, the cellular and molecular mechanism is not well defined. Here, we report that signal transducer and activator of transcription (STAT) 3 is an essential regulator of the antiinflammatory function of ECs in systemic immunity. Because STAT3 deficiency results in early embryonic lethality, we have generated mice with a conditional STAT3 deletion in endothelium (STAT3E−/−). STAT3E−/− mice are healthy and fertile, and isolated ECs initiate normal tube formation in vitro. Conditional endothelial but not organ-specific (i.e., hepatocyte or cardiomyocyte) STAT3 knockout mice show an increased susceptibility to lethality after LPS challenge. The LPS response in STAT3E−/− mice shows exaggerated inflammation and leukocyte infiltration in multiple organs combined with elevated activity of serum alanine aminotransferase and aspartate aminotransferase, indicating organ damage. Concomitantly, proinflammatory cytokines are produced at an exaggerated level and for a prolonged period. This defect cannot be explained by lack of antiinflammatory cytokines, such as interleukin 10 and transforming growth factor β. Instead, we have shown that a soluble activity derived from endothelia and dependent on STAT3 is critical for suppression of interferon γ. These data define STAT3 signaling within endothelia as a critical antiinflammatory mediator and provide new insight to the protective function of ECs in inflammation.
Keywords: transcription factors, Toll-like receptor, cytokines, transgenic/knockout, endotoxin shock
Introduction
Endothelial cells (ECs) provide a large surface area and close contact for pathogens, and compose the interface between hematopoietic cells and organs, mediating various pathological processes. The physiological effects of the immune system and cytokines on ECs have been well documented (1). However, whether ECs are active or passive participants in this process is less well characterized. Both human and murine ECs have been shown to participate in immune recognition by expressing MHC and costimulatory molecules (2, 3), suggesting that they act as antigen-presenting cells. It has been further suggested that ECs would provide signals for immune modulation via cell surface or secreted factors (4), raising the prospect that intracellular signaling in ECs could be manipulated to affect immune responses.
The Janus kinase signal transducer and activator of transcription (STAT) signaling pathway is activated by a diverse array of cytokines and growth factors, and has been implicated in a variety of cellular functions, including inflammatory processes (5). Ligand binding induces receptor dimerization and tyrosine phosphoryalation of receptor-associated Janus kinases, followed by activation of specific STATs. Phosphorylated and dimerized STATs translocate to the nucleus to activate the transcription of target genes. STAT3 is the mediator of many early response genes activated by IL-6 family cytokines (6, 7). The zygotic STAT3 deletion is lethal before gastrulation (8); therefore, we generated mice with a conditional STAT3 allele (9).
Various cell type–specific STAT3 KOs have been established, including keratinocytes (10), T cells (11), or macrophages/neutrophils (12). STAT3 deletion in mice within the macrophage/neutrophil lineage results in chronic inflammation and pathological colitis due to enhanced IFN-γ–producing T cells (12). These mice have an overabundance of proinflammatory cytokines due to a defect in signaling by IL-10, a major antiinflammatory cytokine that requires STAT3 (13). IL-10 KO mice (14) show a similar phenotype as the STAT3 macrophage/neutrophil KO mice, although IL-10 levels are increased in these STAT3-deficient mice (12). We have also shown recently that removal of STAT3 from hematopoietic progenitors results in increased proinflammatory cytokine production, inflammatory bowel disease, and an expanded macrophage population (9). These genetic ablation studies have shown that STAT3 plays a critical role in controlling inflammation as the deletion of STAT3 causes an exacerbated inflammatory response.
The antiinflammatory role of STAT3 is, at least in part, due to the antagonism of STAT1 (15) and inducing genes that inhibit the receptor (e.g., suppressor of cytokine signaling; references 16, 17). Alternative forms of STAT3 have been shown to antagonize inflammatory signaling. STAT3β is an alternatively spliced transcript of STAT3 that lacks the transactivation domain. When STAT3β is knocked out by removal of a cryptic splice site, the animals become sensitive to LPS-induced endotoxic shock (18). The role of STAT3 in directing an antiinflammatory response is clearly defined within immune cells, but less well understood within other cell types, such as endothelia.
We investigated the role of STAT3 cytokine signaling within endothelium to better understand the role that ECs play in inflammation. Innate inflammatory signaling can be initiated through Toll-like receptors (TLRs). TLRs respond to a set of natural pathogenic ligands and induce a primary immune response using the NF-κB Rel family, c-Jun NH2-terminal kinase, phosphatidylinositol 3 kinase, p38, and ERK pathways (19). This primary response leads to a secondary response through type I IFNs that can activate several STAT family members (19–21). LPS from gram-negative bacteria is a potent TLR4 ligand and elicits a strong inflammatory response and a characteristic cytokine burst. The response to LPS is generally self limited at least in part through the actions of IL-10. Activation of the innate immune response to LPS is a measure of the immunological health of an animal. We have generated an endothelial conditional KO of STAT3. Here, we show that endothelial STAT3 signaling plays a critical antiinflammatory role in LPS-induced endotoxic shock by controlling subsequent cytokine-induced tissue damage and lethality. Furthermore, we show that a soluble factor that is dependent on STAT3 plays a critical role in the control of IFN-γ production during LPS-induced inflammation.
Materials and Methods
Animals.
Generation of mice with a conditional STAT3 allele has been described previously (9). Exons 18–20, which contain the SH2 domain of STAT3, were flanked by two loxP sites. Two Tie2-Cre transgenic mouse strains expressing Cre under the control of the TIE2–kinase promoter/enhancer were generated. One was described previously with expression in hematopoietic and ECs (14, 22), and here, we describe another with a more endothelial-specific expression. The TIE2e-Cre;STAT3f/d mutant mice were generated through TIE2e-Cre;STAT3+/d mice crossed to STAT3f/f mice. The genotype was determined by PCR as described previously (9). TIE2e-Cre mice were mated with Z/EG reporter mice, which produce green fluorescence protein (GFP) from a constitutive promoter after removal of LacZ by cre-mediated recombination (23). Transthyretin (TTR) Cre mice were generated in our laboratory (unpublished data) and the α-MHC-Cre mice were from M. Schneider (Baylor College of Medicine, Houston, TX). All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and under the approval of the Yale Medical School Animal Care and Use Committee.
EC Isolation and Flow Cytometry.
Liver sinusoidal ECs (LSECs) were isolated through a modified method (24). Livers were perfused through the portal vein with HBSS with 0.5 mM EGTA briefly and subsequently digested with 0.015% collagenase II and IV (GIBCO BRL) solution. Excised liver was minced and further digested in collagenase solution at 37°C for 30 min. Hepatocytes were removed by brief centrifugation twice at 50 g. The supernatant was collected and centrifuged at 350 g for 5 min. The pellet was resuspended in 30% Percoll-HBSS and loaded on 50% Percoll-HBSS. The density gradient column was centrifuged at 900 g for 30 min. The cell layer from 30 to 50% Percoll-HBSS was collected. This layer contained an enriched fraction of liver ECs.
For further purification, enriched LSECs were blocked with anti-FCR antibody and 5% FCS-PBS, and incubated with anti-CD144 (VE-cadherin) antibody conjugated with R-phycoerythrin (BD Biosciences). Stained cells were sorted on a FACSCalibur™ using CELLQuest™ software (Becton Dickinson). A similar procedure was used in analyzing GFP+ bone marrow and splenocytes.
Tube Formation Assay.
To compare the tube forming ability of control and mutant ECs, cells were plated on a thin layer of Matrigel (Becton Dickinson) at 2 × 105 cells/well of a 24-well plate in 10% FCS DMEM and allowed to form a tubular structure for overnight. Cells were assessed on their ability to form simple tube structures and their morphology.
Western Blotting.
Tissues were washed briefly with PBS and lysed with radioimmunoprecipitation (10 mM Tris-HCl, pH 7.4, 1% NP-40, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 150 mM sodium chloride, 1 mM ethylendiamintetraacetic acid, 100 mM sodium fluoride, 200 μM sodium orthovanadate, 0.5 mM phenylmethanesulfonyl fluoride, 2 μg/ml aprotinin, 1 μg/ml luepeptin, and 1 μg/ml pepstatin). The cell lysates were centrifuged, and the supernatants were removed and assayed for protein content using the bicinchoninic acid protein assay kit (Pierce Chemical Co.). Cell lysates were separated on SDS-PAGEs, transferred to polyvinylidene difluoride membranes, and incubated with antiphospho-specific (Tyr705) STAT3 (New England Biolabs, Inc.), STAT3, and STAT1 (Santa Cruz Biotechnology, Inc.) antibodies. Anti–rabbit antibody conjugated with horseradish peroxidase was used as secondary antibody, and visualized using chemiluminescent substrate (SuperSignal; Pierce Chemical Co.).
LPS Challenge and Serum Chemistry.
Mice were given an intraperitoneal injection of 5 mg/kg LPS and had blood drawn or were killed at indicated times. Serum activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using a transaminase kit according to the manufacturer's instructions (Sigma-Aldrich).
ELISA.
Serum or cell culture supernatant concentrations of IL-4, -6, -10, TNF-α, active TGF-β, and IFN-γ were determined by ELISA according to the manufacturer's instructions (R&D Systems).
Histological Analysis.
Tissue was collected and fixed in 4% paraformaldehyde for 4 h, infused with 9% sucrose for 4 h and 20% sucrose overnight except for liver, which was fixed in ice-cold acetone. Tissue was embedded in optimum cutting temperature, frozen in liquid nitrogen, and stored at −80 C° until sectioning. 10-μM frozen sections were cut and washed in PBS, and GFP was imaged by epifluorescence microscopy. Indirect immunofluorescence staining was done using a platelet-EC adhesion molecule 1 (PECAM-1)/CD31 antibody and rhodamine-conjugated secondary antibody. Alternatively, tissues were fixed in 4% paraformaldehyde and paraffin embedded. 6-μM tissue sections were stained with hematoxylin and eosin using standard techniques.
Results
Endothelial-specific TIE2Cre Transgenic Line.
A line of Cre recombinase–expressing transgenic mice was generated using the TIE2 5′ promoter and first intron enhancer element (Fig. 1 A; reference 22). When these were mated to Z/EG reporter mice that express GFP after Cre-mediated recombination, a widespread expression of GFP was observed within endothelium of brain, heart, and liver (Fig. 1 B), among others. The GFP signal colocalized with CD31/PECAM-1 immunostaining, indicating that GFP+ cells were of endothelial origin (Fig. 1 B).
Figure 1.

Endothelial-specific Cre recombinase. (A) A line of Cre recombinase expressing transgenic mice was generated using the TIE2 5′ promoter and first intron enhancer element. (B) TIE2e-Cre mice were mated to Z/EG reporter mice. Animals were killed, tissue was frozen, and 10 micrometer sections were made. Sections from brain, heart, and liver are shown. Arrow indicates colocalization with CD31/PECAM-1. (C) Flow cytometry of bone marrow and splenotcytes show that ∼1.5% of cells express GFP, indicating that the expression of Cre recombinase is mainly restricted to ECs.
Because ECs are BM derived, we checked the expression of GFP in BM cells to determine the specificity of this TIE2 transgene by flow cytometry. Approximately 1.5% of the cells within BM were positive for GFP (Fig. 1 C). These cells did not segregate into specific populations during subsequent hematopoietic development as judged by flow cytometry analysis of spleen cells (Fig. 1).
Next, we asked if the GFP were associated with any particular cell lineage, such as EC precursors or macrophages. We sorted GFP+ cells with markers for PECAM-1, CD4, CD8, CD44, CD86, Mac-1, and I-AB. No specific association of GFP was seen with any of tested markers (unpublished data). This was supported by a microscopic inspection that revealed that the GFP+ cells were morphologically divergent. Consistently, the BM cell cultures under different cytokines/growth factors, such as macrophage colony–stimulating factor, G–macrophage colony–stimulating factor, granulocyte stimulating factor, vascular endothelial growth factor, and basic fibroblast growth factor, were not populated with GFP+ cells to any particular cell lineage. We conclude that the expression of Cre in this strain of TIE2-Cre mice is restricted to endothelium, and Cre expression is minimally leaky. Therefore, we designated the transgene as TIE2e-Cre.
STAT3-deficient ECs Appear Physiologically Normal.
We crossed the TIE2e-Cre line (C57/BL/6) to the C57/BL/6 mice with a LoxP-targeted disruption in the SH2 domain of STAT3 to obtain TIE2e-Cre;STAT3f/d mice (STAT3E−/−). The deletion results in the loss of the COOH terminus and renders STAT3 transcriptionally inactive. To confirm the KO biochemically, we analyzed LSECs from STAT3E−/− mice and their littermates (TIE2e-Cre;STAT3f/+) along with other tissues. Western blot results showed that STAT3 protein was greatly reduced in the KO ECs, whereas STAT1 was unaffected (Fig. 2, A and B) . The residual STAT3 in the preparation reflects the contamination of other cell types due to LSEC isolation procedure (see Fig. 5 A). As expected, STAT3 was not deleted in hepatocytes, peritoneal macrophages, nor T or B cells (Fig. 2 A). Thus, the deletion of STAT3 in ECs is specific and effective.
Figure 2.

Endothelial STAT3 deletion and endotoxin-induced lethality. (A) Peritoneal macrophages (p-Mac), T cells, B cells, and hepatocytes were isolated from WT and KO mice and total protein was extracted. SDS-PAGE fractionation and Western blotting using a STAT3-specific polyclonal antibody showed robust expression of STAT3 within these cell types. Total STAT1 expression was also unaltered. (B) Isolated ECs from TIE2e-Cre;STAT3f/d, and TIE2e-Cre;STAT3f/+ mice were stimulated with IL-6. Western blot analysis of phospho-specific STAT3 revealed significantly reduced phosphorylated STAT3 in conditional KOs. The membranes were stripped and reblotted with antibodies to STAT3 and STAT1. (C) STAT3 null and wild-type ECs were isolated and plated on Matrigel. Both genotypes formed normal tube structures in vitro, and no differences could be found between groups. (D) Peritoneal macrophages were isolated, stimulated with LPS, and assayed for TNF-α. There was no change in the production of TNF-α, indicating a normal response by macrophages. (E) TIE2e-Cre;STAT3f/d and TIE2e-Cre;STAT3f/+ mice were given an intraperitoneal dose of 5 mg/kg LPS. 60% of TIE2e-Cre;STAT3f/d mice died, whereas all wild-type littermates survived. Lethality was initiated after 16 h. Both cardiomyocyte (αMHC-Cre;STAT3f/f (F) and hepatocyte (TTR-Cre;STAT3f/f) STAT3 conditional KO (G) mice were tested for LPS-induced lethality.
Figure 5.

Endothelial suppression of IFN-γ is STAT3 dependent. (A) ECs were isolated from the liver and purified by FACS® sorting. STAT3 null and WT LSECs were incubated with WT LPS-induced splenocytes and assayed for IFN-γ and TNF-α. (B) IFN-γ was not detected (ND) in cultures without splenocytes regardless of LPS treatment or genetic background. IFN-γ was induced in splenocytes upon LPS treatment and significantly reduced from splenocytes cocultured with WT endothelium (P < 0.05). When STAT3 null LSECs were cocultured with WT splenocytes, IFN-γ was not suppressed. (C) TNF-α was unchanged between groups. (D and E) 10% conditioned media from STAT3 null and WT LSECs were incubated with LPS-stimulated WT splenocytes and assayed for IFN-γ and TNF-α. (D) IFN-γ was significantly reduced from splenocytes incubated with 10% WT LSEC-conditioned media (P < 0.05). This suppression was released when 10% STAT3 null LSECs conditioned media was used. (E) TNF-α was unchanged between groups. (B and C) Shaded bar, without LPS. White bar, LPS treatment. (F) Wild-type EC-conditioned media at concentrations of 0%–20% were incubated with LPS-stimulated splenocytes and measured for IFN-γ and TNF-α, revealing a dose-dependent effect on IFN-γ. ND, not detected.
Mice with a targeted disruption of endothelial STAT3 were born at the expected Mendelian ratio, survived through adulthood, and were fertile, indicating normal developmental angiogenesis. Tube formation on Matrigel is a widely used in vitro assay for a well-defined characteristic of ECs. Isolated LSECs from KOs and controls were assayed for their ability to undergo tube formations on Matrigel. ECs with both genetic backgrounds formed tube structures and we were unable to distinguish differences between groups (Fig. 2 C).
Endothelial STAT3 Protects against Endotoxic Shock.
We asked if mutant mice had normal innate immune function. To test this, we injected STAT3E−/− mice with 5 mg/kg LPS (026:B6 serotype). LPS elicits strong innate immune responses via TLR4 (25, 26) and has a characteristic cytokine response. In STAT3E−/− mice, this response was exaggerated and induced lethality (Fig. 2, E and F). The lethality was evident ∼17 h after LPS administration. By 40 h, 60% of STAT3E−/− mice had died, whereas all of the heterozygous littermates survived. To test if other STAT3 KO lines were susceptible to lethality at this dose, mice from cardiomyocyte-specific αMHC-Cre;STAT3f/d (STAT3H−/−) and hepatocyte-specific TTR-Cre;STAT3f/d (STAT3L−/−) STAT3-deleted mice were subjected to LPS challenge. Both STAT3H−/− and STAT3L−/− mice were not susceptible to endotoxic shock–mediated lethality at this dose, suggesting that ECs play a role in this susceptibility.
STAT3 is known to be important for normal signaling of many cytokines. Removal of STAT3 within cells of the immune system, such as macrophages and neutrophils, results in an exaggerated inflammatory cytokine response and Th1 polarization (12). However, loss of endothelial STAT3 causes a similar, albeit less severe, phenotype as the macrophage KO even though there is normal STAT3 expression within hematopoietic cells. This is a rather surprising finding. To rule out the possibility that the deletion of STAT3 in a limited set of macrophages interfered with the phenotypical readout of STAT3E−/− mice, we assayed TNF-α production in isolated peritoneal macrophages from both mutants and controls (Fig. 2 D). Unlike STAT3-deficient macrophages, which exhibit increased TNF-α expression in response to LPS (12), the macrophages from STAT3E−/− mice behaved identically to controls, suggesting that the deletion of STAT3 in bone marrow cells in STAT3E−/− mice did not alter the function of the macrophages.
LPS Induces Severe Systemic Inflammation in STAT3E−/− Mice.
Upon histological examination of STAT3E−/− mice 17 h after LPS injection, mice had severe infiltration of leukocytes into multiple organs, including the liver (Fig. 3 A) as well as lung and gut (not depicted). We focused on liver pathology as a primary target organ. In the liver, the portal vein area was the most severely affected because leukocytes could be seen invading and accumulating under endothelia around the portal veins. In contrast, individual organs such as the heart in STAT3H−/− and the liver in STAT3L−/− (not depicted) as well as in control mice did not show obvious infiltration of leukocytes (Fig. 3 A), suggesting a role for endothelial STAT3 in controlling the influx of leukocytes.
Figure 3.
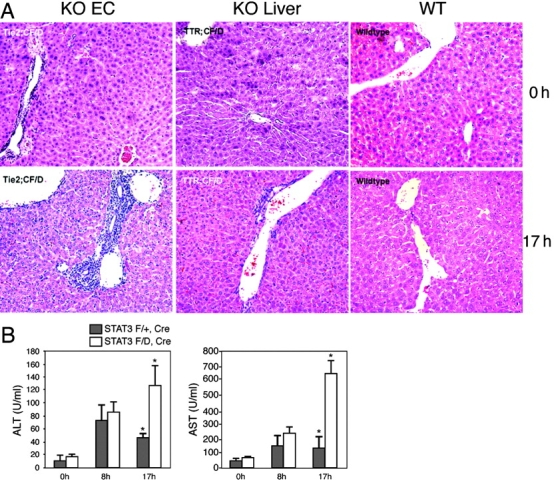
Endothelial STAT3 mutant mice have liver and tissue damage after LPS administration. (A) Tissue was collected at 0 and 17 h after LPS administration and stained with hematoxylin and eosin. Leukocyte infiltration can be seen in the liver of LPS-treated TIE2e-Cre;STAT3f/d mice. Specifically, the portal vein had the most severe leukocyte accumulation. TTR-Cre;STAT3f/d and control mice do not show severe infiltration. (B) Serum ALT and AST activity was measured in TIE2e-Cre;STAT3f/d and TIE2e-Cre;STAT3f/+ mice. At 17 h, both were higher in TIE2e-Cre;STAT3f/d mice than controls (P < 0.05).
To further evaluate the systemic effect of LPS on STAT3E−/− mice, we measured the serum levels of ALT and AST. Serum ALT is an indicator of liver damage, whereas AST is expressed in numerous organs, including liver, heart, skeletal muscle, kidney, brain, and pancreas. Therefore, it is an indicator of multiorgan damage (27). The ALT and AST activities in serum were increased at comparable levels in both STAT3E−/− and control mice initially (8 h after LPS challenge). By 17 h, the ALT and AST levels were reduced or remained at low in the control mice, but both ALT (P < 0.05) and AST (P < 0.01) levels further increased in STAT3E−/− mice (Fig. 3 B).
LPS Elicits an Exaggerated Cytokine Response in STAT3E−/− Mutants.
To examine the immunological status and the potential causes of lethality in these mice, we measured serum levels of known inflammatory mediators. Serum was taken at different time points after LPS treatment (Fig. 4) . The kinetics of cytokine induction were measured. STAT3E−/− mice exhibited significantly elevated inflammatory cytokines. TNF-α showed a rapid and significant increase of fourfold as early as 1.5 h (P < 0.05), but dropped rapidly as expected. In STAT3E−/− mice, the rise of IL-6 was both exaggerated at 1.5 h (P < 0.05) and prolonged at 17 h (P < 0.05). As expected, IFN-γ was delayed in response to LPS challenge. It increased slightly at 8 h in control animals. However, in STAT3E−/− mice, IFN-γ levels were significantly increased by 7.5-fold at 8 h (P < 0.05) and sustained through 17 h, a time when the controls had no detectable level of IFN-γ.
Figure 4.

Serum cytokines have an exaggerated and prolonged expression. Serum was collected from mutant and wild-type animals after LPS treatment and measured by ELISA at 0-, 1.5-, 8-, and 17-h time points (animals in each group, n = 4). Mutant TNF-α was significantly increased by fourfold over controls at 1.5 h but was reduced at 8 h. IL-10 had a similar pattern as TNF-α, increasing by twofold. In contrast, IFN-γ was increased at 8 h (7.5-fold) and remained high at 17 h, whereas IL-6 was increased at 1.5 and 17 h. Shaded bar, TIE2e-Cre;STAT3f/+. White bar, TIE2e-Cre;STAT3f/d. ND, not detected. *, P < 0.05.
Next, we asked if IL-10 production was compromised in STAT3E−/− mice, which could account for the exaggerated inflammatory phenotype induced by LPS, as IL-10 is a potent antiinflammatory cytokine. Inconsistent with this notion, STAT3E−/− mice exhibited significantly increased IL-10 levels after LPS treatment (P < 0.05) and this was sustained longer than in controls (Fig. 4). Because STAT3 signaling within immune cells is not compromised genetically in STAT3E−/− mutants, the loss of antiinflammatory cytokines in these animals cannot be explained by a block of IL-10 signaling, a result that is different from the macrophage/neutrophil STAT3 KO reported previously (12). Similarly, TGF-β and IL-4 levels were unchanged between control and KO (unpublished data). Together, these data suggest a yet-undefined STAT3-dependent endothelial factor that is able to regulate the expression of proinflammatory cytokines.
LSEC Inhibit IFN-γ via a STAT3 Dependent Mechanism.
Our in vivo studies have demonstrated that STAT3 mediates interplay between ECs and the immune system in systemic immunity. To confirm the in vivo observation in a simpler system, we studied the effect of isolated LSECs on inflammatory cytokine production by leukocytes. By using FACS® with a CD144 antibody, we were able to purify LSECs to >98% (Fig. 5 A). At such purity, LPS did not induce either IFN-γ or TNF-α in ECs from either genetic background (Fig. 5, B and C). In contrast, the purified splenocytes responded to LPS with production of both IFN-γ and TNF-α. These results indicate that it is the splenocytes that were the primary LPS-responding and cytokine-producing cells.
We tested the regulatory function of ECs on cytokine production by splenocytes. Purified LSECs were incubated with freshly isolated wild-type (C57BL/6) splenocytes with 1 μg/ml LPS. Cytokine levels were assayed after 17 h of coculture. A strong suppression of IFN-γ production by control LSECs was observed (Fig. 5 B). However, IFN-γ was not suppressed by STAT3E−/− LSECs (Fig. 5 B). In contrast, TNF-α induction was not affected by the LSEC coculture (Fig. 5 C). Thus, STAT3 mediates its antiinflammatory effect, at least in part, by providing a signal to antagonize IFN-γ production.
LSEC Inhibit IFN-γ through Soluble Factors.
Next, we asked if the suppressive effect of ECs on inflammation is mediated through a soluble or cell surface molecule, as it has been postulated that LSECs can suppress IFN-γ from Th1 cells via class II MHC (28). We incubated 10% endothelial conditioned media with splenocytes under LPS stimulation. The media from control ECs significantly reduced IFN-γ production, whereas STAT3E−/− media could not (Fig. 5 D). TNF-α was minimally affected in both media (Fig. 5 E).
Several control experiments indicated that the inhibitory effects of conditioned media and LSECs on IFN-γ production are rather specific and STAT3 dependent. First, STAT1−/− LSECs retained the ability to suppress IFN-γ production, whereas both KO and WT hepatocytes could not, suggesting that both the molecular regulator and the cellular origin of the suppressor are specific (unpublished data). The suppression of the IFN-γ production was potent and dose dependent (Fig. 5 F). Again, the suppressive effects of conditioned media on TNF-α were minimal, and did not follow the same pattern of IFN-γ suppression. These findings suggest that the effector is soluble, constitutive, and dependent on STAT3, and showed a degree of specificity to IFN-γ.
It is important to determine EC production of antiinflammatory cytokines such as IL-10 and TGF-β in our system because both molecules suppress the production of IFN-γ from splenocytes. We measured IL-10 from LSEC-conditioned media from both control and STAT3E−/− LSECs. IL-10 is constitutively expressed in wild-type ECs. If IL-10 is the soluble effector, one would expect to see a reduction of IL-10 levels in the media from STAT3E−/− ECs. However, the IL-10 levels in wild-type and mutant LSEC-conditioned mediums were equivalent (unpublished data). Similarly, the TGF-β levels were unaffected by STAT3 deletion (unpublished data). Evidently, there is a different IFN-γ regulator produced by LSECs under the control of STAT3. The nature of this suppressor is under investigation.
Discussion
The interplay between ECs and the immune system is fundamental for systemic immunity. Here, we have shown that ECs control LPS-induced toxicity by controlling the cytokine response via STAT3. This demonstrates the importance of ECs during endotoxic shock and underscores the importance of STAT3 in the control of systemic inflammation.
The KO of STAT3 is embryonically lethal at E6.5, and the cause of lethality is not fully understood (8). STAT3 is expressed early in embryonic postimplantation development when angiogenesis is critical. We assume that STAT3 is not an essential angiogenic factor because STAT3 endothelial conditional KOs are born at a normal Mendelian ratio, and pups are healthy and indistinguishable from littermates. Furthermore, in vitro assays for tube formation show no obvious difference between control and STAT3 null ECs.
LPS elicits a strong and predictable immune response. Therefore, we used LPS as an indicator of the immunological vigor of these animals. Under threshold levels of LPS, control mice survived, but 60% of STAT3E−/− mice died as a result of the treatment. These data are further validated by the normal response of LPS in other conditional STAT3 KOs. We used both cardiomyocyte- and hepatocyte-specific STAT3 conditional KO strains. Although the endothelial-specific strain had massive leukocyte infiltration into the liver, the hepatocyte and cardiomyocyte conditional KOs had a less severe phenotype. Analysis of macrophages from STAT3E−/− animals showed no contribution to the phenotype, suggesting a specific role for ECs. This underscores the importance of endothelia in systemic inflammation and points to STAT3 as a central control molecule for this inflammation.
Serum chemistry from LPS-challenged STAT3E−/− mice was consistent with hepatitis and other tissue damage as evidenced by elevated activity of serum ALT and AST, hallmarks of liver and systemic tissue damage respectively (27). These data cumulatively show that LPS induces severe tissue damage in the conditional KO.
We did not directly investigate the cause of lethality due to LPS. However, we suspect the pro-inflammatory burst. Although TNF-α and IL-6 were elevated significantly in these animals, both are not the mediators of lethality as shown by the genetic disruption of these cytokines. Recently, a double KO of macrophage STAT3 and TNF-α did not relieve the inflammatory bowel disease in these animals. However, deletion of IL-12, a cytokine essential for IFN-γ production, did relieve the phenotype (29). Genetic ablation of IFN-γ or its receptor confers resistance to LPS-mediated lethality. Together, these data with the increased and prolonged expression of IFN-γ in the endothelial STAT3 KO strongly suggest a role for IFN-γ in promoting the LPS-induced lethality.
IL-10 is a strong immunosuppressant and is expressed in endothelium. The significant increase of IL-10 in LPS-treated STAT3E−/− mice is puzzling because it would be expected that such high levels of IL-10 would be adequate to inhibit IFN-γ expression. However, in the absence of STAT3, suppressor of cytokine signaling 3 levels could be reduced, resulting in higher IL-10 levels produced in ECs (30). These data would suggest that there is another important mediator of cytokine expression that is absent in STAT3E−/− mice.
We found that ECs could inhibit LPS-induced IFN-γ expression from splenocytes, and that STAT3E−/− ECs could not. IFN-γ is a critical mediator of lethality during endotoxin challenge (31). It has been postulated that class II MHC present on LSECs could inhibit IFN-γ production by TH1 cells in vitro. Alternatively, our data suggest that a soluble factor, dependent on STAT3, is present that can selectively suppress the production of IFN-γ from splenocytes (Fig. 5, B–E). Our preliminary characterization suggests that the factor is a large protein that differs in size from known inhibitory cytokines (unpublished data).
The discovery of an endothelial-derived mediator of suppression of systemic endotoxin shock and cytokine production could be of clinical relevance. Whether ECs control the expression of IFN-γ at the transcriptional level or if they control differentiation and polarization of T cells has not been determined. Because STAT3 has been shown to regulate its expression in vivo and in vitro, one may be able to modulate STAT3 signaling in patients exhibiting symptoms of bacterial shock to limit tissue damage.
Acknowledgments
We thank Dr. J. Madri for helpful discussions.
R. Flavell is an investigator of the Howard Hughes Medical Institute and X.-Y. Fu was a recipient of a Career Development Award from National Institutes of Health (NIH). This work is supported by NIH grants AI34522 (to X.-Y. Fu) and HL51014 (to J.S. Pober).
A. Kano, M.J. Wolfgang, and Q. Gao contributed equally to this work.
The present address of Y. Iwamoto is the Division of Urology and Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, WI 53226.
Abbreviations used in this paper: ALT, alanine aminotransferase; AST, aspartate aminotransferase; EC, endothelial cell; GFP, green fluorescent protein; LSEC, liver sinusoidal EC; PECAM-1, platelet-EC adhesion molecule 1; STAT, signal transducer and activator of transcription; TLR, Toll-like receptor; TTR, transthyretin.
References
- 1.Pober, J.S., and R.S. Cotran. 1990. The role of endothelial cells in inflammation. Transplantation. 50:537–544. [DOI] [PubMed] [Google Scholar]
- 2.Hughes, C.C., C.O. Savage, and J.S. Pober. 1990. The endothelial cell as a regulator of T-cell function. Immunol. Rev. 117:85–102. [DOI] [PubMed] [Google Scholar]
- 3.Lohse, A.W., P.A. Knolle, K. Bilo, A. Uhrig, C. Waldmann, M. Ibe, E. Schmitt, G. Gerken, and K.H. Meyer Zum Buschenfelde. 1996. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology. 110:1175–1181. [DOI] [PubMed] [Google Scholar]
- 4.Limmer, A., J. Ohl, C. Kurts, H.G. Ljunggren, Y. Reiss, M. Groettrup, F. Momburg, B. Arnold, and P.A. Knolle. 2000. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 6:1348–1354. [DOI] [PubMed] [Google Scholar]
- 5.Leonard, W.J., and J.J. O'Shea. 1998. Jaks and STATs: biological implications. Annu. Rev. Immunol. 16:293–322. [DOI] [PubMed] [Google Scholar]
- 6.Akira, S., Y. Nishio, M. Inoue, X.J. Wang, S. Wei, T. Matsusaka, K. Yoshida, T. Sudo, M. Naruto, and T. Kishimoto. 1994. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 77:63–71. [DOI] [PubMed] [Google Scholar]
- 7.Zhong, Z., Z. Wen, and J.E. Darnell, Jr. 1994. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 264:95–98. [DOI] [PubMed] [Google Scholar]
- 8.Takeda, K., K. Noguchi, W. Shi, T. Tanaka, M. Matsumoto, N. Yoshida, T. Kishimoto, and S. Akira. 1997. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA. 94:3801–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welte, T., S.S.M. Zhang, T. Wang, Z. Zhang, A. Hesslein, A. Yin, A. Kano, Y. Iwamoto, E. Li, J.E. Craft, et al. 2003. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality, revealing a role of STAT3 in regulation of innate immunity. Proc. Natl. Acad. Sci. USA. 100:1879–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sano, S., S. Itami, K. Takeda, M. Tarutani, Y. Yamaguchi, H. Miura, K. Yoshikawa, S. Akira, and J. Takeda. 1999. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 18:4657–4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeda, K., T. Kaisho, N. Yoshida, J. Takeda, T. Kishimoto, and S. Akira. 1998. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 161:4652–4660. [PubMed] [Google Scholar]
- 12.Takeda, K., B.E. Clausen, T. Kaisho, T. Tsujimura, N. Terada, I. Forster, and S. Akira. 1999. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 10:39–49. [DOI] [PubMed] [Google Scholar]
- 13.Benkhart, E.M., M. Siedlar, A. Wedel, T. Werner, and H.W. Ziegler-Heitbrock. 2000. Role of Stat3 in lipopolysaccharide-induced IL-10 gene expression. J. Immunol. 165:1612–1617. [DOI] [PubMed] [Google Scholar]
- 14.Kuhn, R., J. Lohler, D. Rennick, K. Rajewsky, and W. Muller. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 75:263–274. [DOI] [PubMed] [Google Scholar]
- 15.Costa-Pereira, A.P., S. Tininini, B. Strobl, T. Alonzi, J.F. Schlaak, H. Is'harc, I. Gesualdo, S.J. Newman, I.M. Kerr, and V. Poli. 2002. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc. Natl. Acad. Sci. USA. 99:8043–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong, F., B. Jaruga, W.H. Kim, S. Radaeva, O.N. El-Assal, Z. Tian, V.A. Nguyen, and B. Gao. 2002. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J. Clin. Invest. 110:1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakagawa, R., T. Naka, H. Tsutsui, M. Fujimoto, A. Kimura, T. Abe, E. Seki, S. Sato, O. Takeuchi, K. Takeda, et al. 2002. SOCS-1 participates in negative regulation of LPS responses. Immunity. 17:677–687. [DOI] [PubMed] [Google Scholar]
- 18.Yoo, J.Y., D.L. Huso, D. Nathans, and S. Desiderio. 2002. Specific ablation of Stat3beta distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell. 108:331–344. [DOI] [PubMed] [Google Scholar]
- 19.Ardeshna, K.M., A.R. Pizzey, S. Devereux, and A. Khwaja. 2000. The PI3 kinase, p38 SAP kinase, and NF-kappaB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood. 96:1039–1046. [PubMed] [Google Scholar]
- 20.Jacobs, A.T., and L.J. Ignarro. 2001. Lipopolysaccharide-induced expression of interferon-beta mediates the timing of inducible nitric-oxide synthase induction in RAW 264.7 macrophages. J. Biol. Chem. 276:47950–47957. [DOI] [PubMed] [Google Scholar]
- 21.Doyle, S., S. Vaidya, R. O'Connell, H. Dadgostar, P. Dempsey, T. Wu, G. Rao, R. Sun, M. Haberland, R. Modlin, and G. Cheng. 2002. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 17:251–263. [DOI] [PubMed] [Google Scholar]
- 22.Koni, P.A., S.K. Joshi, U.A. Temann, D. Olson, L. Burkly, and R.A. Flavell. 2001. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J. Exp. Med. 193:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novak, A., C. Guo, W. Yang, A. Nagy, and C.G. Lobe. 2000. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 28:147–155. [PubMed] [Google Scholar]
- 24.Braet, F., R. De Zanger, T. Sasaoki, M. Baekeland, P. Janssens, B. Smedsrod, and E. Wisse. 1994. Assessment of a method of isolation, purification, and cultivation of rat liver sinusoidal endothelial cells. Lab. Invest. 70:944–952. [PubMed] [Google Scholar]
- 25.Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 26.Qureshi, S.T., L. Lariviere, G. Leveque, S. Clermont, K.J. Moore, P. Gros, and D. Malo. 1999. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J. Exp. Med. 189:615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amacher, D.E. 1998. Serum transaminase elevations as indicators of hepatic injury following the administration of drugs. Regul. Toxicol. Pharmacol. 27:119–130. [DOI] [PubMed] [Google Scholar]
- 28.Knolle, P.A., T. Germann, U. Treichel, A. Uhrig, E. Schmitt, S. Hegenbarth, A.W. Lohse, and G. Gerken. 1999. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J. Immunol. 162:1401–1407. [PubMed] [Google Scholar]
- 29.Kobayashi, M., M.N. Kweon, H. Kuwata, R.D. Schreiber, H. Kiyono, K. Takeda, and S. Akira. 2003. Toll-like receptor-dependent production of IL-12 p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J. Clin. Invest. 111:1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berlato, C., M.A. Cassatella, I. Kinjyo, L. Gatto, A. Yoshimura, and F. Bazzoni. 2002. Involvement of suppressor of cytokine signaling-3 as a mediator of the inhibitory effects of IL-10 on lipopolysaccharide-induced macrophage activation. J. Immunol. 168:6404–6411. [DOI] [PubMed] [Google Scholar]
- 31.Salkowski, C.A., G. Detore, R. McNally, N. van Rooijen, and S.N. Vogel. 1997. Regulation of inducible nitric oxide synthase messenger RNA expression and nitric oxide production by lipopolysaccharide in vivo: the roles of macrophages, endogenous IFN-gamma, and TNF receptor-1-mediated signaling. J. Immunol. 158:905–912. [PubMed] [Google Scholar]