

Abstract
Integrin-mediated adhesion and B cell antigen receptor (BCR) signaling play a critical role in B cell development and function, including antigen-specific B cell differentiation. Here we show that the BCR controls integrin α4β1 (VLA-4)-mediated adhesion of B cells to vascular cell adhesion molecule-1 and fibronectin. Molecular dissection of the underlying signaling mechanism by a combined biochemical, pharmacological, and genetic approach demonstrates that this BCR-controlled integrin-mediated adhesion requires the (consecutive) activation of Lyn, Syk, phosphatidylinositol 3-kinase, Bruton's tyrosine kinase (Btk), phospholipase C (PLC)γ2, IP3R-mediated Ca2+ release, and PKC. In contrast, activation of mitogen-activated protein kinase kinase (MEK) or extracellular signal–regulated kinase (ERK) is not required, and simultaneous activation of MEK, ERK, and PKB is not sufficient either. Furthermore, Btk is also involved in the control of integrin-mediated adhesion of preB cells. The control of integrin α4β1-mediated B cell adhesion by the BCR involves cytoskeletal reorganization and integrin clustering. These results reveal a novel function for the BCR and Btk, i.e., regulation of integrin α4β1 activity, thereby providing new insights into the control of B cell development and differentiation, as well as into the pathogenesis of the immunodeficiency disease X-linked agammaglobulineamia (XLA).
Keywords: lymphocyte adhesion, B-cell development, X-linked agammaglobulinaemia, germinal center, VCAM-1
Introduction
Integrin-mediated cell–cell and cell–matrix adhesion, as well as migration, play an important role in a wide variety of processes controlling B-lymphocyte development and function. During early B cell development in the BM, hematopoietic progenitor cells successively develop into pro-, pre-, and immature B cells, which will emigrate into the bloodstream. This development occurs in defined microenvironments and is controlled by integrin α4β1-mediated interactions with fibronectin (FN) in the ECM and with vascular cell adhesion molecule (VCAM)-1 expressing BM stromal cells (1–4). Furthermore, integrins mediate the transendothelial migration of mature B cells required for their recirculation and homing (5–8), and play a key role in antigen-specific B cell differentiation.
Antigen-specific B cell differentiation takes place within germinal centers (GCs), specialized microenvironments in the B cell area of secondary lymphoid organs (9–11). Here, T cell-dependent humoral immune responses are initiated by the activation of naive B cells in the T cell (paracortical) area, which leads to their migration into B cell follicles where they initiate the formation of GCs. In the GC, the B cells undergo rapid clonal expansion and somatic hypermutation of their immunoglobulin (Ig) genes. The mutated B cells reencounter antigen, presented by follicular dendritic cells (FDCs), and undergo affinity maturation: B cells expressing high affinity B cell antigen receptor (BCR) mutants will be rescued from apoptosis by their interaction with the FDC (11, 12), expand, undergo Ig-isotype switching, and mature into memory B cells or plasma cells. The interaction with FDCs is mediated by the integrins α4β1 and αLβ2 (LFA-1) expressed on the GC B cell, which engage VCAM-1 and ICAM-1 on the FDC, respectively (13–15). Interestingly, apart from establishing physical contact, outside-in signaling by α4β1 and LFA-1 has been shown to suppress apoptosis of GC B cells (16, 17). Hence, integrin-mediated interactions of GC B cells with FDCs play a key role in antigen-specific B cell differentiation (11). This notion is further supported by the observations that the T cell-dependent humoral immune response is impaired in mice lacking expression of VCAM-1 on FDCs (2) or of integrin β1 in the hematopoietic system (18).
Despite the important role of integrins in B cell development and antigen-specific B cell differentiation, the stimuli and underlying signal transduction mechanisms that control integrin activity of B cells are as yet poorly defined. Given the critical role of the (pre)BCR in B cell development and differentiation (19–22), we asked whether the BCR may control integrin-mediated adhesion of B cells and, if so, what is the signal transduction mechanism involved. BCR signal transduction involves a wide variety of signaling molecules (20, 23, 24), some of which, e.g., phosphatidylinositol 3-kinase (PI3K), phospholipase C (PLC), protein kinase C (PKC), extracellular signal–regulated kinase (ERK) and several members of the Ras family, have been implicated in the control of integrin activity by other receptors (25, 26). Here, we demonstrate that stimulation of the BCR induces integrin α4β1-mediated adhesion of B cells to VCAM-1 and FN. Furthermore, our data reveal a critical role for the consecutive activation of Lyn, Syk, PI3K, Bruton's tyrosine kinase (Btk), PLCγ2, IP3R-mediated Ca2+ release, and PKC in the BCR-controlled inside-out signaling mechanism underlying α4β1 activation, which involves cytoskeletal reorganization and integrin clustering. These results explain how the concerted action of both BCR signaling and integrin activation, as well as outside-in signaling, is controlled and contributes to antigen-specific B cell differentiation. In addition, our study reveals a novel function for Btk, which suggests that a loss of control of integrin activity contributes to the developmental and functional B cell defects observed in the immunodeficiency disease X-linked agammaglobulinemia (XLA).
Materials and Methods
Materials.
The following reagents were used in this study: the phosphorylation state-specific antibodies phospho-p44/42 MAP kinase [T202/Y204] against ERK1 and 2 and phospho-Akt [Ser473] against PKB/Akt (New England Biolabs); anti-ERK2 (C-14) and anti-Akt1/2 (H-136; Santa Cruz Biotechnology); the mouse monoclonal IgG1 antibodies HP2/1 against integrin subunit α4 (Immunotech); Act-1 against integrin subunit β7 (kindly provided by Dr. A. Lazarovits); TS2/16 against integrin subunit β1 (kindly provided by Dr. F. Sanchez-Madrid); TS1/22 against integrin subunit αL (ATCC); CSAT IgG2b against the chicken integrin subunit β1 (DSHB, University of Iowa); and the rat monoclonal antibodies OXM718-FITC and PS/2-biotin against mouse integrin α4 and β1, respectively (Cymbus Biotechnology). Mouse anti-T7-tag (Novagen); anti-CD45R(B220) and anti-mouse IgM microbeads (Miltenyi Biotec); mouse anti-human IgM (MH15; kindly provided by the Central Laboratory of the Netherlands Red Cross Blood Transfusion Service, Amsterdam, the Netherlands). Streptavidin-PE, Streptavidin-FITC, goat-anti mouse-Biotin, and (Fab)2-fragments of goat anti-human IgM (DAKO); goat anti-mouse IgM and goat anti-mouse-PE (Southern Biotechnology Associates), (Fab)2-fragments of goat anti-mouse IgM and goat anti-human Fc-FITC (Jackson Immunoresearch Laboratories), goat anti-chicken IgM (Bethyl Laboratories), and rabbit anti-goat-Biotin (Vector). The pharmacological inhibitors PD-98059, LY-294002, Wortmannin, U-73122, Chelerythrine, cytochalasin D, Jasplakinolide, and Calpeptin (Biomol). Recombinant human sVCAM-1 (R&D Systems), human plasma Fibronectin, BSA (fraction V) and poly-l-Lysine (PLL) (Sigma-Aldrich).
Cell Lines and Culture.
The Burkitt's lymphoma cell line Namalwa clone V3M (27) and the human preB cell line Nalm6 (28) were cultured as described. The chicken bursal lymphoma B cell line DT40 and DT40 cells deficient for both Lyn and Syk (29), Btk (29), PLCγ2 (30), and for all three types of IP3R (31), obtained from Riken Cell Bank (Tsukuba Science City, Japan) with permission from Dr. T. Kurosaki (Kansai Medical University, Moriguchi, Japan), were cultured at 39.5°C as described (32). All DT40 cells, i.e., WT, gene-deficient and reconstituted (see below), showed similar expression of surface IgM and integrin β1, as determined by FACS analysis, using goat anti-chicken IgM (10 μg/ml) and rabbit anti-goat-Biotin/Streptavidin-PE, or CSAT (3 μg/ml) and goat anti-mouse-PE, respectively, analyzed with a FACSCaliburTM (Beckton Dickinson) using CELLQuest software.
Isolation of Primary Tonsillar and Splenic B Cells.
Human tonsillar B cells were isolated essentially as previously described (16). Typically, the obtained cell population contains >97% B cells as determined by FACS analysis. Isolated B cells were maintained in RPMI containing 10% FCS and were used immediately or after overnight storage at 4°C.
Mouse splenic B cells from C57BL/6 mice, which were bred and maintained at the animal care facility of the Academic Medical Center, were obtained using the MACS system (Miltenyi Biotec) by positive selection with anti-CD45R (B220) microbeads, essentially according to manufacturer's instruction.
Mice and BM Cultures.
Btk−/LacZ mice were crossed onto a C57BL/6 background for over 6 generations, and Btk genotyped as described (33). 3–83 μδ Ig transgenic mice which bear 3–83 Ig heavy and κ-light chain transgenes (anti-H-2Kkb) on a B10.D2 background (34), were crossed with WT or Btk-deficient mice. These mice were bred and maintained at the animal care facility of the Erasmus MC (Rotterdam, the Netherlands).
To obtain mouse preB cells or B cells, IL-7-driven primary BM cultures of WT and Btk-deficient mice, or of 3–83 transgenic WT and Btk-deficient mice, respectively, were performed essentially as described (35, 36). In brief, BM cells were erythrocyte depleted and cultured at 107 cells/4 ml/well in 6-well plates in IMDM supplemented with 10% FCS and 100 U/ml murine IL-7 (R&D) for 5 d. Naive B cells from IL-7-driven BM cultures of 3–83 μδ Ig transgenic WT and Btk-deficient mice were used since mature B cells from Btk-deficient mice have an aberrant immature IgMhigh phenotype. The IL-7 driven BM cultures of the nontransgenic WT or Btk-deficient mice typically resulted in >98% B220+ cells and ∼80% and 95% preB cells (B220+/IgM−), respectively. Cultures from 3–83 transgenic mice contained >95% B220+/IgM+ B cells. Expression levels of integrin α4 and β1 on mice preB cells were determined by FACS analysis using OXM718-FITC (1:20) and PS/2-biotin (1:80) antibodies, respectively.
Generation of Reconstituted DT40 Cells.
The reconstitution of the Btk- or PLCγ2-deficient DT40 cell lines was performed by electroporation, using pApuro expression vectors containing the T7-tagged cDNAs for human Btk (29) and rat PLCγ2 (30), which were provided by Dr. T. Kurosaki (Kansai Medical University, Moriguchi, Japan). Briefly, 25 μg linearized plasmid DNA (pApuro-T7-Btk or T7-PLCγ2) was added to 107 cells in 0.5 ml culture medium in a 0.4 cm electrode gap Gene Pulser Cuvette (BioRad) and electroporated using a Gene Pulser Apparatus with Capacitance Extender (BioRad) at 250 V, 960 μF. After 24 h recovery at 39.5°C in DT40 medium, cells were selected in DT40 medium containing 0.5 μg/ml Puromycin (Sigma). Puromycin-resistant clones were screened for expression of the transgene by immunoblotting with anti-T7. At least three clones of each reconstituted stable cell line were tested and found to be functionally restored in BCR-induced adhesion.
Immunoblotting.
After stimulation of 107 cells /ml RPMI with 10 μg/ml anti-IgM antibody, cells were directly lysed in SDS-PAGE sample buffer. 2 × 105 cells were applied on a 10% SDS-PAGE gel and blotted with anti-phospho-mitogen-activated protein kinase (MAPK)1/2 and anti-phospho-PKB, followed by HRP-conjugated goat anti-rabbit and developed by enhanced chemiluminescence (Amersham Pharmacia). To confirm equal expression (WT versus deficient DT40 cells) and loading, the blots were stripped or occasionally blots were generated in a parallel fashion, and incubated with the antibodies anti-ERK2 and anti-PKB.
Cell Adhesion Assay.
Adhesion assays were done in triplicate on round-bottom 96-well plates (Costar) coated overnight at 4°C with PBS containing, unless otherwise indicated, 1 μg/ml sVCAM-1, 10 μg/ml Fibronectin, 4% BSA, or for 15 min at 37°C with 1 mg/ml poly-l-lysine (PLL), and blocked for 2 h at 37°C with 4% BSA in RPMI 1640. Cells were pretreated with the pharmacological inhibitors at 37°C or anti-integrin antibodies TS1/22 (1:1000), Act-1 (1:100), TS2/16 (1:5), and HP2/1 (1:250) at 4°C for 30 min in RPMI with 1% BSA. Subsequently, cells were stimulated with either 10 μg/ml mouse anti-human IgM MH15, goat anti-chicken IgM, (Fab)2 fragments of anti-human or anti-mouse IgM, anti-mouse IgM microbeads (1:300) or 50 ng/ml PMA, and 1.5 × 105 Namalwa or DT40 cells, or 3 × 105 primary B cells, were plated in 100 μl/well and incubated at 37°C (39.5°C for DT40) for 30 min, unless otherwise indicated. After extensive washing of the plate with RPMI containing 1% BSA to remove nonadhering cells, the adherent cells were fixed for 10 min with 10% glutaraldehyde in PBS and stained for 45 min with 0.5% crystal violet in 20% methanol. After extensive washing with water, the dye was eluted in methanol and absorbance was measured after 5 to 40 min at 570 nm on a spectrophotometer (Microplate Reader 450, Biorad). Background absorbance (no cells added) was subtracted. Absorbance due to nonspecific adhesion, as determined in wells coated with 4% BSA, was always less than 10% of the absorbance of anti-IgM-stimulated cells. Maximal (100%) adhesion was determined by applying the PMA-stimulated cells to wells coated with the positively charged PLL, resulting in nonspecific adhesion of all cells, without washing the wells before fixation (see also Fig. 1, A-D) . Unless otherwise indicated, adhesion of the nonpretreated anti-IgM-stimulated cells was normalized to 100% and the bars represent the means ±SD of a triplicate experiment representative of at least three independent experiments.
Figure 1.
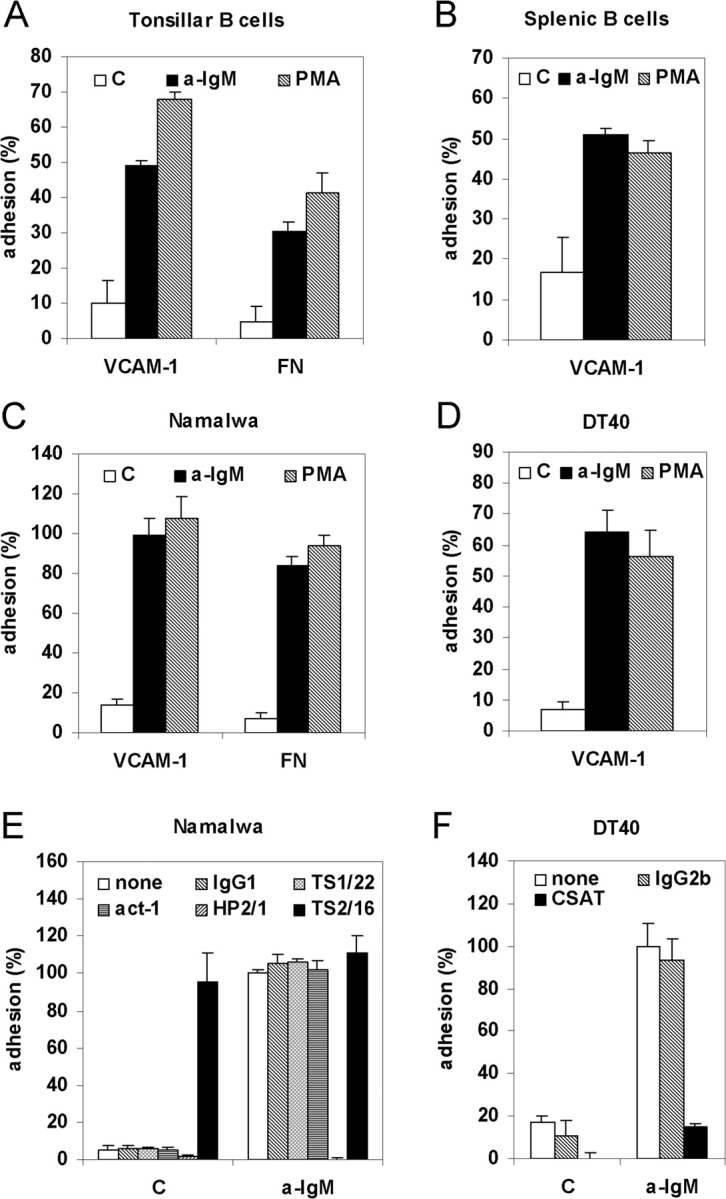
BCR activation induces integrin α4β1-mediated B cell adhesion to VCAM-1 and FN. (A) Primary tonsillar B cells were not stimulated (C) or stimulated with (Fab)2-fragments of anti-IgM (a-IgM) or PMA and plated for 20 min on a surface coated with either VCAM-1 or FN, as indicated. (B) Mouse splenic B cells were not stimulated (C) or stimulated with (Fab)2-fragments of anti-IgM (a-IgM), or PMA and plated on a surface coated with 0.3 μg/ml VCAM-1. (C) Namalwa cells were not stimulated (C) or stimulated with anti-IgM (a-IgM) or PMA and plated on a surface coated with either VCAM-1 or FN, as indicated. (D) DT40 cells were not stimulated (C) or stimulated with anti-IgM (a-IgM) or PMA and plated on a surface coated with VCAM-1. (E) Namalwa cells were preincubated for 1 h at 4°C with medium alone (none), 10 μg/ml of an IgG1 isotype control (IgG1), or antibody TS1/22 blocking LFA-1 (TS1/22), Act-1 blocking α4β7 (Act-1), HP2/1 blocking α4β1 (HP2/1), or antibody TS2/16 activating integrin α4β1 (TS2/16). Subsequently, cells were not stimulated (C) or stimulated with anti-IgM (a-IgM), as indicated, and plated on a surface coated with VCAM-1. (F) DT40 cells were preincubated for 1 h at 4°C with medium alone (none), or 10 μg/ml of an IgG2b isotype control (IgG2b) or antibody CSAT against integrin subunit β1 (CSAT). Subsequently, cells were stimulated with anti-IgM and plated on a surface coated with VCAM-1. (A-D) The adhesion is presented as absolute adhesion, with input being determined by adhesion of all cells to PLL (= 100% adhesion). (E and F) The adhesion was normalized to 100% for the anti-IgM-stimulated cells.
Soluble VCAM-1 Binding Assay.
DT40 cells were incubated with or without 10 μg/ml goat anti-chicken IgM in RPMI + 1% BSA for 10 min at 39.5°C. Subsequently 5 × 104 cells were incubated in a 96-well V-shaped bottom plate without or with 2 or 10 μg/ml soluble VCAM-1-Fc (kindly provided by Y. van Kooyk) in the absence or presence of 3 mM MnCl2 in a final volume of 25 μl for 30 min at 4°C (similar results were obtained when anti-IgM or MnCl2 and VCAM-1-Fc were added simultaneously and cells were incubated for 30 min at 39.5°C). After washing the cells twice, the cells were incubated with goat anti-human Fc-FITC (1:200) in RPMI + 1% BSA for 30 min at 4°C, washed twice, and analyzed by FACS. Mean fluorescence intensity (MFI) values are depicted after subtraction of the MFI values obtained in the absence of soluble VCAM-1-Fc.
Analysis of Integrin α4β1 Clustering by Confocal Microscopy.
DT40 cells (1.5 × 105) were incubated with or without 10 μg/ml goat anti-chicken IgM in RPMI for different time periods at 39.5°C. Subsequently, cells were fixed with 4% paraformaldehyde in PBS for 15 min at RT, incubated with 7.5 μg/ml CSAT for 30 min at 4°C, washed, incubated with biotinylated goat-anti mouse (1:400) for 30 min at 4°C, washed, incubated with Streptavidin-FITC (1:200) for 15 min at 4°C, and washed again. Finally, the cell suspension was spotted onto 0.1% PLL-coated slides, and after 5 min the slides were rinsed and cells were mounted in Vectashield for confocal laser scanning microscopy, using a Leica TCS SP2 with a krypton/argon laser at 488 nm and a 63x lens. To evaluate integrin clustering, horizontal 0.6 μm optical serial sections were taken throughout the cell. Clustering was inspected at midheight optical sections and by whole cell projections along the z-axis using maximum fluorescence values.
Statistical Analysis.
The unpaired two-tailed Student's t test was used to determine the significance of differences between means. All relevant comparisons (e.g., control versus inhibitors or WT versus deficient DT40) were significantly different (P < 0.01).
Results
BCR Activation Induces Integrin α4β1-mediated B Cell Adhesion to VCAM-1 and FN.
To investigate whether the BCR is able to control B cell adhesion, freshly isolated human tonsillar and murine splenic B cells were stimulated with anti-IgM antibody to ligate and activate the BCR, or with the phorbol ester PMA to activate PKC, and plated on a surface coated with either VCAM-1 or FN. As shown in Fig. 1, A and B, BCR activation, as well as PMA treatment, resulted in a strong stimulation of adhesion to both VCAM-1 and FN. Anti-IgM stimulation, as well as PMA-treatment of the human B cell line Namalwa, which resembles GC B cells, also resulted in strongly enhanced adhesion to both VCAM-1 and FN (Fig. 1 C). Similar results were obtained using chicken DT40 B cells (Fig. 1 D).
As shown in Fig. 1 E, antibody HP2/1, known to block α4β1 function, specifically and completely blocked both basal as well as BCR-induced adhesion of Namalwa cells to VCAM-1, whereas no effect was observed with either an isotype control IgG1, or the specific antibodies TS1/22, blocking LFA-1, or Act-1, blocking integrin α4β7. On the other hand, antibody TS2/16, known to activate integrin α4β1, strongly promoted adhesion to VCAM-1. Furthermore, the antibody CSAT, directed against the chicken integrin β1 subunit, blocked the BCR-controlled adhesion of DT40 cells to VCAM-1 (Fig. 1 F). Taken together, our data demonstrate that stimulation of the BCR of either primary B cells or B cell lines results in their enhanced adhesion to VCAM-1, which is mediated by integrin α4β1.
BCR-controlled Adhesion Requires Activation of Lyn, Syk, and PI3K, but Not MAPK Kinase or ERK.
Given the proximal and prominent role in BCR signaling for Lyn, a Src-family tyrosine kinase, and Syk, a member of the ZAP70/Syk family of tyrosine kinases, we investigated their possible role in the regulation of integrin-mediated adhesion by the BCR. As shown in Fig. 2 A, in Lyn/Syk double-deficient DT40 cells the BCR-induced adhesion to VCAM-1 was completely abolished.
Figure 2.

BCR-controlled adhesion is dependent on Lyn and Syk and requires activation of PI3K, but not of MEK or ERK. (A) WT or Lyn/Syk double-deficient DT40 cells (Lyn−/Syk−) were not stimulated (C) or stimulated with anti-IgM (a-IgM) and plated on a surface coated with VCAM-1. (B) Activation of PKB and ERK in WT or Lyn/Syk double-deficient DT40 cells (Lyn−/Syk−) after stimulation with anti-IgM for the indicated period of time (min). The immunoblots are probed with anti-phospho-PKB (P-PKB) and anti-phospho-MAPK (P-ERK) and reprobed with anti-ERK2 and anti-PKB, as indicated. (C) Activation of PKB and ERK in Namalwa cells (left) or DT40 cells (right) which were pretreated with 20 μM LY294002 (LY), 100 nM Wortmannin (WM), 50 μM PD98059 (PD), or left untreated (−) for 30 min, and stimulated with anti-IgM (a-IgM) or not (C) for 5 min, as indicated. The blots are probed as in B. (D) Namalwa cells (left) or DT40 cells (right) were pretreated with 50 μM LY294002 (LY), 100 nM Wortmannin (WM), 50 μM PD98059 (PD), or left untreated for 30 min, and not stimulated (C) or stimulated with anti-IgM (a-IgM) and plated on a surface coated with VCAM-1.
As shown in Fig. 2, B and C, BCR stimulation of DT40 and Namalwa cells results in a strong activation of PKB/Akt-1 and the MAP kinase ERK2/MAPK1 and, in Namalwa cells, also of ERK1. In agreement with previous studies (37–41), and confirming the prominent role of Lyn and Syk in BCR signaling, the BCR-induced activation of PKB and ERK2 was completely abolished in the Lyn/Syk-deficient cells (Fig. 2 B), whereas their activation by PMA was not affected (data not depicted). Furthermore, the activation of ERK1 and 2 is completely abolished by pretreatment of the cells with the MAPK kinase (MEK) inhibitor PD-98059 (PD), but not in DT40, and only partially in Namalwa, by treatment with the PI3K inhibitors LY-294002 (LY) or Wortmannin (WM) (Fig. 2 C). By contrast, PKB activation was completely abrogated by pretreatment with the PI3K inhibitors LY and WM, but was not affected by the MEK inhibitor PD (Fig. 2 C). Thus, BCR stimulation results in activation of PI3K, which is required for the activation of PKB and, in Namalwa, is also partially involved in the activation of ERK.
PI3K, MEK, and ERK have previously been implicated in the regulation of integrin activity by other stimuli (25). As shown in Fig. 2 D, the BCR-induced integrin-mediated adhesion to VCAM-1 of both Namalwa and DT40 cells was completely abolished upon treatment of the cells with the PI3K inhibitors LY or WM, whereas treatment with the MEK inhibitor PD had no effect. Taken together, these data demonstrate that Lyn- and Syk-dependent activation of PI3K is required for BCR-induced α4β1 activation, whereas activation of MEK or ERK is not involved.
Btk Is Required for BCR-controlled Adhesion.
PI3K activation results in the formation of phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 mediates the plasma membrane translocation and activation of several signaling molecules, including the cytoplasmic tyrosine kinase Btk, the serine/threonine kinase PKB, or the exchange factor Vav, as a consequence of the interaction of their PH domains with PIP3. Interestingly, using Btk-deficient DT40 cells, we observed a complete loss of BCR-, but not PMA-, induced adhesion to VCAM-1 (Fig. 3 A). This defect is not due to clonal variation since it could be completely restored by stable transfection of an expression construct encoding Btk (Fig. 3 A). In agreement with previous data, we observed that, whereas ERK2 activation was reduced in the Btk-deficient DT40 cells (40), PKB activation remained unaffected (37, 41; however see 39) (Fig. 3 B). Thus, whereas (PI3K-mediated) activation of PKB is not sufficient, Btk is required for activation of integrin α4β1 by the BCR.
Figure 3.

Btk is required for BCR-controlled adhesion. (A) WT, Btk-deficient (Btk−), or Btk-deficient DT40 cells reconstituted with Btk (Btk+) were not stimulated (C) or stimulated with anti-IgM (a-IgM) or PMA and plated on a surface coated with VCAM-1. The inset shows expression of Btk in the Btk-reconstituted DT40 cell-line as detected by immunoblotting with anti-T7. (B) Activation of PKB and ERK in WT or Btk-deficient (Btk−) DT40 cells after stimulation with anti-IgM for the indicated period of time (min). The blots are probed as in Fig. 2 B. (C) B cells from WT or Btk-deficient (Btk−) mice were not stimulated (C) or stimulated with anti-IgM (a-IgM) or PMA and plated for 15 min on a surface coated with 0.3 μg/ml VCAM-1. The experiment shown is representative for two independent experiments. (D) Nalm6 (circles), DT40 (squares), or Namalwa (triangles) cells were not stimulated (open symbols) or stimulated with PMA (closed symbols) and plated for 20 min on a surface coated with different concentrations of VCAM-1. The adhesion was normalized to 100% for the PMA-stimulated cells plated on 1 μg/ml VCAM-1. The experiment shown is representative for two independent experiments. (E) Pre-B cells from WT or Btk-deficient (Btk−) mice were not stimulated (C) or stimulated with PMA and plated for 15 min on a surface coated with 0.3 μg/ml VCAM-1. The adhesion was normalized to 100% for the unstimulated WT preB cells. The bars represent the means ±SD of 2 WT and 2 Btk-deficient mice, each assayed in triplicate. In a total of three independent experiments (6 WT and 6 Btk-deficient mice), the basal adhesion of the Btk-deficient mice was 54% ± 10% in comparison to WT preB cells.
These observations are particularly interesting since Btk-deficiency causes the B cell immunodeficiency disease XLA in humans and X-linked immunodeficiency (Xid) in mice. Therefore, we further studied the role of Btk in BCR-induced adhesion using B cells derived from Btk-deficient mice. As shown in Fig. 3 C, Btk-deficiency did not affect BCR-controlled integrin-mediated adhesion of mouse B cells. This difference with the chicken DT40 cells may be due to species-specific functional redundancy of Btk for other tec-family kinases in mouse B cells (see Discussion).
Pre-B cell differentiation, which is controlled by the presumably constitutively active ligand-independent preBCR (19, 21, 22, 42), is impaired in Btk-deficient mice (33, 43) and almost completely arrested in XLA patients (44, 45). Interestingly, using mouse preB cells and the human preB cell line Nalm6, in which Btk is constitutively phosphorylated (46, 47), we observed high basal integrin-mediated adhesion which, in contrast to DT40 or Namalwa cells, could not be further enhanced by PMA (Fig. 3, D and E). Noteworthy, the PI3K inhibitor Wortmannin reduced the high basal adhesion of the Nalm6 cells (on 0.125 μg/ml VCAM-1; data not depicted). Moreover, preB cells derived from Btk-deficient mice showed an ∼50% lower basal adhesion in comparison to WT preB cells, which could be enhanced by PMA up to the level of adhesion of the WT preB cells (Fig. 3 E). The WT and Btk-deficient preB cells display similar expression levels of integrin α4 and β1, as determined by FACS analysis (data not depicted). Since the preBCR signals in a presumably constitutively active ligand-independent fashion involving Btk (42, 46, 48, 49), these results suggest that Btk mediates integrin activation by the preBCR. In conclusion, our data reveal a critical regulatory function for Btk in the (pre)BCR-controlled integrin-mediated adhesion.
BCR-controlled Adhesion Requires PLCγ2 Activation, IP3R-mediated Ca2+ Release, and PKC Activation.
Given the intriguing critical role of Btk in BCR-induced integrin activation, we wished to further extend and strengthen our observations by exploring which signaling molecules may control this process downstream from Btk. A major candidate is PLCγ2, since it has been implicated in BCR signaling, and has been identified as a substrate for both Btk and Syk (29). Indeed, as shown in Fig. 4 A, treatment of Namalwa or DT40 cells with the PLC inhibitor U73122 abolished BCR-induced VCAM-1 adhesion. Noteworthy, U73122 also strongly reduced the high basal adhesion of the Nalm6 cells (on 0.125 μg/ml VCAM-1) (data not depicted). Moreover, PLCγ2-deficient DT40 cells showed a complete loss of BCR-, but not PMA-, induced VCAM-1 adhesion (Fig. 4 B), which could be completely restored by gene complementation with a PLCγ2 expression construct (Fig.4 B).
Figure 4.

BCR-controlled adhesion requires activation of PLCγ2, IP3R-mediated Ca2+ release, and PKC. (A) Namalwa cells (left) or DT40 cells (right) were pretreated with 1 or 2.5 μM U73122 (U), respectively, or left untreated for 30 min, and subsequently not stimulated (C) or stimulated with anti-IgM (a-IgM) and plated on a surface coated with VCAM-1. (B) WT, PLCγ2-deficient (PLCγ2−), or PLCγ2-deficient DT40 cells reconstituted with PLCγ2 (PLCγ2+) were not stimulated (C) or stimulated with anti-IgM (a-IgM) or PMA and plated on a surface coated with VCAM-1. The inset shows expression of PLCγ2 in the PLCγ2-reconstituted DT40 cell-line as detected by immunoblotting with anti-T7. (C) WT or IP3R-deficient DT40 cells (IP3R−) were not stimulated (C) or stimulated with anti-IgM (a-IgM) and plated on a surface coated with VCAM-1. (D) Activation of PKB and ERK in WT, PLCγ2(PLCγ2−)-, or IP3R(IP3R−)-deficient DT40 cells after stimulation with anti-IgM for the indicated period of time (min). The blots are probed as in Fig. 2 B. (E) Namalwa cells (left) or DT40 cells (right) were pretreated with 5 or 10 μM Chelerythrine (CE), respectively, or left untreated for 30 min, and subsequently not stimulated (C) or stimulated with anti-IgM (a-IgM) and plated on a surface coated with VCAM-1.
PLC activation results in the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) into inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DG). IP3 binds to IP3Rs on the endoplasmic reticulum resulting in the release of Ca2+, whereas DG activates PKC. Using IP3R-triple-deficient DT40 cells, in which the genes encoding all three types of IP3Rs expressed in these cells have been inactivated resulting in a loss of BCR-mediated Ca2+ release (31), we observed a complete loss of BCR-induced VCAM-1 adhesion (Fig. 4 C). Furthermore, in agreement with previous reports (32, 37, 39, 41), PKB activation does not require any PLCγ2- or IP3R-mediated signals, whereas ERK activation involves DG-dependent activation of PKC but not IP3R-mediated Ca2+-release (Fig. 4 D). Thus, the IP3R-deficient B cells reveal that simultaneous activation of both PKB and ERK is not sufficient for integrin α4β1 activation (Fig. 4, C and D). Finally, treatment of Namalwa or DT40 cells with the PKC inhibitor Chelerythrine abolished PMA- (data not depicted) and BCR-induced VCAM-1 adhesion (Fig. 4 E), whereas treatment with the Ca2+-ionophore Ionomycin did not result in enhanced integrin-mediated adhesion (data not depicted). Combined, these data indicate that activation of PLCγ2 and subsequently the concomitant IP3R-mediated release of calcium and DG-mediated activation of PKC are required for BCR-controlled integrin α4β1-mediated adhesion.
BCR-controlled Integrin α4β1-mediated Adhesion Involves Cytoskeletal Reorganization and Integrin Clustering.
Integrin activation can be accomplished by a conformational change resulting in enhanced ligand affinity, by an increase in lateral mobility and clustering resulting in enhanced avidity, or both (25, 26). As shown in Fig. 5 A, whereas MnCl2 treatment resulted in enhanced binding of soluble VCAM-1 to DT40 cells, no change of α4β1 affinity for VCAM-1 was observed after stimulation with anti-IgM. In contrast, confocal microscopy analysis revealed enhanced α4β1 clustering after anti-IgM stimulation of DT40 cells. Stimulation with anti-IgM for 15 min results in the formation of many small clusters, whereas after 30 min rather large clusters of α4β1 are observed (Fig. 5 B). PMA stimulation results in the formation of larger and more diffuse α4β1 clusters in WT and Btk-deficient DT40 cells (Fig. 5 B). In Btk-deficient DT40 cells, however, consistent with their disability to adhere to VCAM-1 upon BCR activation (Fig. 3), no anti-IgM-induced α4β1 clustering is observed (Fig. 5 B).
Figure 5.
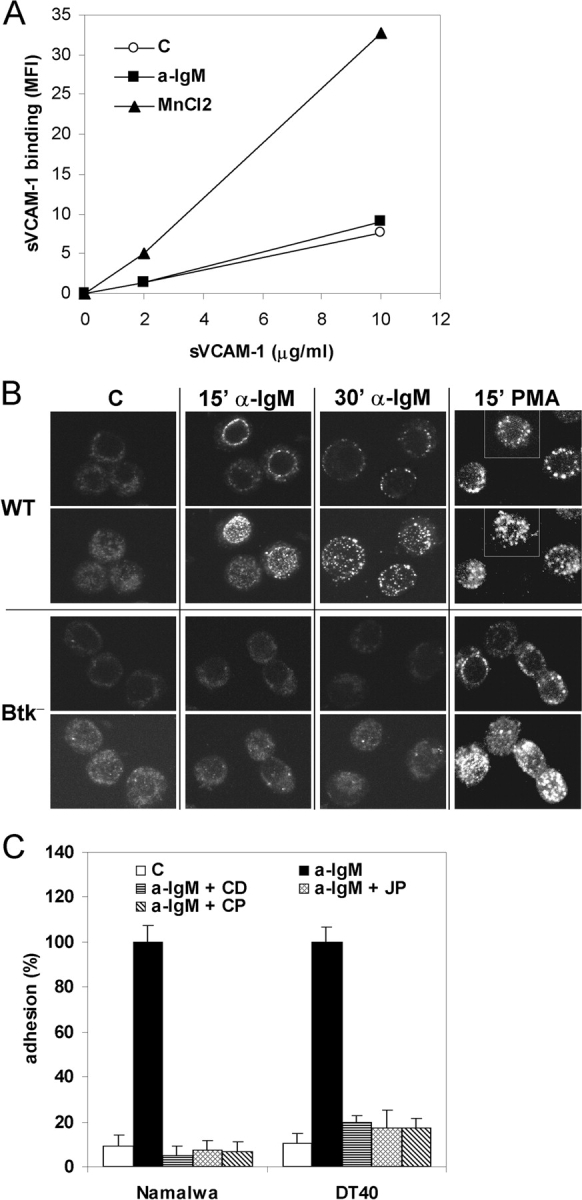
BCR-controlled integrin α4β1-mediated adhesion involves integrin clustering and cytoskeletal reorganization. (A) DT40 cells were incubated without (open circles) or with 10 μg/ml anti-IgM (closed squares) or 3 mM MnCl2 (closed triangles) and 0, 2, or 10 μg/ml soluble VCAM-1-Fc, as indicated. Binding of soluble VCAM-1-Fc was measured by FACS analysis. Mean fluorescence intensity (MFI) values of a representative experiment are depicted. (B) DT40 cells, either WT or Btk-deficient (Btk−), were not stimulated (C) or stimulated for 15 or 30 min with anti-IgM (a-IgM) or PMA, as indicated. Clustering of integrin α4β1 was analyzed by means of confocal microscopy. For each condition, representative images of optical midheight sections (1st and 3rd row), and corresponding whole cell projections along the z-axis using maximum fluorescence values (2nd and 4th row), are shown. (C) Namalwa cells (left) or DT40 cells (right) were pretreated for 30 min with 1 μg/ml Cytochalasin D (CD), 1 μM Jasplakinolide (JP), 100 μg/ml Calpeptin (CP), or left untreated, and subsequently not stimulated (C) or stimulated with anti-IgM (a-IgM) and plated on a surface coated with VCAM-1.
The actin cytoskeleton has been implicated in the control of clustering of integrins, including of LFA-1 and α4β1, and in integrin-mediated adhesion (26, 50–54). To investigate whether (re)organization of the actin cytoskeleton contributes to the BCR-induced α4β1-mediated adhesion, and to provide further support for the role of α4β1 clustering in this process, we examined the effect of Cytochalasin D (CD), which prevents actin filament polymerization and organization, and Jasplakinolide (JP), which prevents actin depolymerization and stabilizes preexisting actin filaments. Since the Ca2+-dependent protease Calpain has previously been implicated in TCR-, chemokine-, and PMA-induced LFA-1 clustering and LFA-1-mediated T cell adhesion (53, 55), and given the observed critical role of IP3R-mediated Ca2+-release in BCR-controlled α4β1 activation (Fig. 4 C), we also studied the effect of an inhibitor of Calpain, Calpeptin (CP). As shown in Fig. 5 C, pretreatment of DT40 and Namalwa cells with CD, JP, or CP completely abolished the BCR-induced α4β1-mediated adhesion to VCAM-1. Taken together, these data indicate that the control of integrin α4β1-mediated B cell adhesion by the BCR involves cytoskeletal (re)organization and stimulation of integrin avidity.
Discussion
The Inside-out Signaling Mechanism Underlying BCR-controlled Integrin α4β1 Activation.
In this study we demonstrate that stimulation of the BCR results in inside-out activation of integrin α4β1 and consequently in strongly enhanced adhesion of B cells to FN and VCAM-1 (Fig. 1). Using a combined biochemical, pharmacological, and genetic approach, we also show that the tyrosine kinases Lyn and Syk, as well as PI3K, are required for BCR-controlled α4β1-mediated adhesion (Fig. 2). By contrast, activation of MEK or the MAP kinases ERK1 or 2 is not involved (Fig. 2 D). In concordance with the observed critical role of Lyn, Syk, and PI3K in α4β1 activation, activation of PI3K by the BCR can be mediated by Lyn (56, 57), and, via the adaptor protein BCAP, by Syk (24, 38, 39). The use of Btk-, PLCγ2-, and IP3R-deficient B cells revealed that PI3K-controlled activation of PKB, either alone or in combination with ERK2, is not sufficient for α4β1 activation (Figs. 3 and 4). Interestingly, we found that the PI3K-controlled tyrosine kinase Btk is a critical mediator of BCR-induced adhesion (Fig. 3). Btk activation requires its translocation to the plasma membrane by the interaction of its PH domain with the PI3K product PIP3, and phosphorylation of Y551 in the kinase domain activation loop (58). Indeed, reflecting their role in PI3K activation or Y551 phosphorylation, BCR-controlled Btk activation is mediated by Lyn and Syk (23, 24). We also found that the Btk substrate PLCγ2 mediates the BCR-controlled adhesion (Fig. 4, A and B). PLCγ2 activation by the BCR requires phosphorylation of the adaptor BLNK by Syk, thus creating docking sites for the SH2 domains of Btk and PLCγ2. This brings PLCγ2 into close proximity of Btk, resulting in its phosphorylation and activation by Btk (24). The apparent bifurcation of the signaling cascade underlying α4β1 activation downstream of PLCγ2, i.e., concomitant IP3-mediated Ca2+ release and DG-mediated activation of PKC (Fig. 4, C and E), either indicates the involvement of a Ca2+-dependent DG-activated PKC isotype (i.e., PKC α or β), or the requirement for PKC activation and Ca2+ release in a parallel fashion. Taken together, we have identified a (linear) cascade of essential signaling components underlying BCR-controlled α4β1 activation, i.e., the (consecutive) activation of Lyn, Syk, PI3K, Btk, PLCγ2, IP3R-mediated Ca2+ release and PKC (Fig. 6) , and although any alternative or branching pathways may also play a role, activation of those pathways alone will not be sufficient for α4β1 activation by the BCR.
Figure 6.

Molecular dissection of the signaling cascade underlying BCR-controlled integrin α4β1-mediated adhesion. A schematic representation of the signaling pathway underlying the inside-out activation mechanism of integrin α4β1 upon stimulation of the BCR is shown. The proteins encoded for by the inactivated genes in the DT40 cells and the pharmacological inhibitors used in this study are boxed and italicized, respectively. For sake of clarity, the role of actin cytoskeleton (re)organization is not included. See the Discussion section for further detail.
α4β1-mediated adhesion can be controlled by integrin clustering (avidity) (50, 51, 54), or by active regulation of α4β1 affinity (59, 60). Our data revealed that the stimulation of the BCR results in enhanced α4β1 avidity, whereas no change in affinity was observed (Fig. 5). The role for α4β1 avidity regulation in the control of adhesion is further supported by our observation that BCR-induced α4β1-mediated adhesion is controlled by Ca2+-release, activation of Calpain and (re)organization of the actin cytoskeleton (Figs. 4 and 5), since these processes have previously been implicated in clustering of integrins, in particular of LFA-1, underlying TCR- and chemokine-induced LFA-1-mediated adhesion (26, 51–55). In addition, recent studies also implicated membrane lipid rafts in the positive control of clustering and activation of integrins, indicating that the role of the cytoskeleton may involve stabilization of raft structures as well (61, 62). Combining our data with these studies, we propose that upon BCR stimulation α4β1 may initially be released from a cytoskeletal constraint by Ca2+-mediated Calpain activation (CP-sensitive) or actin depolymerization (JP-sensitive), followed by mobilization to lipid rafts which are stabilized by the cytoskeleton (JP- and CD-sensitive), and the subsequent formation of α4β1 clusters which may become tethered to the (repolymerized) actin cytoskeleton again (JP- and CD-sensitive), resulting in enhanced α4β1 avidity and adhesion.
Integrin α4β1-mediated Adhesion in B Cell Development and Differentiation.
During B cell development both the preBCR (19, 21, 22), and integrin-mediated adhesion to VCAM-1-expressing BM stromal cells and ECM (1, 3, 4), play a critical role in the survival of preB cells and their differentiation into immature B cells. The preBCR, which presumably signals in a ligand-independent fashion, controls signaling molecules similar to the BCR (42, 48). Therefore, it is tempting to speculate that expression of the preBCR controls integrin activity (see Fig. 3, D and E), which may be required for retention of the preB cells in the proper BM microenvironment and their transition into immature B cells.
Previously, we and others have shown that the interaction of GC B cells with FDCs, which plays a major role in antigen-specific B cell differentiation, is mediated by α4β1 (2, 13, 14), and that adhesion through α4β1 suppresses apoptosis of GC B cells (16, 17). Combined with our present findings, we propose that antigen-specific B cell differentiation is controlled by stimulation of the BCR by the FDC-presented antigen, generating signals that result in inside-out integrin activation. This will “lock” the interaction of the GC B cell with the FDC. The enhanced α4β1-mediated adhesion to FDCs may counteract GC B cell migration along a chemokine gradient, e.g., of BLC. After the initial migration arrest, the integrin-mediated formation of clusters of B cells and FDCs will give rise to the structural microenvironment of the GC. The retention of the GC B cells in these GCs and their contact with the FDC plays an important role in affinity maturation: without the proper signals provided by the GC microenvironment in general, and by the FDCs in particular, GC B cells will not proliferate and undergo apoptosis (12, 15). Moreover, the interaction with the FDC also generates integrin-mediated anti-apoptotic outside-in signals which may directly contribute to the clonal selection of the GC B cells (16, 17). Thus, by competing for antigen and integrin-mediated survival signals, B cells expressing a high affinity BCR will prevent or terminate the BCR signal in a B cell of lower affinity, which will consequently “unlock” from the FDC, resulting in apoptosis.
A Critical Role for Btk in the Control of Integrin Activity: Implications for XLA and Xid.
This study is the first to demonstrate a critical role for Btk (and PLCγ2 and IP3Rs) in the control of integrin activity (Fig. 3). Interestingly, loss-of-function germline mutations in the BTK gene give rise to the B cell immunodeficiency disease XLA in humans and Xid in mice. XLA patients show a severe reduction in mature B cell numbers (>99%) and consequently in Ig serum levels (44, 45). In Xid and the phenotypically identical Btk-deficient mice, however, mature B cell numbers are only 50% reduced (63). This may be due to functional redundancy of Btk with other Tec family members in murine B cells, as illustrated by the ability to overcome signaling defects in Btk-deficient DT40 cells and XLA patient B cells by expression of other Tec family members (64, 65), and the observation that, in contrast to the Btk- or Tec-deficient mice, mature B-cells are almost absent in Btk/Tec double-deficient mice (66). In particular since, in contrast to Btk-deficient chicken DT40 cells (29) and XLA patient B cells (65), Xid mice mature B cells also show (near) normal BCR-induced activation of PLCγ2 (67), PI hydrolysis (68), and Ca2+ release (67–69), this species-specific redundancy may explain why Btk-deficient murine naive B cells exhibit normal BCR-induced adhesion as well (Fig. 3 C).
The earliest role for Btk in B cell development occurs at the switch from preB cells to immature B cells, which is severely impaired in XLA patients and, to a lesser extent, in Xid and Btk − / − mice (33, 43, 44). In XLA this is due to a proliferation defect of the μH-chain positive preB cells (45). Supported by the observation that the proliferation of preB cells from α4 integrin-deficient mice is also reduced (4), and because the preBCR signals through Btk (46, 48, 49), our results suggest that the loss of control of integrin-mediated adhesion by the preBCR may contribute to this proliferation defect in XLA preB cells (Fig. 3, D and E; see also discussion above). In addition, both Btk and integrins are involved in the retention (2, 70) and positive selection (63, 71) of immature B cells in the BM, as well as in the development, survival and follicular entry of long-lived recirculating follicular cells (33, 63, 72). Furthermore, besides a loss of T-independent antigen responses, Xid and Btk-deficient mice show a strongly reduced primary and a variably affected secondary T-dependent antigen response, whereas both responses are absent in Btk/Tec double-deficient, which is accompanied by smaller follicles and less GCs (63, 66, 73, 74). Gain-of-function E41K-Btk transgenic mice also revealed a loss of the T-dependent antigen response, smaller follicles and the absence of marginal zones and GCs (72). This may reflect a defect in (BCR-controlled) α4β1 activity of GC B cells, which, as discussed above, has an important role in GC formation and antigen-specific B cell differentiation. Finally, it is noteworthy that mice deficient in the p85 subunit of PI3K, BLNK, PLCγ2, or PKCβ, which like Btk play a critical role in the signaling cascade underlying BCR-controlled integrin activity (Figs. 2, 4, and 6), reveal a Xid-like phenotype as well (75).
In conclusion, whereas the control of integrin activity by the (pre)BCR plays an important role in B cell development and antigen-specific B cell differentiation, the loss of control of integrin activity, due to loss-of-function germline mutations of BTK, may contribute to the developmental and functional B cell defects as observed in Xid mice and XLA patients.
Acknowledgments
We thank Lia Smit, Robbert van der Voort, and Esther Schilder-Tol for their valuable technical assistance and helpful comments; Tomohiro Kurosaki for kindly providing the DT40 cells and various constructs; Michelle Klitsie, Peter van Rijn, Nico de Vries, and coworkers of the Department of Otolaryngology of the Sint Lucas/Andreas Hospital Amsterdam for providing the tonsils; Ronald van der Neut (University of Amsterdam) for providing mouse spleens and for critical reading of the manuscript; and Yvette van Kooyk and Sandra van Vliet for providing VCAM-1-Fc and for helpful suggestions on the sVCAM-1 binding assay and confocal microscopy integrin clustering analysis.
Abbreviations used in this paper: BCR, B cell antigen receptor; Btk, Bruton's tyrosine kinase; CD, cytochalasin D; CP, Calpeptin; DG, diacylglycerol; ERK, extracellular signal–regulated kinase; FDC, follicular dendritic cell; FN, fibronectin; GC, germinal center; IP3, inositol-1,4,5-trisphosphate; JP, jasplakinolide; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase; MFI, mean fluorescence intensity; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PLC, phospholipase C; PLL, poly-l-lysine; VCAM, vascular cell adhesion molecule; Xid, X-linked immunodeficiency; XLA, X-linked agammaglobulinaemia.
References
- 1.Miyake, K., I.L. Weissman, J.S. Greenberger, and P.W. Kincade. 1991. Evidence for a role of the integrin VLA-4 in lympho-hemopoiesis. J. Exp. Med. 173:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leuker, C.E., M. Labow, W. Muller, and N. Wagner. 2001. Neonatally induced inactivation of the vascular cell adhesion molecule 1 gene impairs B cell localization and T cell-dependent humoral immune response. J. Exp. Med. 193:755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arroyo, A.G., J.T. Yang, H. Rayburn, and R.O. Hynes. 1996. Differential requirements for alpha4 integrins during fetal and adult hematopoiesis. Cell. 85:997–1008. [DOI] [PubMed] [Google Scholar]
- 4.Arroyo, A.G., J.T. Yang, H. Rayburn, and R.O. Hynes. 1999. Alpha4 integrins regulate the proliferation/differentiation balance of multilineage hematopoietic progenitors in vivo. Immunity. 11:555–566. [DOI] [PubMed] [Google Scholar]
- 5.Koni, P.A., S.K. Joshi, U.A. Temann, D. Olson, L. Burkly, and R.A. Flavell. 2001. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J. Exp. Med. 193:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berlin-Rufenach, C., F. Otto, M. Mathies, J. Westermann, M.J. Owen, A. Hamann, and N. Hogg. 1999. Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1-deficient mice. J. Exp. Med. 189:1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butcher, E.C., and L.J. Picker. 1996. Lymphocyte homing and homeostasis. Science. 272:60–66. [DOI] [PubMed] [Google Scholar]
- 8.Kunkel, E.J., and E.C. Butcher. 2002. Chemokines and the tissue-specific migration of lymphocytes. Immunity. 16:1–4. [DOI] [PubMed] [Google Scholar]
- 9.Liu, Y.J., and C. Arpin. 1997. Germinal center development. Immunol. Rev. 156:111–126. [DOI] [PubMed] [Google Scholar]
- 10.MacLennan, I.C. 1994. Germinal centers. Annu. Rev. Immunol. 12:117–139. [DOI] [PubMed] [Google Scholar]
- 11.Lindhout, E., G. Koopman, S.T. Pals, and C. de Groot. 1997. Triple check for antigen specificity of B cells during germinal centre reactions. Immunol. Today. 18:573–577. [DOI] [PubMed] [Google Scholar]
- 12.van Eijk, M., T. Defrance, A. Hennino, and C. de Groot. 2001. Death-receptor contribution to the germinal-center reaction. Trends Immunol. 22:677–682. [DOI] [PubMed] [Google Scholar]
- 13.Freedman, A.S., J.M. Munro, G.E. Rice, M.P. Bevilacqua, C. Morimoto, B.W. McIntyre, K. Rhynhart, J.S. Pober, and L.M. Nadler. 1990. Adhesion of human B cells to germinal centers in vitro involves VLA-4 and INCAM-110. Science. 249:1030–1033. [DOI] [PubMed] [Google Scholar]
- 14.Koopman, G., H.K. Parmentier, H.J. Schuurman, W. Newman, C.J. Meijer, and S.T. Pals. 1991. Adhesion of human B cells to follicular dendritic cells involves both the lymphocyte function-associated antigen 1/intercellular adhesion molecule 1 and very late antigen 4/vascular cell adhesion molecule 1 pathways. J. Exp. Med. 173:1297–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosco, M.H., E. Pflugfelder, and D. Gray. 1992. Follicular dendritic cell-dependent adhesion and proliferation of B cells in vitro. J. Immunol. 148:2331–2339. [PubMed] [Google Scholar]
- 16.Koopman, G., R.M. Keehnen, E. Lindhout, W. Newman, Y. Shimizu, G.A. van Seventer, C. de Groot, and S.T. Pals. 1994. Adhesion through the LFA-1 (CD11a/CD18)-ICAM-1 (CD54) and the VLA-4 (CD49d)-VCAM-1 (CD106) pathways prevents apoptosis of germinal center B cells. J. Immunol. 152:3760–3767. [PubMed] [Google Scholar]
- 17.Koopman, G., R.M. Keehnen, E. Lindhout, D.F. Zhou, C. de Groot, and S.T. Pals. 1997. Germinal center B cells rescued from apoptosis by CD40 ligation or attachment to follicular dendritic cells, but not by engagement of surface immunoglobulin or adhesion receptors, become resistant to CD95-induced apoptosis. Eur. J. Immunol. 27:1–7. [DOI] [PubMed] [Google Scholar]
- 18.Brakebusch, C., S. Fillatreau, A.J. Potocnik, G. Bungartz, P. Wilhelm, M. Svensson, P. Kearney, H. Korner, D. Gray, and R. Fassler. 2002. Beta1 integrin is not essential for hematopoiesis but is necessary for the T cell-dependent IgM antibody response. Immunity. 16:465–477. [DOI] [PubMed] [Google Scholar]
- 19.Meffre, E., R. Casellas, and M.C. Nussenzweig. 2000. Antibody regulation of B cell development. Nat. Immunol. 1:379–385. [DOI] [PubMed] [Google Scholar]
- 20.Niiro, H., and E.A. Clark. 2002. Regulation of B-cell fate by antigen-receptor signals. Nat. Rev. Immunol. 2:945–956. [DOI] [PubMed] [Google Scholar]
- 21.Papavasiliou, F., Z. Misulovin, H. Suh, and M.C. Nussenzweig. 1995. The role of Ig beta in precursor B cell transition and allelic exclusion. Science. 268:408–411. [DOI] [PubMed] [Google Scholar]
- 22.Torres, R.M., H. Flaswinkel, M. Reth, and K. Rajewsky. 1996. Aberrant B cell development and immune response in mice with a compromised BCR complex. Science. 272:1804–1808. [DOI] [PubMed] [Google Scholar]
- 23.Kurosaki, T. 1999. Genetic analysis of B cell antigen receptor signaling. Annu. Rev. Immunol. 17:555–592. [DOI] [PubMed] [Google Scholar]
- 24.Kurosaki, T. 2002. Regulation of B-cell signal transduction by adaptor proteins. Nat. Rev. Immunol. 2:354–363. [DOI] [PubMed] [Google Scholar]
- 25.Hughes, P.E., and M. Pfaff. 1998. Integrin affinity modulation. Trends Cell Biol. 8:359–364. [DOI] [PubMed] [Google Scholar]
- 26.van Kooyk, Y., and C.G. Figdor. 2000. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr. Opin. Cell Biol. 12:542–547. [DOI] [PubMed] [Google Scholar]
- 27.van der Voort, R., T.E. Taher, V.J. Wielenga, M. Spaargaren, R. Prevo, L. Smit, G. David, G. Hartmann, E. Gherardi, and S.T. Pals. 1999. Heparan sulfate-modified CD44 promotes hepatocyte growth factor/scatter factor-induced signal transduction through the receptor tyrosine kinase c-Met. J. Biol. Chem. 274:6499–6506. [DOI] [PubMed] [Google Scholar]
- 28.van der Voort, R., T.E. Taher, R.M. Keehnen, L. Smit, M. Groenink, and S.T. Pals. 1997. Paracrine regulation of germinal center B cell adhesion through the c-met-hepatocyte growth factor/scatter factor pathway. J. Exp. Med. 185:2121–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takata, M., and T. Kurosaki. 1996. A role for Bruton's tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J. Exp. Med. 184:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takata, M., Y. Homma, and T. Kurosaki. 1995. Requirement of phospholipase C-gamma 2 activation in surface immunoglobulin M-induced B cell apoptosis. J. Exp. Med. 182:907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugawara, H., M. Kurosaki, M. Takata, and T. Kurosaki. 1997. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 16:3078–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashimoto, A., H. Okada, A. Jiang, M. Kurosaki, S. Greenberg, E.A. Clark, and T. Kurosaki. 1998. Involvement of guanosine triphosphatases and phospholipase C-gamma2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J. Exp. Med. 188:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hendriks, R.W., M.F. de Bruijn, A. Maas, G.M. Dingjan, A. Karis, and F. Grosveld. 1996. Inactivation of Btk by insertion of lacZ reveals defects in B cell development only past the pre-B cell stage. EMBO J. 15:4862–4872. [PMC free article] [PubMed] [Google Scholar]
- 34.Russell, D.M., Z. Dembic, G. Morahan, J.F. Miller, K. Burki, and D. Nemazee. 1991. Peripheral deletion of self-reactive B cells. Nature. 354:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rolink, A., M. Streb, and F. Melchers. 1991. The kappa/lambda ratio in surface immunoglobulin molecules on B lymphocytes differentiating from DHJH-rearranged murine pre-B cell clones in vitro. Eur. J. Immunol. 21:2895–2898. [DOI] [PubMed] [Google Scholar]
- 36.Dingjan, G.M., S. Middendorp, K. Dahlenborg, A. Maas, F. Grosveld, and R.W. Hendriks. 2001. Bruton's tyrosine kinase regulates the activation of gene rearrangements at the lambda light chain locus in precursor B cells in the mouse. J. Exp. Med. 193:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gold, M.R., M.P. Scheid, L. Santos, M. Dang-Lawson, R.A. Roth, L. Matsuuchi, V. Duronio, and D.L. Krebs. 1999. The B cell antigen receptor activates the Akt (protein kinase B)/glycogen synthase kinase-3 signaling pathway via phosphatidylinositol 3-kinase. J. Immunol. 163:1894–1905. [PubMed] [Google Scholar]
- 38.Beitz, L.O., D.A. Fruman, T. Kurosaki, L.C. Cantley, and A.M. Scharenberg. 1999. SYK is upstream of phosphoinositide 3-kinase in B cell receptor signaling. J. Biol. Chem. 274:32662–32666. [DOI] [PubMed] [Google Scholar]
- 39.Craxton, A., A. Jiang, T. Kurosaki, and E.A. Clark. 1999. Syk and Bruton's tyrosine kinase are required for B cell antigen receptor-mediated activation of the kinase Akt. J. Biol. Chem. 274:30644–30650. [DOI] [PubMed] [Google Scholar]
- 40.Jiang, A., A. Craxton, T. Kurosaki, and E.A. Clark. 1998. Different protein tyrosine kinases are required for B cell antigen receptor-mediated activation of extracellular signal-regulated kinase, c-Jun NH2-terminal kinase 1, and p38 mitogen-activated protein kinase. J. Exp. Med. 188:1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pogue, S.L., T. Kurosaki, J. Bolen, and R. Herbst. 2000. B cell antigen receptor-induced activation of Akt promotes B cell survival and is dependent on Syk kinase. J. Immunol. 165:1300–1306. [DOI] [PubMed] [Google Scholar]
- 42.Bannish, G., E.M. Fuentes-Panana, J.C. Cambier, W.S. Pear, and J.G. Monroe. 2001. Ligand-independent signaling functions for the B lymphocyte antigen receptor and their role in positive selection during B lymphopoiesis. J. Exp. Med. 194:1583–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Middendorp, S., G.M. Dingjan, and R.W. Hendriks. 2002. Impaired precursor B cell differentiation in Bruton's tyrosine kinase-deficient mice. J. Immunol. 168:2695–2703. [DOI] [PubMed] [Google Scholar]
- 44.Conley, M.E., and M.D. Cooper. 1998. Genetic basis of abnormal B cell development. Curr. Opin. Immunol. 10:399–406. [DOI] [PubMed] [Google Scholar]
- 45.Nomura, K., H. Kanegane, H. Karasuyama, S. Tsukada, K. Agematsu, G. Murakami, S. Sakazume, M. Sako, R. Tanaka, Y. Kuniya, et al. 2000. Genetic defect in human X-linked agammaglobulinemia impedes a maturational evolution of pro-B cells into a later stage of pre-B cells in the B-cell differentiation pathway. Blood. 96:610–617. [PubMed] [Google Scholar]
- 46.Aoki, Y., K.J. Isselbacher, and S. Pillai. 1994. Bruton tyrosine kinase is tyrosine phosphorylated and activated in pre-B lymphocytes and receptor-ligated B cells. Proc. Natl. Acad. Sci. USA. 91:10606–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitanaka, A., H. Mano, M.E. Conley, and D. Campana. 1998. Expression and activation of the nonreceptor tyrosine kinase Tec in human B cells. Blood. 91:940–948. [PubMed] [Google Scholar]
- 48.Guo, B., R.M. Kato, M. Garcia-Lloret, M.I. Wahl, and D.J. Rawlings. 2000. Engagement of the human pre-B cell receptor generates a lipid raft-dependent calcium signaling complex. Immunity. 13:243–253. [DOI] [PubMed] [Google Scholar]
- 49.Kouro, T., K. Nagata, S. Takaki, S. Nisitani, M. Hirano, M.I. Wahl, O.N. Witte, H. Karasuyama, and K. Takatsu. 2001. Bruton's tyrosine kinase is required for signaling the CD79b-mediated pro-B to pre-B cell transition. Int. Immunol. 13:485–493. [DOI] [PubMed] [Google Scholar]
- 50.Grabovsky, V., S. Feigelson, C. Chen, D.A. Bleijs, A. Peled, G. Cinamon, F. Baleux, F. Arenzana-Seisdedos, T. Lapidot, Y. van Kooyk, et al. 2000. Subsecond induction of alpha4 integrin clustering by immobilized chemokines stimulates leukocyte tethering and rolling on endothelial vascular cell adhesion molecule 1 under flow conditions. J. Exp. Med. 192:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leitinger, B., and N. Hogg. 2000. Effects of I domain deletion on the function of the beta2 integrin lymphocyte function-associated antigen-1. Mol. Biol. Cell. 11:677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Kooyk, Y., S.J. van Vliet, and C.G. Figdor. 1999. The actin cytoskeleton regulates LFA-1 ligand binding through avidity rather than affinity changes. J. Biol. Chem. 274:26869–26877. [DOI] [PubMed] [Google Scholar]
- 53.Stewart, M.P., A. McDowall, and N. Hogg. 1998. LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J. Cell Biol. 140:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yauch, R.L., D.P. Felsenfeld, S.K. Kraeft, L.B. Chen, M.P. Sheetz, and M.E. Hemler. 1997. Mutational evidence for control of cell adhesion through integrin diffusion/clustering, independent of ligand binding. J. Exp. Med. 186:1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Constantin, G., M. Majeed, C. Giagulli, L. Piccio, J.Y. Kim, E.C. Butcher, and C. Laudanna. 2000. Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 13:759–769. [DOI] [PubMed] [Google Scholar]
- 56.Buhl, A.M., C.M. Pleiman, R.C. Rickert, and J.C. Cambier. 1997. Qualitative regulation of B cell antigen receptor signaling by CD19: selective requirement for PI3-kinase activation, inositol-1,4,5-trisphosphate production and Ca2+ mobilization. J. Exp. Med. 186:1897–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pleiman, C.M., W.M. Hertz, and J.C. Cambier. 1994. Activation of phosphatidylinositol-3′ kinase by Src-family kinase SH3 binding to the p85 subunit. Science. 263:1609–1612. [DOI] [PubMed] [Google Scholar]
- 58.Rawlings, D.J., A.M. Scharenberg, H. Park, M.I. Wahl, S. Lin, R.M. Kato, A.C. Fluckiger, O.N. Witte, and J.P. Kinet. 1996. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science. 271:822–825. [DOI] [PubMed] [Google Scholar]
- 59.Rose, D.M., P.M. Cardarelli, R.R. Cobb, and M.H. Ginsberg. 2000. Soluble VCAM-1 binding to alpha4 integrins is cell-type specific and activation dependent and is disrupted during apoptosis in T cells. Blood. 95:602–609. [PubMed] [Google Scholar]
- 60.Chigaev, A., A.M. Blenc, J.V. Braaten, N. Kumaraswamy, C.L. Kepley, R.P. Andrews, J.M. Oliver, B.S. Edwards, E.R. Prossnitz, R.S. Larson, and L.A. Sklar. 2001. Real time analysis of the affinity regulation of alpha 4-integrin. The physiologically activated receptor is intermediate in affinity between resting and Mn(2+) or antibody activation. J. Biol. Chem. 276:48670–48678. [DOI] [PubMed] [Google Scholar]
- 61.Leitinger, B., and N. Hogg. 2002. The involvement of lipid rafts in the regulation of integrin function. J. Cell Sci. 115:963–972. [DOI] [PubMed] [Google Scholar]
- 62.Krauss, K., and P. Altevogt. 1999. Integrin leukocyte function-associated antigen-1-mediated cell binding can be activated by clustering of membrane rafts. J. Biol. Chem. 274:36921–36927. [DOI] [PubMed] [Google Scholar]
- 63.Khan, W.N., F.W. Alt, R.M. Gerstein, B.A. Malynn, I. Larsson, G. Rathbun, L. Davidson, S. Muller, A.B. Kantor, L.A. Herzenberg, et al. 1995. Defective B cell development and function in Btk-deficient mice. Immunity. 3:283–299. [DOI] [PubMed] [Google Scholar]
- 64.Tomlinson, M.G., T. Kurosaki, A.E. Berson, G.H. Fujii, J.A. Johnston, and J.B. Bolen. 1999. Reconstitution of Btk signaling by the atypical tec family tyrosine kinases Bmx and Txk. J. Biol. Chem. 274:13577–13585. [DOI] [PubMed] [Google Scholar]
- 65.Fluckiger, A.C., Z. Li, R.M. Kato, M.I. Wahl, H.D. Ochs, R. Longnecker, J.P. Kinet, O.N. Witte, A.M. Scharenberg, and D.J. Rawlings. 1998. Btk/Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. EMBO J. 17:1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ellmeier, W., S. Jung, M.J. Sunshine, F. Hatam, Y. Xu, D. Baltimore, H. Mano, and D.R. Littman. 2000. Severe B cell deficiency in mice lacking the tec kinase family members Tec and Btk. J. Exp. Med. 192:1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Forssell, J., A. Nilsson, and P. Sideras. 2000. Reduced formation of phosphatidic acid upon B-cell receptor triggering of mouse B-lymphocytes lacking Bruton's tyrosine kinase. Scand. J. Immunol. 52:30–38. [DOI] [PubMed] [Google Scholar]
- 68.Rigley, K.P., M.M. Harnett, R.J. Phillips, and G.G. Klaus. 1989. Analysis of signaling via surface immunoglobulin receptors on B cells from CBA/N mice. Eur. J. Immunol. 19:2081–2086. [DOI] [PubMed] [Google Scholar]
- 69.Fujimoto, M., J.C. Poe, A.B. Satterthwaite, M.I. Wahl, O.N. Witte, and T.F. Tedder. 2002. Complementary roles for CD19 and Bruton's tyrosine kinase in B lymphocyte signal transduction. J. Immunol. 168:5465–5476. [DOI] [PubMed] [Google Scholar]
- 70.Cariappa, A., T.J. Kim, and S. Pillai. 1999. Accelerated emigration of B lymphocytes in the Xid mouse. J. Immunol. 162:4417–4423. [PubMed] [Google Scholar]
- 71.Martin, F., and J.F. Kearney. 2000. Positive selection from newly formed to marginal zone B cells depends on the rate of clonal production, CD19, and btk. Immunity. 12:39–49. [DOI] [PubMed] [Google Scholar]
- 72.Dingjan, G.M., A. Maas, M.C. Nawijn, L. Smit, J.S. Voerman, F. Grosveld, and R.W. Hendriks. 1998. Severe B cell deficiency and disrupted splenic architecture in transgenic mice expressing the E41K mutated form of Bruton's tyrosine kinase. EMBO J. 17:5309–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ridderstad, A., G.J. Nossal, and D.M. Tarlinton. 1996. The xid mutation diminishes memory B cell generation but does not affect somatic hypermutation and selection. J. Immunol. 157:3357–3365. [PubMed] [Google Scholar]
- 74.Vinuesa, C.G., Y. Sunners, J. Pongracz, J. Ball, K.M. Toellner, D. Taylor, I.C. MacLennan, and M.C. Cook. 2001. Tracking the response of Xid B cells in vivo: TI-2 antigen induces migration and proliferation but Btk is essential for terminal differentiation. Eur. J. Immunol. 31:1340–1350. [DOI] [PubMed] [Google Scholar]
- 75.Fruman, D.A., A.B. Satterthwaite, and O.N. Witte. 2000. Xid-like phenotypes: a B cell signalosome takes shape. Immunity. 13:1–3. [DOI] [PubMed] [Google Scholar]