

Abstract
Interleukin (IL)-4–secreting tumors are rejected in mice, an effect that is thought to be immune mediated. However, solid tumors are embedded in a stroma that often contains tumor-promoting fibroblasts, a cell population whose function is also affected by IL-4. Here we show that IL-4–secreting tumors grew undiminished in IL-4 receptor (R)–deficient (IL-4R−/−) mice. In IL-4R+/+ mice they were long-term suppressed in the absence of T cells but complete rejection required T cells, compatible with the assumption that hematopoietic cells needed to respond to IL-4. Surprisingly, bone marrow (BM) chimeric mice revealed that IL-4R expression exclusively on non-BM–derived cells was sufficient for tumor rejection. Fibroblasts in the tumor stroma were identified as a target cell type for IL-4 because they accumulated in IL-4–secreting tumors and displayed an activated phenotype. Additionally, coinjection of IL-4R+/+ but not IL-4R−/− fibroblasts was sufficient for the rejection of IL-4–secreting tumors in IL-4R−/− mice. Our data demonstrate a novel mechanism by which IL-4 contributes to tumor rejection and show that the targeted modulation of tumor-associated fibroblasts can be sufficient for tumor rejection.
Keywords: IL-4 receptor, knockout mice, bone marrow transplantation, angiogenesis, collagen
Introduction
The antitumor effect of IL-4 was discovered more than a decade ago (1) and many studies suggested that IL-4–secreting tumors were rejected in an immunological manner (2, 3). The importance of particular cell types for IL-4–mediated tumor rejection was concluded from their accumulation within the tumor or from impaired tumor rejection after their depletion in vivo. These experiments demonstrated that tumor rejection occurred in a biphasic manner. Long-term suppression but usually not complete rejection was observed in the absence of T cells. Granulocytes were made responsible, at least in part, for inhibition of tumor burden (4) and CD8+ T cells for complete rejection of IL-4–secreting tumors (5–7). However, granulocyte depletion by antibodies only partially restored tumor growth and experiments were terminated too early to decide whether IL-4–secreting tumors would have been rejected despite the depletion of granulocytes. This indicated that there might be other cell types that inhibited tumor growth. Importantly, it has not been analyzed whether tumor rejection resulted from a direct or indirect action of IL-4 on immune cells and whether nonhematopoietic cells, e.g., tumor stroma cells, were involved.
Solid tumors are embedded in a stroma that is usually composed of inflammatory cells as well as non-BM–derived cells. Fibroblasts are the predominant stromal cell in primary carcinomas (8, 9). Their almost ubiquitous presence in tumors suggests that they might support tumor growth. Indeed, in a number of tumor transplantation models it was shown that coinjection of fibroblasts with tumor cells of different malignancies consistently accelerated tumor growth or facilitated tumor take (10–15). This emphasizes that in addition to genetic alterations of tumor cells, interactions between tumor cells and fibroblasts are critically important for tumor growth (8). Fibroblasts express IL-4R (16, 17) and respond to IL-4 with increased proliferation (17, 18) and synthesis of extracellular matrix proteins (17, 19–22). Given that (a) IL-4Rs are expressed on most, if not all, cell types (16), and (b) that IL-4 mediates its antitumor effect locally rather than systemically (1), we asked whether non-BM–derived stromal cells contribute to IL-4–mediated tumor rejection. Surprisingly, IL-4 responsiveness exclusively by tumor stroma fibroblasts was sufficient for rejection of IL-4–secreting tumors.
Materials and Methods
Mice, Cell Lines, Tumor Cell Injection, and Histology.
BALB/c mice were purchased from Charles River Laboratories. RAG2−/− (C.129(B6)-Rag2 tm1 N12) BALB/c mice were purchased from Taconic. IL-4–deficient (IL-4−/−) IL-4R α chain–deficient (IL-4R−/−; reference 23) and RAG2−/−/IL-4R−/− double knockout BALB/c mice were provided by N. Noben-Trauth (George Washington University, Washington, DC) and bred in our animal facility. J558L is a BALB/c plasmacytoma cell line. Its Il-4–secreting derivative (J558-IL-4) has been described (6). Within 24 h of culture, 106 J558-IL-4 cells secrete 6.3 ng/ml IL-4 as determined by ELISA (BD Biosciences). The tumor cell line was cultured in RPMI 1640 plus 10% FCS, penicillin/streptomycin, MEM, and 2-ME (50 μM; complete RPMI) with 1 mg/ml G418. For the analysis of tumor growth in vivo, mice were injected s.c. with 1–2 × 106 cells. The tumor diameter was determined as the mean of the largest diameter and the diameter at right angle. Mice that developed a tumor of 1 cm in diameter were scored as tumor positive. Mice shown as tumor free at the end of the experiment had completely rejected the tumor. For the immunohistological analysis of tumor tissues from WT and IL-4R−/− mice, 107 tumor cells were injected s.c. 6 d later, tumor tissues were removed and cryo sectioned. To detect fibroblasts/extracellular matrix and collagen I, the monoclonal antibodies ER-TR7 (BMA Biomedicals; reference 24) and anti–collagen I (Dunn Labortechnik) were used, respectively. Subsequent alkaline phosphatase staining was performed by standard technique using enzyme-coupled secondary antibodies from Jackson ImmunoResearch Laboratories.
Generation of BM Chimeras.
BM was harvested from femurs of WT and IL-4R−/− BALB/c mice and washed twice in PBS. 5 × 106 BM cells in PBS were injected i.v. into lethally irradiated (9 Gy) WT or IL-4R−/− recipient mice on the same day. After reconstitution, all mice were maintained on antibiotic water for 4 wk. Chimeric mice were injected with tumor cells 10 and 17 wk after reconstitution. To test whether peripheral lymphocytes were derived from donor or recipient BM, MHC class II up-regulation in response to IL-4 was determined on splenic B cells. For this purpose, single cell suspensions were prepared from spleens of the indicated mice and cultured for 16 h at 2 × 106/ml with or without 2 ng/ml recombinant mouse IL-4 (BD Biosciences) in complete RPMI. Cells were incubated with anti–B220-PE (RA3-6B2) and anti–I-Ad/I-Ed–FITC (2G9) and analyzed using Coulter EPICS-XL (Coulter Electronics). Flow cytometry of spleen cells of chimeric mice using an IL-4R–specific mAb confirmed the results. Additionally, il4r-specific PCR analysis with genomic DNA from peripheral blood cells confirmed the results obtained by flow cytometry. Reconstituted mice had normal CD4+, CD8+, and B220+ cell numbers compared with untreated WT and IL-4R−/− mice (unpublished data).
Generation of Fibroblasts and Coinjection with J558-IL-4 Cells.
Lungs from WT or IL-4R−/− mice were minced and incubated for 1 h at 37°C in RPMI plus 5% FCS with 1 mg/ml collagenase D (Roche Diagnostics). Cells were further separated by meshing through a 70-μM nylon filter. Single cell suspensions were washed twice with PBS and cultured in DMEM plus 10% FCS, penicillin/streptomycin, MEM, and 2-ME (50 μM) for 24 h. Nonadherent cells were removed and adherent cells were passaged every 3–5 d. After eight passages, 85–95% of the cultured cells were positive for fibronectin and collagen I, suggesting that the majority of the cells were fibroblasts (unpublished data). For the coinjection with J558-IL-4 cells, 106 WT or IL-4R−/− fibroblasts, respectively, were mixed with 106 J558-IL-4 cells, injected s.c. in the abdominal region of IL-4R−/− mice and tumor growth was measured.
Online Supplemental Material.
All experiments related to Fig. 1 are shown in Table SI, which is available at http://www.jem.org/cgi/content/full/jem.20030849/DC1.
Figure 1.

Biphasic rejection of IL-4–producing tumor cells requires host IL-4R but not IL-4. BALB/c WT (wt; ▵), Rag2−/− (•), IL-4−/− (⋄), IL-4R−/− (▪), and RAG2−/−/IL-4R−/− mice (□) were injected with 2 × 106 J558-IL-4 cells and tumor growth was measured. One representative experiment per experimental group (five to six mice per group) is shown. In total, 30 BALB/c (six experiments), 11 RAG2−/− (two experiments), 17 IL-4−/− (two experiments), 33 IL-4R−/− (six experiments), and 11 RAG2−/−/IL-4R−/− mice (two experiments) were analyzed. In three of the experiments with J558-IL-4 cells in IL-4R−/− mice, a group with parental J558L cells in WT mice (n = 13) was included showing no significant difference in growth kinetics between the two groups. All experiments are presented individually in Table SI, available at http://www.jem.org/cgi/content/full/jem.20030849/DC1.
Results
Biphasic Rejection of IL-4–secreting Tumors.
First, we confirmed and extended that long-term suppression of IL-4–secreting tumors occurred in the absence of T cells but that complete rejection required their presence. As shown in Fig. 1 and published previously (6), IL-4–secreting BALB/c plasmacytoma J558L cells (J558-IL-4) were rejected in WT BALB/c mice. IL-4R−/− BALB/c and IL-4R−/−/RAG2−/− mice developed J558-IL-4 tumors within 8–10 d after tumor cell injection on the average. To exclude that IL-4R–independent effects inhibited the growth of IL-4–producing tumors, the growth kinetics of parental J558L tumors in WT mice (n = 13) was compared with that of J558-IL-4 cells in IL-4R−/− mice (n = 17). Except for one J558L tumor that required 31 d to grow to 1 cm in diameter, J558L and J558-IL-4 tumors reached this size in 8.6 ± 1.3 and 8.5 ± 1.5 d, respectively (Table SI, available at http://www.jem.org/cgi/content/full/jem.20030849/DC1). This suggested that the antitumor effect of IL-4 was entirely dependent on IL-4R expression by host cells and, thus, resulted from a paracrine effect on host cells rather than from an autocrine effect on tumor cells. IL-4 expression by the tumor cells was sufficient for tumor rejection because IL-4−/− mice rejected J558-IL-4 cells (Fig. 1). In RAG2−/− mice, J558-IL-4 tumor growth was strongly delayed. However, all mice finally developed tumors (Fig. 1). This is in line with previous results showing that CD8+ T cells are essential for the rejection of J558-IL-4 tumors (6). This shows that IL-4 mediated its antitumor effect in two phases. Within the first weeks after tumor cell injection, IL-4R expression by nonlymphoid cells was sufficient to prevent rapid tumor formation, whereas lymphocytes were required in the later phase to allow complete tumor rejection.
Generation of IL-4R BM Chimeric Mice.
To identify the cell types that contributed to IL-4–mediated tumor rejection, we generated BM chimeric mice expressing IL-4R selectively on BM- or non-BM–derived cells. Lethally irradiated WT mice were reconstituted with BM cells from WT (WT > WT) or IL-4R−/− mice (IL-4R−/− > WT) and lethally irradiated IL-4R−/− mice were reconstituted with BM cells from IL-4R−/− (IL-4R−/− > IL-4R−/−) or WT (WT > IL-4R−/−) mice. To ensure complete donor cell reconstitution, spleen cells from BM chimeric mice 10 wk after BM transplantation and untreated WT and IL-4R−/− mice were cultured for 16 h with or without IL-4. The up-regulation of MHC class II molecules on B cells in response to IL-4 was determined by flow cytometry as an indication for IL-4R expression. The histograms in Fig. 2, A–F , show MHC class II expression profiles of B cells after culture without (red lines) or with IL-4 (blue lines). B cells from WT > IL-4R−/− mice up-regulated MHC class II expression (histogram in Fig. 2 E), similar to B cells from normal WT mice (histogram in Fig. 2 A) and WT > WT chimeras (histogram in Fig. 2 C). In IL-4R−/− > WT mice, peripheral lymphocytes were also derived from donor BM, since their B cells were unable to respond to IL-4 (Fig. 2 F, histogram) as observed for B cells from IL-4R−/− mice (Fig. 2 B, histogram) and IL-4R−/− > IL-4R−/− chimeric mice (Fig. 2 D, histogram). The results were confirmed by flow cytometry for IL-4R expression on splenocytes and by PCR analysis with genomic DNA from peripheral blood that discriminated between donor and recipient il4r alleles (unpublished data). This indicated that the hematopoietic system was almost completely of donor cell origin 10 wk after BM transplantation.
Figure 2.
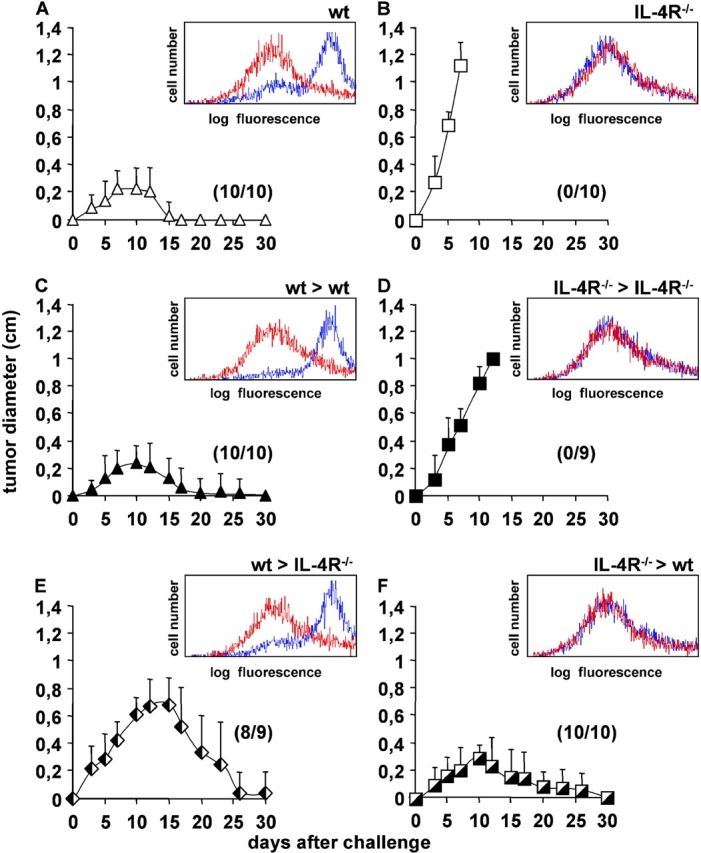
IL-4R expression on non-BM–derived cells is sufficient for the rejection of J558-IL-4 cells. For the generation of BM chimeras, irradiated (9 Gy) BALB/c WT (wt) mice were reconstituted with BM cells from (C) WT (WT > WT) or (F) IL-4R−/− mice (IL-4R−/− > WT). Irradiated IL-4R−/− mice were reconstituted with BM cells from (D) IL-4R−/− (IL-4R−/− > IL-4R−/−) or (E) WT (WT > IL-4R−/−) mice. (C–F) 2 × 106 J558-IL-4 cells were injected s.c. in the indicated BM chimeras, (A) untreated WT, and (B) IL-4R−/− mice. Pooled results from two independent experiments performed 10 and 17 wk after reconstitution are shown. In total, 9–10 mice per group were analyzed. Numbers in the graph indicate the number of mice that rejected the tumor per number of analyzed mice. (A–F, histograms) To analyze whether the peripheral lymphocytes were derived from donor or recipient BM, the up-regulation of MHC class II on splenic B cells in response to IL-4 was tested. Spleen cells from untreated mice of each experimental group were stimulated with 2 ng/ml recombinant IL-4 for 16 h or were left untreated. The expression of MHC class II on B220+ cells was determined by flow cytometry. Red lines show MHC II expression on untreated B220+ cells and blue lines show MHC II expression after incubation with IL-4. An isotype-matched control antibody was used to verify staining specificity (unpublished data). One representative experiment out of two is shown.
IL-4R Expression on Non-BM–derived Cells Is Sufficient for Rejection of J558-IL-4 Cells.
Next, we determined whether the chimeric mice were able to reject J558-IL-4 tumors. WT > WT chimeras (Fig. 2 C) and normal WT mice (Fig. 2 A) were injected with 2 × 106 J558-IL-4 cells and tumor growth was measured. As shown in Fig. 2 C, all WT > WT mice rejected tumors (10/10) as efficiently as normal WT mice (10/10; Fig. 2 A), showing that tumor rejection was not impaired in BM chimeras. To exclude that tumors were rejected in BM chimeras due to the experimental procedure rather than IL-4R responsiveness, IL-4R−/− > IL-4R−/− chimeras (Fig. 2 D) and normal IL-4R−/− mice (Fig. 2 B) were injected with J558-IL-4 cells. As shown in Fig. 2, B and D, tumors grew rapidly and with similar kinetics in both experimental groups (tumor rejection in 0/10 mice in both experimental groups). This demonstrated that (a) tumor growth was not impaired in BM chimeras, and that (b) IL-4R expression by host cells was still required for the rejection of J558-IL-4 tumors.
Perhaps not surprising, IL-4R expression on BM-derived cells was sufficient for tumor rejection. As shown in Fig. 2 E, WT > IL-4R−/− mice rejected J558-IL-4 tumors (eight out of nine), showing that IL-4R expression by BM-derived cells was sufficient for IL-4–mediated tumor rejection in most cases. However, it is important to note that tumors in WT > IL-4R−/− chimeras reached an average size of ∼0.7 cm in diameter before rejection. This was in contrast to WT > WT mice (Fig. 2 C), in which the tumors remained smaller before regression. These differences suggested that IL-4R expression by non-BM–derived cells contributed to tumor growth suppression. This interpretation was supported by the fact that tumor formation was completely prevented in IL-4R−/− > WT chimeras expressing IL-4R exclusively on non-BM–derived cells (10/10; Fig. 2 F). This demonstrated that IL-4R expression by non-BM–derived cells was sufficient for rejection of IL-4–secreting tumors. It should be mentioned that several tumor-free WT > IL-4R−/− mice developed a wasting syndrome that finally led to their death between days 30 and 66 after tumor cell injection. This mortality was not seen in this form in the other experimental groups and requires further investigation.
Tumor Stroma Fibroblasts Are Differently Activated in IL-4–secreting Tumors of WT and IL-4R−/− Mice.
Next, we aimed to identify the non-BM–derived cell population responsible for the rejection of IL-4–secreting tumors. Assuming that IL-4 exerted its antitumor effect locally and has pronounced effects on fibroblasts, we analyzed tumor stroma fibroblasts. WT and IL-4R−/− mice were injected with 107 J558-IL-4 or parental J558L cells and tumors were removed 6 d later. Tumor sections were stained with the mAb ER-TR7 that is specific for fibroblasts/extracellular matrix (24). As shown in Fig. 3 , ER-TR7 staining was visible in J558L and J558-IL-4 tumors of both WT and IL-4R−/− mice, indicating that fibroblasts were recruited by the tumor independently of IL-4. However, J558-IL-4 tumors derived from WT mice clearly showed the strongest ER-TR7 staining (Fig. 3 A) compared with J558-IL-4 tumors from IL-4R−/− mice (Fig. 3 B) and J558L tumors from WT (Fig. 3 C) and IL-4R−/− mice (Fig. 3 D). Interestingly, ER-TR7 staining in J558-IL-4 tumors from IL-4R−/− mice and J558L tumors from WT and IL-4R−/− mice was seen as a lining completely surrounding lumen-containing structures typical for blood vessels (Fig. 3, B–D, arrows). Despite the more intense staining, these structures were lacking in J558-IL-4 tumors of WT mice. It is known that solid tumors need to induce angiogenesis to receive nutrients and oxygen and that mesenchymal cells contribute to angiogenesis by intima formation (25). The more intensive ER-TR7 staining in J558-IL-4 tumors from WT mice and its absence from the luminal structures suggested that tumor infiltrating fibroblasts responding to IL-4 were in a different activation state than those not responding to IL-4. To support this interpretation, RAG2−/− and RAG2−/−/IL-4R−/− mice were injected with J558-IL-4 cells. Tumors were removed and sectioned when they had reached a size of 1 cm in diameter. As shown in Fig. 3 E, a local accumulation of collagen I forming regular structures in tumors derived from RAG2−/− but not in those derived from RAG2−/− × IL-4R−/− mice, could be observed. When consecutive sections were stained with ER-TR7, a similar staining pattern as observed with collagen I–specific antibodies was observed (unpublished data). ER-TR7 and collagen staining in parental J558L tumors of WT and IL-4R−/− mice was similar to that of J558-IL-4 tumors in IL-4R−/− mice (unpublished data). Together, the data suggested that the tumor stroma fibroblasts were differently activated in the absence or presence of IL-4 or IL-4R.
Figure 3.
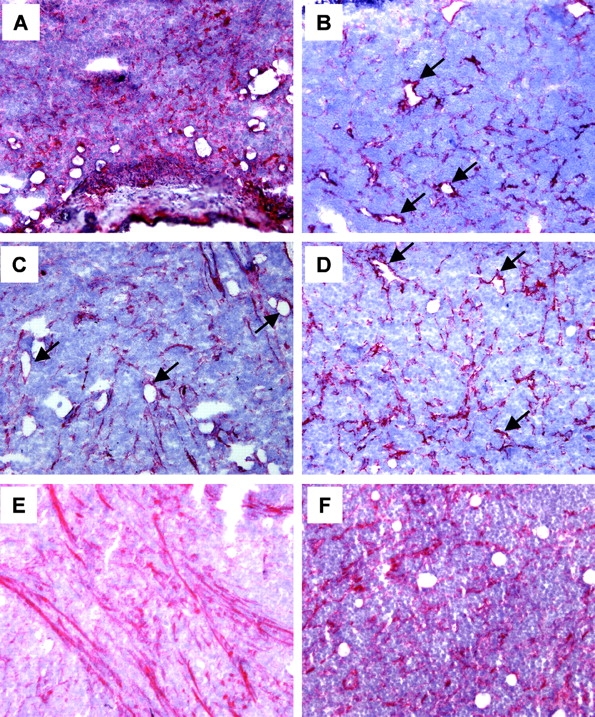
Fibroblasts in J558-IL-4 tumors of WT mice have an altered phenotype. WT (A and C) and IL-4R−/− mice (B and D) were injected s.c. with 107 J558-IL-4 (A and B) or J558L cells (C and D) and tumors were removed 6 d later. Tumor tissue sections were stained with a mAb that stains fibroblasts/extracellular matrix (ER-TR7). Arrows indicate staining around lumen-containing structures typical for blood vessels. (E and F) 2 × 106 J558-IL-4 cells were injected s.c. into (E) RAG2−/− and (F) RAG2−/−/IL-4R−/− mice and tumors were removed at 1 cm in diameter. Because the tumors grew with different kinetics in IL-4R–deficient and IL-4R–competent mice (see Fig. 1), they were removed from RAG2−/−/IL-4R−/− mice at day 8 and from RAG2−/− mice at day 30. Tumor tissue sections were stained with a mAb specific for collagen I. Representative sections for four tumors analyzed per group are shown. ×100.
IL-4R Expression by Tumor Stroma Fibroblasts Is Sufficient for Tumor Rejection.
To analyze whether there was a causal relationship between the altered activation state of tumor stroma fibroblasts in response to IL-4 and tumor rejection, fibroblasts were isolated from lungs of WT and IL-4R−/− mice. 85–95% of the cells were positive for fibronectin and collagen I after eight passages in vitro (unpublished data). 106 J558-IL-4 cells were coinjected with either 106 WT or IL-4R−/− fibroblasts into IL-4R−/− mice and tumor growth was measured. As shown in Fig. 4 A, 50% (three out of six) of IL-4R−/− mice rejected tumors when WT fibroblasts were coinjected with the tumor cells. In contrast, all tumors (five out of five) grew when the fibroblasts were derived from IL-4R−/− mice (Fig. 4 B). The fact that IL-4R−/− mice receiving WT fibroblasts in addition to tumor cells rejected tumors less efficiently than IL-4R−/− > WT BM chimeric mice indicates that either the number of fibroblasts used for the mixing experiment was not optimal or that IL-4R expression by other, non-BM–derived cells contributed to tumor rejection. Together, our data demonstrate that IL-4 responsiveness of tumor stroma fibroblasts is sufficient for rejection of IL-4–secreting tumors.
Figure 4.

IL-4 responsiveness of fibroblasts in the tumor is sufficient for rejection of J558-IL-4 cells. 106 lung fibroblasts from (A) WT or (B) IL-4R−/− mice were mixed with 106 J558-IL-4 cells, injected s.c. into IL-4R−/− mice, and tumor growth was measured. Combined data from two independent experiments with two to three mice/group are shown.
Discussion
Fibroblasts are consistently described as tumor promoting (10–15). Therefore, the major finding in this study was that the modulation of tumor stroma fibroblasts was sufficient for tumor rejection. Our data also challenge the current view concerning the mechanism of tumor rejection by IL-4. We think that the mechanism to interfere with the tumor's effort to establish the necessary stroma follows a general scheme during immune-mediated rejection of tumor cells injected as a cell suspension that is not only observed during IL-4–mediated tumor rejection. However, the target cell type(s) may differ in different models, which can be best illustrated if tumor rejection by IL-4 and IFNγ are compared. Both cytokines, even though often described as counter-players or antiinflammatory and proinflammatory, respectively, can mediate tumor rejection if they are expressed by adoptively transferred CD4+ T cells (26–29), CD8+ T cells (30–33), or transfected tumor cells (2, 3). Because the latter model somehow mimics the local production of cytokines by adoptively transferred T cells homing to the tumor site, it was used here. In a redundant fashion, for tumor rejection induced by either cytokine, responsiveness of non-BM–derived stroma cells is sufficient (Fig. 2 F and 28). However, IFNγ acts (directly or indirectly) on endothelial (CD31+) cells and inhibits tumor-induced angiogenesis by arresting CD31+ cells at the border between tumor and adjacent tissue in an IFNγ-dependent fashion (28, 34). The IL-4 produced by the tumor, in contrast, appears to act primarily on fibroblasts. Fibroblast subpopulations and in particular those in tumors are still poorly characterized (7, 8) but the participation of mesenchymal cells in angiogenesis, e.g., by provision of extracellular matrix, is known. The immunohistochemical stainings in Fig. 3, B–D, suggest that those fibroblasts attracted by tumor cells in IL-4R−/− mice contributed to blood vessel formation. This function was impaired in IL-4–producing tumors of WT mice, shown by the absence of lumen-containing blood vessels that has been described by Di Carlo et al. (35). The assumption that IL-4–activated fibroblasts do not participate in tumor blood vessel formation is supported by the observation that IL-4 inhibits angiogenesis (36, 37). Therefore, it appears that the tumor rejection mechanisms induced by IL-4 and IFNγ culminate through different cellular targets in the same pathway. However, a deeper understanding of the role of fibroblasts during tumor growth or rejection requires a better characterization of the phenotype of IL-4R+/+ versus IL-4R−/− tumor stroma fibroblasts. In any case, there is growing evidence from different models that the primary target during immune-mediated tumor rejection is the stroma and not the tumor. In this regard, our data could explain how adoptively transferred, IL-4–expressing T cells reject tumors (27, 29, 30, 32, 38), namely by targeting fibroblasts and interfering with the establishment of a tumor stroma. Whether IL-4 can inhibit the pro-tumorigenic function of fibroblasts only in the early phase of tumor stroma formation or also at later time points is not known. Furthermore, if data based on tumor transplantation experiments can be used to explain phenomena in spontaneous tumors, our results could explain why spontaneous solid tumors virtually never express substantial amounts of IL-4.
It is important to emphasize that we do not want to exclude other non-BM–derived stromal cell types in addition to fibroblasts that have to respond to IL-4 to support tumor rejection. Interestingly, it has been shown that endothelial cells in IL-4–producing tumors express adhesion molecules like vascular cell adhesion molecule and CD62E that are not expressed on endothelial cells in the parental tumor (35). The lack of their contribution could explain why only 50% of the IL-4R−/− mice coinjected with WT fibroblasts and J558-IL-4 cells rejected the tumor (Fig. 4 A), whereas all BM chimeric mice expressing IL-4R only on non-BM–derived cells rejected the IL-4–producing tumor (Fig. 2 F). However, we do not know whether the number of coinjected fibroblasts was optimal and how long the fibroblasts were functional and survived in vivo. Because IL-13 also uses the IL-4R, induces rejection of transfected tumor cells, and can be induced by IL-4 (39), we cannot exclude that host IL-13 contributed to IL-4–dependent tumor rejection.
It has been known for a while that the growth of IL-4–producing tumors is, depending on the level of IL-4 expression, long-term suppressed in T cell–deficient or CD8+ T cell–depleted mice. However, such tumors finally grow out showing that the presence of T cells is critical in the later phase of the IL-4–mediated antitumor response (5–7 and Fig. 1). The comparison of the WT > WT and WT > IL-4R−/− mice (Fig. 2, C and E) demonstrated that the rapid formation of tumor burden was inhibited by IL-4–responsive, non-BM–derived cells in the very early phase of the antitumor response. This firmly establishes a hypothesis originally put forward by Hock et al. (6). They suggested that T cell–independent inhibition of tumor burden of IL-4–secreting tumors allowed T cells to become activated and effective without necessarily being stimulated by IL-4 that is produced by the tumor. One cannot exclude, however, that IL-4 acted on T cells because WT > IL-4R−/− chimeras also rejected the tumor. Which BM-derived cells had to express IL-4R is not known. However, BM cells are a complementary source of smooth muscle-like cells during neointimal formation after BM transplantation (40) and fibroblast-like cells isolated from peripheral blood, termed fibrocytes, which rapidly enter sites of tissue injury and promote angiogenesis, have been described (41–43). Which physiological response does IL-4/fibroblast-mediated tumor rejection resemble? Fibroblasts are critically involved in wound healing, a process that is supported by IL-4 (44). Because tumors have remarkable similarities with healing wounds (45), we suggest that IL-4–mediated tumor rejection, at least in part, resembles a wound healing response during which IL-4–activated fibroblasts control normal tissue homeostasis that is defective in progressing tumors (8, 9). Together, our data suggest that IL-4–activated fibroblasts do not contribute to formation of a pro-tumorigenic environment, probably because they cannot participate in tumor-induced angiogenesis. This prevents rapid tumor burden and provides sufficient time for the activation of T cells that are required for complete tumor rejection but must not necessarily be able to respond to IL-4.
Acknowledgments
We thank R. Willebrand and G. Baukus for excellent technical assistance and T. Kammertoens for discussion. We thank N. Noben-Trauth for the provision of IL-4R−/− and Rag2−/−/IL-4R−/− mice.
This work was supported by grants from the Deutsche Krebshilfe, Dr. Mildred-Scheel-Stiftung e.V. (10-1535-BL2), the Deutsche Forschungsgemeinschaft (SFB 506), and the Bundesministerium für Bildung und Forschung (BMBF-01KV9911).
The online version of this article contains supplemental material.
References
- 1.Tepper, R.I., P.K. Pattengale, and P. Leder. 1989. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell. 57:503–512. [DOI] [PubMed] [Google Scholar]
- 2.Blankenstein, T., S. Cayeux, and Z. Qin. 1996. Genetic approaches to cancer immunotherapy. Rev. Physiol. Biochem. Pharmacol. 129:1–49. [DOI] [PubMed] [Google Scholar]
- 3.Musiani, P., A. Modesti, M. Giovarelli, F. Cavallo, M.P. Colombo, P.L. Lollini, and G. Forni. 1997. Cytokines, tumour-cell death and immunogenicity: a question of choice. Immunol. Today. 18:32–36. [DOI] [PubMed] [Google Scholar]
- 4.Noffz, G., Z. Qin, M. Kopf, and T. Blankenstein. 1998. Neutrophils but not eosinophils are involved in growth suppression of IL-4-secreting tumors. J. Immunol. 160:345–350. [PubMed] [Google Scholar]
- 5.Golumbek, P.T., A.J. Lazenby, H.I. Levitsky, L.M. Jaffee, H. Karasuyama, M. Baker, and D.M. Pardoll. 1991. Treatment of established renal cancer by tumor cells engineered to secrete interleukin-4. Science. 254:713–716. [DOI] [PubMed] [Google Scholar]
- 6.Hock, H., M. Dorsch, U. Kunzendorf, Z. Qin, T. Diamantstein, and T. Blankenstein. 1993. Mechanisms of rejection induced by tumor cell-targeted gene transfer of interleukin 2, interleukin 4, interleukin 7, tumor necrosis factor, or interferon gamma. Proc. Natl. Acad. Sci. USA. 90:2774–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pericle, F., M. Giovarelli, M.P. Colombo, G. Ferrari, P. Musiani, A. Modesti, F. Cavallo, F. Di Pierro, F. Novelli, and G. Forni. 1994. An efficient Th2-type memory follows CD8+ lymphocyte-driven and eosinophil-mediated rejection of a spontaneous mouse mammary adenocarcinoma engineered to release IL-4. J. Immunol. 153:5659–5673. [PubMed] [Google Scholar]
- 8.Tlsty, T.D., and P.W. Hein. 2001. Know thy neighbor: stromal cells can contribute oncogenic signals. Curr. Opin. Genet. Dev. 11:54–59. [DOI] [PubMed] [Google Scholar]
- 9.Elenbaas, B., and R.A. Weinberg. 2001. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 264:169–184. [DOI] [PubMed] [Google Scholar]
- 10.Picard, O., Y. Rolland, and M.F. Poupon. 1986. Fibroblast-dependent tumorigenicity of cells in nude mice: implication for implantation of metastases. Cancer Res. 46:3290–3294. [PubMed] [Google Scholar]
- 11.Horgan, K., D.L. Jones, and R.E. Mansel. 1987. Mitogenicity of human fibroblasts in vivo for human breast cancer cells. Br. J. Surg. 74:227–229. [DOI] [PubMed] [Google Scholar]
- 12.Camps, J.L., S.M. Chang, T.C. Hsu, M.R. Freeman, S.J. Hong, H.E. Zhau, A.C. von Eschenbach, and L.W. Chung. 1990. Fibroblast-mediated acceleration of human epithelial tumor growth in vivo. Proc. Natl. Acad. Sci. USA. 87:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noel, A., M.C. De Pauw-Gillet, G. Purnell, B. Nusgens, C.M. Lapiere, and J.M. Foidart. 1993. Enhancement of tumorigenicity of human breast adenocarcinoma cells in nude mice by matrigel and fibroblasts. Br. J. Cancer. 68:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olumi, A.F., G.D. Grossfeld, S.W. Hayward, P.R. Carroll, T.D. Tlsty, and G.R. Cunha. 1999. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 59:5002–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elenbaas, B., L. Spirio, F. Koerner, M.D. Fleming, D.B. Zimonjic, J.L. Donaher, N.C. Popescu, W.C. Hahn, and R.A. Weinberg. 2001. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15:50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelms, K., A.D. Keegan, J. Zamorano, J.J. Ryan, and W.E. Paul. 1999. The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17:701–738. [DOI] [PubMed] [Google Scholar]
- 17.Sempowski, G.D., M.P. Beckmann, S. Derdak, and R.P. Phipps. 1994. Subsets of murine lung fibroblasts express membrane-bound and soluble IL-4 receptors. Role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J. Immunol. 152:3606–3614. [PubMed] [Google Scholar]
- 18.Trautmann, A., G. Krohne, E.B. Brocker, and C.E. Klein. 1998. Human mast cells augment fibroblast proliferation by heterotypic cell-cell adhesion and action of IL-4. J. Immunol. 160:5053–5057. [PubMed] [Google Scholar]
- 19.Postlethwaite, A.E., M.A. Holness, H. Katai, and R. Raghow. 1992. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J. Clin. Invest. 90:1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makhluf, H.A., J. Stepniakowska, S. Hoffman, E. Smith, E.C. LeRoy, and M. Trojanowska. 1996. IL-4 upregulates tenascin synthesis in scleroderma and healthy skin fibroblasts. J. Invest. Dermatol. 107:856–859. [DOI] [PubMed] [Google Scholar]
- 21.Ong, C., C. Wong, C.R. Roberts, H.S. Teh, and F.R. Jirik. 1998. Anti-IL-4 treatment prevents dermal collagen deposition in the tight-skin mouse model of scleroderma. Eur. J. Immunol. 28:2619–2629. [DOI] [PubMed] [Google Scholar]
- 22.Kodera, T., T.L. McGaha, R. Phelps, W.E. Paul, and C.A. Bona. 2002. Disrupting the IL-4 gene rescues mice homozygous for the tight-skin mutation from embryonic death and diminishes TGF-beta production by fibroblasts. Proc. Natl. Acad. Sci. USA. 99:3800–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noben-Trauth, N., L.D. Shultz, F. Brombacher, J.F. Urban, Jr., H. Gu, and W.E. Paul. 1997. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc. Natl. Acad. Sci. USA. 94:10838–10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Vliet, E., M. Melis, J.M. Foidart, and W. Van Ewijk. 1986. Reticular fibroblasts in peripheral lymphoid organs identified by a monoclonal antibody. J. Histochem. Cytochem. 34:883–890. [DOI] [PubMed] [Google Scholar]
- 25.Lusis, A.J. 2000. Atherosclerosis. Nature. 407:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mumberg, D., P.A. Monach, S. Wanderling, M. Philip, A.Y. Toledano, R.D. Schreiber, and H. Schreiber. 1999. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc. Natl. Acad. Sci. USA. 96:8633–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishimura, T., K. Iwakabe, M. Sekimoto, Y. Ohmi, T. Yahata, M. Nakui, T. Sato, S. Habu, H. Tashiro, M. Sato, et al. 1999. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 190:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin, Z., and T. Blankenstein. 2000. CD4+ T cell–mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 12:677–686. [DOI] [PubMed] [Google Scholar]
- 29.Mattes, J., M. Hulett, W. Xie, S. Hogan, M.E. Rothenberg, P. Foster, and C. Parish. 2003. Immunotherapy of cytotoxic T cell–resistant tumors by T helper 2 cells: an eotaxin and STAT6-dependent process. J. Exp. Med. 197:387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobrzanski, M.J., J.B. Reome, and R.W. Dutton. 1999. Therapeutic effects of tumor-reactive type 1 and type 2 CD8+ T cell subpopulations in established pulmonary metastases. J. Immunol. 162:6671–6680. [PubMed] [Google Scholar]
- 31.Becker, C., H. Pohla, B. Frankenberger, T. Schüler, M. Assenmacher, D.J. Schendel, and T. Blankenstein. 2001. Adoptive tumor therapy with T lymphocytes enriched through an IFN-gamma capture assay. Nat. Med. 7:1159–1162. [DOI] [PubMed] [Google Scholar]
- 32.Dobrzanski, M.J., J.B. Reome, and R.W. Dutton. 2001. Role of effector cell-derived IL-4, IL-5, and perforin in early and late stages of type 2 CD8 effector cell-mediated tumor rejection. J. Immunol. 167:424–434. [DOI] [PubMed] [Google Scholar]
- 33.Schüler, T., and T. Blankenstein. 2003. Cutting edge: CD8(+) effector T cells reject tumors by direct antigen recognition but indirect action on host cells. J. Immunol. 170:4427–4431. [DOI] [PubMed] [Google Scholar]
- 34.Qin, Z., J. Schwartzkopff, F. Pradera, T. Kammertöns, B. Seliger, H. Pircher, and T. Blankenstein. 2003. A critical requirement of IFNγ-mediated angiostasis for tumor rejection by CD8+ T cells. Cancer Res. 63:4095–4100. [PubMed] [Google Scholar]
- 35.Di Carlo, E., A. Modesti, A. Coletti, M.P. Colombo, M. Giovarelli, G. Forni, M.G. Diodoro, and P. Musiano. 1998. Interaction between endothelial cells and the secreted cytokine drives the fate of an IL4- or IL5-transduced tumour. J. Pathol. 186:390–397. [DOI] [PubMed] [Google Scholar]
- 36.Saleh, M., I.D. Davis, and A.F. Wilks. 1997. The paracrine role of tumour-derived mIL-4 on tumour-associated endothelium. Int. J. Cancer. 72:664–672. [DOI] [PubMed] [Google Scholar]
- 37.Volpert, O.V., T. Fong, A.E. Koch, J.D. Peterson, C. Waltenbaugh, R.I. Tepper, and N.P. Bouck. 1998. Inhibition of angiogenesis by interleukin 4. J. Exp. Med. 188:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobrzanski, M.J., J.B. Reome, and R.W. Dutton. 2000. Type 1 and type 2 CD8+ effector T cell subpopulations promote long-term tumor immunity and protection to progressively growing tumor. J. Immunol. 164:916–925. [DOI] [PubMed] [Google Scholar]
- 39.Wynn, T.A. 2003. IL-13 effector functions. Annu. Rev. Immunol. 21:425–456. [DOI] [PubMed] [Google Scholar]
- 40.Han, C.I., G.R. Campbell, and J.H. Campbell. 2001. Circulating bone marrow cells can contribute to neointimal formation. J. Vasc. Res. 38:113–119. [DOI] [PubMed] [Google Scholar]
- 41.Bucala, R., L.A. Spiegel, J. Chesney, M. Hogan, and A. Cerami. 1994. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 42.Chesney, J., C. Metz, A.B. Stavitsky, M. Bacher, and R. Bucala. 1998. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J. Immunol. 160:419–425. [PubMed] [Google Scholar]
- 43.Abe, R., S.C. Donnelly, T. Peng, R. Bucala, and C.N. Metz. 2001. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J. Immunol. 166:7556–7562. [DOI] [PubMed] [Google Scholar]
- 44.Salmon-Ehr, V., L. Ramont, G. Godeau, P. Birembaut, M. Guenounou, P. Bernard, and F.X. Maquart. 2000. Implication of interleukin-4 in wound healing. Lab. Invest. 80:1337–1343. [DOI] [PubMed] [Google Scholar]
- 45.Dvorak, H.F. 1986. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315:1650–1659. [DOI] [PubMed] [Google Scholar]