

Abstract
Although Toll-like receptors (TLRs) are critical mediators of the immune response to pathogens, the influence of polymorphisms in this gene family on human susceptibility to infection is poorly understood. We demonstrated recently that TLR5 recognizes flagellin, a potent inflammatory stimulus present in the flagellar structure of many bacteria. Here, we show that a common stop codon polymorphism in the ligand-binding domain of TLR5 (TLR5392STOP) is unable to mediate flagellin signaling, acts in a dominant fashion, and is associated with susceptibility to pneumonia caused by Legionella pneumophila, a flagellated bacterium. We also show that flagellin is a principal stimulant of proinflammatory cytokine production in lung epithelial cells. Together, these observations suggest that TLR5392STOP increases human susceptibility to infection through an unusual dominant mechanism that compromises TLR5's essential role as a regulator of the lung epithelial innate immune response.
Keywords: inflammation, immunity, genetic predisposition to disease, genetic markers, bacterial infections
Introduction
Pneumonia is the sixth leading cause of death in the United States and the most common cause of death due to infectious disease (1). Despite the seriousness of pneumonia, very little is known about genetic factors that predispose individuals to acquire lower respiratory tract infections. Recent advances in innate immunity and genetics are opening avenues to examine the immunologic and genetic factors that predispose individuals to acquire common infections such as pneumonia (2). The innate immune system enables the host to differentiate itself from invading microbes, discriminate among pathogens, and initiate a cascade of inflammatory molecules that influence formation of the acquired immune response as well as host survival. Toll-like receptors (TLRs) constitute a family of transmembrane proteins that differentially recognize pathogen-associated molecular patterns (PAMPs) through an extracellular domain and initiate inflammatory signaling pathways through an intracellular domain (3–6). Due to the central role of TLRs in the innate immune response, genetic variation in this gene family is predicted to alter susceptibility to infections in a PAMP-specific manner based on the ligand recognition of the individual TLR. Although initial papers have supported this hypothesis for TLR4, the degree to which other TLRs affect susceptibility to infections is unknown (7–11).
We demonstrated recently that TLR5 recognizes the flagellin protein, a potent inflammatory stimulus present in the flagellar structure of many bacteria (12, 13). Herein, we provide the first evidence that TLR5 influences human susceptibility to infection as well as describe an important role for TLR5 in the innate immune response of the lung epithelium.
Materials and Methods
Materials.
RPMI 1640 medium, l-glutamine, penicillin, and streptomycin were obtained from Life Technologies, DMEM was obtained from JRH Biosciences, and Ham's F12 was obtained from BioWhittaker. Ultrapure LPS was from Salmonella minnesota R595 (List Biological Labs, Inc.). Flagellin was purified from Salmonella typhimurium as described previously from strain TH4778, which is fljB−/fliC+ (a gift from K. Hughes, University of Washington, Seattle, WA; references 12, 14).
Cells, Mice, and Bacteria.
Chinese hamster ovary (CHO) cells (CHO-K1), A549 cells, HEK 293 cells, and Calu-3 cells were grown according to American Type Culture Collection recommendations. PBMCs were isolated from 50 ml of blood from individuals with a Ficoll gradient separation. MyD88−/− and TLR4−/− mice (a gift from S. Akira, Research Institute for Microbial Diseases, Osaka University, Japan) were derived on a 129SvJ × C57Bl/6 background, backcrossed for six generations to C57Bl/6, and used for experiments along with littermate controls (15, 16). Bone marrow was harvested from these mice and grown in RPMI 1640 supplemented with 10% heat-inactivated FCS (Hyclone Laboratories), 20% L cell–conditioned media, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine. Bone marrow–derived macrophages were used after 4–10 d of culture. The University of Washington and Institute for Systems Biology's Institutional Animal Care and Use Committees approved all animal protocols. L. pneumophila Corby (serogroup 1) and L. pneumophila Corby FlaA− were gifts from K. Heuner (University of Wurzburg, Wurzburg, Germany), and the JR32 strain was a gift from H. Shuman (Columbia University, New York, NY; references 17, 18). The Bovenkarspel strain was a gift from R. van Ketel (Academic Medical Center, Amsterdam, Netherlands). Knoxville-1 and Philadelphia-1 strains were obtained from American Type Culture Collection. For stimulation assays, bacteria were grown on buffered charcoal yeast extract–agar at 35°C (with kanamycin selection for the FlaA− mutant), resuspended in PBS, and heat killed for 65°C for 15 min (17).
Human Subjects and Data Collection.
Approval for human study protocols was obtained from the human subjects review boards at the University of Amsterdam Medical Center, the University of Washington Medical Center, and the Institute for Systems Biology. All participants gave written informed consent. Genomic DNA was purified from peripheral blood leukocytes from 10 ml of blood. The first study group included 54 healthy volunteers from the Seattle, WA area. Enrollment of the cases and controls from the Legionnaires' disease (LD) outbreak in the Netherlands has been described previously (19–21). Of the 188 cases identified in the original investigation of the outbreak, 141 consented for the study. 18 individuals died and no DNA was available for genotyping. 108 cases were available (93 definite LD, 15 probable LD) with both DNA and epidemiologic data for analysis. Controls were drawn from the exhibitioners who worked at the flower show and were at high risk for exposure to Legionella pneumophila. 1,616 controls were contacted by letter to be in the analysis. The first 508 who completed the questionnaire had blood drawn for genetic analysis.
Statistics.
Univariate analysis was performed for categorical variables with chi-square test, and for continuous variables using a Student's t test. Single nucleotide polymorphism (SNP) data were analyzed using a recently developed methodology based on an estimating equation technique, the details of which have been described previously (22, 23). The basic idea is to estimate the distribution of haplotypes from SNP genotype data, to correlate all common haplotypes with LD phenotype with adjustment for demographic variables, and to assess interactions of haplotypes with exposure factors. The penetrance of paired haplotypes (H,h) and exposure (E) to LD outcome (D = 1) is quantified via a logistic regression model: 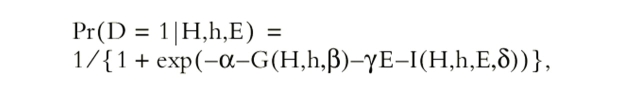 where unknown parameter β in the function G(H,h,β) quantifies disease associations with haplotypes, γ quantifies the disease association with exposure, and δ in I(H,h,E,δ) quantifies the disease associations with the interaction of haplotypes and exposure. Estimating regression parameters (β,δ) is of primary interest because their relevant statistics can be used to make scientific inferences. The key challenge to the estimation is that the haplotype structure for many individuals is not observed (i.e., phases of SNP alleles are not observed).
where unknown parameter β in the function G(H,h,β) quantifies disease associations with haplotypes, γ quantifies the disease association with exposure, and δ in I(H,h,E,δ) quantifies the disease associations with the interaction of haplotypes and exposure. Estimating regression parameters (β,δ) is of primary interest because their relevant statistics can be used to make scientific inferences. The key challenge to the estimation is that the haplotype structure for many individuals is not observed (i.e., phases of SNP alleles are not observed).
Molecular Biology.
SNP discovery and genotyping was performed by PCR amplification of TLR5 from genomic DNA followed by sequencing. To generate templates for sequencing, the coding region of TLR5 was amplified from 25 ng of genomic DNA in two fragments with Amplitaq DNA polymerase (Applied Biosystems). Primers for fragment A were as follows: T5-5, forward, 5′-GTATCTGCCAACAGCCACGTCTGTGG-3′; and T5-23, reverse, 5′-GAGAATCTGGAGATGAGGTACCCG-3′. Primers for fragment B were as follows: T5-3, forward, 5′-GCATACCCGATATCTTCTTGAGTGGC-3′; and T5-21, reverse, 5′-CTGGAAACCTCTAAGGGCAGTTGC-3′. For genotyping by DdeI restriction digestion, a 276-bp PCR product was first generated with primers T5-11, forward, 5′-GGTAGCCTACATTGATTTGC-3′; and T5-23, digested with DdeI (New England Biolabs, Inc.), and separated on a 1.8% agarose gel. For functional studies, the coding region of TLR5 was amplified from genomic DNA by PCR using Pfu Turbo polymerase (Stratagene) and cloned into the pEF6/V5-His-TOPO vector (Invitrogen). PCR primers for the full length gene included T5-10, forward, 5′-ATGGGAGACCACCTGGACCTTCTCC-3′; and T5-30, reverse, 5′-GGAGATGGTTGCTACAGTTTGCAACGG-3′. PCR primers for TLR5392STOP were T5-10, forward; and T5-31, reverse, 5′-GAGATCCAAGGTCTGTAATTTTTCCAGG-3′. To obtain the different polymorphic variants of TLR5, genomic DNA was selected from individuals with the desired genotype. The cloned variants of TLR5 were sequenced for verification with Big Dye Terminator v3.0 and analyzed on a capillary sequencer (model ABI PRISM 3700; Applied Biosystems). Sequence was aligned and analyzed with the programs PHRED/PHRAP and CONSED (24).
Transfections.
CHO cells were transfected as described previously (12). HEK293 cells were transfected as described previously in a 96-well plate format with calcium phosphate, using 5 × 104 cells per well with 8 ng of TLR5, 8 ng of pRL-TK, and 80 ng of endothelial leukocyte adhesion molecule (ELAM)–luciferase per well (25). For some experiments, HEK293 cells were alternatively transfected with Polyfect (QIAGEN) according to the manufacturer's instructions with 5 × 104 cells per well in a 96-well plate with 175 ng TLR5, 8 ng pRL-TK, and 80 ng of ELAM-luciferase per well. Stimulated cells were processed as described previously (12).
Protein Analysis.
For the immunoblot, cells were lysed with 1% Triton X-100/150 mM NaCl/10 mM Hepes, pH 8.0, or passive lysis buffer (Promega) with a cocktail of protease inhibitors (Sigma-Aldrich). An immunoblot with proteins from cell lysates was probed with an anti-V5 epitope antibody (Serotec) followed by HRP-conjugated rabbit anti–mouse IgG (Zymed Laboratories) and developed with chemiluminescent reagents (Roche). Whole blood cytokine assays were prepared by diluting venous blood 1:5 with RPMI 1640, plating in a 96-well dish, stimulating for 18 h, and harvesting supernatants. For Calu-3 and A549 assays, cells were plated at a density of 2 × 104 per well in a 96-well dish, stimulated for 18 h, and supernatants were harvested. Cytokine levels were determined with a sandwich ELISA technique (Duoset; R&D Systems).
Results
TLR5 SNP Discovery.
To discover novel SNPs with disease association, we PCR amplified and sequenced the TLR5 coding region in 40 healthy individuals. TLR5 is a type I transmembrane protein with a 642–amino acid leucine-rich extracellular domain, an 18-residue transmembrane domain, and a 198-residue cytoplasmic Toll homology signaling domain. Four SNPs were detected in TLR5. Most dramatic was a cytosine–thymidine transition at base pair 1174 that changed the arginine at amino acid 392 to a stop codon (Fig. 1 A, TLR5392STOP). This change is predicted to prematurely truncate TLR5 in the extracellular domain and cause the loss of the transmembrane domain and the entire signaling cytoplasmic tail. We verified this polymorphism with a DdeI restriction enzyme digestion, which selectively cut a 276-bp PCR product encompassing the 1174T variant (Fig. 1 B). Two additional nonsynonymous SNPs (A1775G [amino acid N592S] and T1846C [F616L]) alter residues in the ectodomain, whereas a fourth synonymous SNP (A2523G [K841K]) was in the cytoplasmic tail. The frequency of subjects who possessed the stop codon variant as heterozygotes (1174 CT) and homozygotes (1174 TT) was 7.5 and 0%, respectively. The serine variant at amino acid 592 and the leucine variant at 616 were present as heterozygote frequencies of 35.0 (1775 AG) and 45.0% (1846TC), and homozygote frequencies of 5.0 (1775 GG) and 15.0% (1846 CC), respectively. The synonymous SNP at amino acid 841 (A2523G) was present as a heterozygote frequency of 20.0% (2523 AG) and a homozygote frequency of 0% (2523 GG). Except for minor deviations, the observed allelic frequencies of these four SNPs were consistent with expected allelic frequencies under the Hardy–Weinberg equilibrium.
Figure 1.
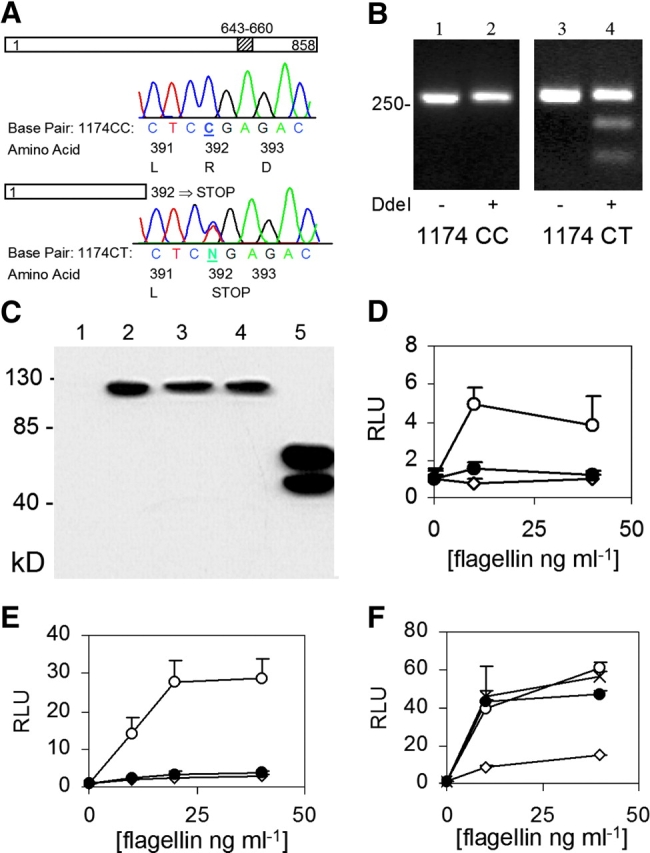
TLR5 polymorphisms and FliC signaling. (A) Sequence chromatogram indicating a polymorphism at base pair 1174. Amino acids are numbered for the respective protein depictions. The diagonal stripes at amino acid 643–660 indicate the transmembrane domain. (B) DdeI restriction digest to verify C1174T SNP. Lanes 1 and 2, genotype 1174 CC; lanes 3 and 4, genotype 1174 CT. Lanes 1 and 3, no DdeI; lanes 2 and 4, with DdeI. (C) Western immunoblot. CHO cells were transfected with control vector (lane 1, pEF6 without insert), TLR5RNF (lane 2), TLR5RNL (lane 3), TLR5RSF (lane 4), or TLR5392STOP (lane 5). Immunoblot probed with an anti-V5 antibody. (d and e) Luciferase assay in CHO (D) and HEK293 (E) cells. Cells were transfected with pEF6 control (⋄), TLR5RNF (○), or TLR5–392STOP (•), an NF-κB luciferase reporter, and pRL-TK as a transfection control. (F) Luciferase assay in HEK293 cells transfected with: pEF6 control (⋄), TLR5RNF (○), TLR5RNL (•), or TLR5RSF (×). RLU, relative luciferase unit. Assays performed in triplicate with standard deviations indicated. (D–F) Transfected cells were stimulated for 4 h with S. typhimurium FliC at the indicated concentration before determining luminescence levels.
cDNAs encoding the TLR5 polymorphisms were amplified by PCR and cloned into the pEF6-TOPO expression vector in frame with a COOH-terminal V5-epitope tag. The full-length constructs were labeled TLR5RNF, TLR5RNL, and TLR5RSF, using the single letter designations of the amino acids at the three polymorphic amino acid positions (392, 592, and 616). When assessed by Western blot, TLR5RNF, TLR5RNL, and TLR5RSF protein expression levels were similar in transfected CHO cells (Fig. 1 C). TLR5392STOP was expressed at higher levels in comparison to the full-length versions and was detected as a doublet band, which may be secondary to proteolysis or glycosylation. Next, we examined whether TLR5392STOP was able to mediate flagellin signaling. We transfected CHO cells with TLR5 variants along with an NF-κB firefly luciferase reporter plasmid (ELAM) and stimulated with purified S. typhimurium flagellin for 4 h (12). Flagellin induced NF-κB activity in cells expressing the full length TLR5 (Fig. 1 D). In contrast, cells expressing TLR5392STOP were unable to respond to flagellin. Identical results were obtained in HEK293 cells, demonstrating that the inability of TLR5392STOP to mediate flagellin signaling was not cell specific (Fig. 1 E). Incubation with LPS at 100 ng/ml did not induce any luciferase activity in TLR5-transfected CHO cells and polymyxin B did not alter the results of the flagellin stimulation (unpublished data). These controls demonstrated that the TLR5-mediated NF-κB response was flagellin specific. In contrast to the results obtained with TLR5392STOP, the other variants of TLR5 were fully functional in mediating flagellin signaling. When introduced into HEK293 cells, TLR5-RNL and TLR5-RSF were no different from TLR5-RNF in mediating flagellin signaling (Fig. 1 F). As seen in CHO cells, Western blot analysis revealed similar levels of expression of the three variants in HEK293 cells (Fig. 1 C, not depicted).
SNPs in TLR5 Are Associated with Susceptibility to LD.
Next, we examined whether these TLR5 SNPs were associated with susceptibility to human infection in a PAMP-specific manner. L. pneumophila, first described in 1976 as the agent of LD, is a flagellated gram-negative bacteria that causes anywhere from 1 to 30% of cases of community-acquired pneumonia (26–30). We found that heat-killed L. pneumophila (serogroup 1, Corby strain) induced NF-κB activity in TLR5-transfected CHO cells (Fig. 2 A). This stimulatory activity was abolished in flagellin mutant strains of L. pneumophila (FlaA− Corby strain; reference 17). In addition, we found that several other strains of L. pneumophila stimulated TLR5 activity, including the Bovenkarspel strain, which caused a recent epidemic of LD at a flower show in the Netherlands in 1999 (19). We also stimulated bone marrow–derived macrophages from wild-type, TLR4−/−, and MyD88−/− mice with heat-killed L. pneumophila. Although TNF-α and IL-6 levels were abolished in MyD88−/− macrophages, there was no decrease in the TLR4−/− cells in comparison to wild-type controls (Fig. 2, B and C). In contrast, as a control, cytokine production was abolished in both TLR4−/− and MyD88−/− macrophages stimulated with S. typhimurium LPS. These results demonstrate that TLR4 is not involved in recognition of L. pneumophila by murine macrophages.
Figure 2.

(A) L. pneumophila activates TLR5 through FliC. CHO cells were stably transfected with an NF-κB luciferase promoter and either a control vector (pEF6, shaded bars) or TLR5RNF (white bars). Cells were stimulated with S. typhimurium FliC at 10 ng/ml−1, S. minnesota LPS at 100 ng/ml−1, or heat-killed L. pneumophila (multiplicity of infection = 250:1). Strains of L. pneumophila are as follows: LpWT, wild-type Corby strain; LpFlaA−, Corby strain FliC mutant; Boven, Bovenkarspel; Phil, Philadelphia; Knox, Knoxville. RLU, relative luciferase unit. (B and C) L. pneumophila stimulates macrophages through MyD88, but not TLR4. Macrophages were derived from mouse bone marrow and stimulated with PBS (white bars), S. minnesota LPS at 10 ng ml−1 (shaded bars), or LpWT (multiplicity of infection = 100:1; black bar). After 16 h of stimulation, supernatants were assayed by ELISA for TNF-α (B) or IL-6 (C). WT, wild type. T4, TLR4−/−. M88, MyD88−/− mouse strains. (A–C) Assays performed in triplicate with standard deviations indicated. (A) Transfected cells were stimulated for 4 h before determining luminescence levels.
To determine whether TLR5 is associated with susceptibility to LD, we examined genotypes in a case-control cohort from the Bovenkarspel epidemic (19–21). Cases included individuals with radiologically confirmed pneumonia occurring during the epidemic time period around the West Friese Flower Show in the town of Bovenkarspel in the Netherlands. Controls were drawn from a pool of exhibitioners who were likely to be exposed to the contaminated water product that caused the epidemic. All of the cases and controls were from the Netherlands and >95% of both groups were Caucasian Dutch. To provide an additional control for population admixture, 89 of the 508 controls were matched to cases for their place of residence ± 25 km, as well as age and sex. The baseline characteristics of the case and control groups are shown in Table I. The mean age and the prevalence of diabetes were statistically different between cases and controls. In addition, smoking appeared to be marginally different between cases and controls. To formally assess the associations of age, sex, diabetes, and smoking status, our null hypothesis is that these variables do not associate with LD when comparing cases and controls (odds ratio [OR] = 1), in contrast to an alternative hypothesis of OR > 1 or OR < 1. A two-sided test was used for the purpose of evaluating statistical significance. Next, we adjusted the analysis by logistic regression with covariates for age (stratified into <40, 40–49, 50–60, and >60), gender, diabetes status, and smoking status. In the adjusted analysis, smoking status (OR = 2.84; 95% confidence interval [CI] = 1.55–5.21; P < 0.05) and age (OR in reference to 40–49 age group: OR = 0.071 (age, 0–39); CI = 0.01–0.57; P < 0.05; OR = 2.30 (age, 50–60); CI = 1.0–5.29; P = 0.05; and OR = 21.80 (age, >60); CI = 9.26–51.28; P < 0.05) had a significant association with LD. In contrast, gender (OR = 1.09; CI = 0.62–1.91) and diabetes status were not associated with LD (OR = 1.43; CI = 0.51–4.03).
Table I.
Baseline Clinical Characteristics of Cases and Controls
| Cases n = 108 |
All controls n = 508 |
Matched controls n = 89 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| n (%) | n (%) | OR | (95% CI) | P | n (%) | OR | (95% CI) | P | |
| Male | 65 (60.2) | 261 (52.4) | 1.37 | (0.90, 2.10) | 0.14 | 53 (60.2) | 1.00 | (0.56, 1.77) | 1.00 |
| Smoking | 47 (49.0) | 172 (40.6) | 1.41 | (0.90, 2.19) | 0.13 | 32 (42.7) | 1.29 | (0.70, 2.37) | 0.41 |
| History of | |||||||||
| COPD | 8 (7.8) | 39 (7.8) | 1.01 | (0.46, 2.23) | 0.98 | 4 (4.5) | 1.81 | 0.39a | |
| DM | 10 (10.1) | 14 (2.8) | 3.92 | (1.69, 9.09) | <0.05 | 5 (5.6) | 1.89 | 0.29a | |
| Malignancy | 4 (4.0) | 12 (2.5) | 1.64 | 0.50a | 2 (2.3) | 1.77 | 0.69a | ||
| Rheum | 2 (2.0) | 13 (2.6) | 0.78 | 1.00a | 4 (4.5) | 0.44 | 0.43a | ||
| Mean ± SD | Mean ± SD | P | Mean ± SD | P | |||||
| Age | 63.8 ± 10.8 | 44.9 ± 13.6 | <0.05b | 54.6 ± 11.2 | <0.05b | ||||
Prevalence of baseline demographic and clinical information is indicated. Statistics are reported for each control population in reference to the cases. The odds ratios (OR) with 95% confidence intervals is indicated (CI). Chi-square test used to calculate two-sided significance values. Not all variables were known in all patients.
For subgroups with small sample numbers, a Fisher's exact test was used.
Mean ages compared with an independent sample t test.
%, number of patients tested for that variable; Rheum, rheumatologic disorder; COPD, chronic obstructive pulmonary disease; DM, diabetes mellitus.
Next, we determined the genotype and allele frequencies of individuals for the three nonsynonymous TLR5 SNPs (Tables II and III). There was no significant departure from the Hardy–Weinberg equilibrium of the observed and expected frequencies of cases or controls for these SNPs. The 1174T (stop codon variant) and 1775G alleles were both associated with susceptibility to LD. The allele frequency of 1174T was 8.3% in the cases compared with 5.0% in the entire control group and 4.5% in the matched control group. For the purpose of assessing genetic association, our formal null hypothesis is that individual alleles or their haplotypes do not associate with case-control status (i.e., OR = 1). The alternative hypothesis is that there is some association, with OR > 1 or OR < 1. Two-sided testing was used again for evaluating statistical significance. The OR for the association of allele 1174T with developing LD was 1.75 (95% CI = 1.00–3.05; P = 0.05) when compared with the entire control group. The association of 1174T with LD is even more striking because this stop codon variant was almost exclusively present as a heterozygote genotype, as there were only two homozygote individuals in the entire cohort (1174TT). The presence of both homozygote stop codon individuals in the control group is likely due to the larger control sample size (n = 508) as compared with the cases (n = 108). In addition, although the controls were selected for an increased risk of contact with L. pneumophila, the actual exposure dose of each individual is not known. The OR for the comparison with the matched control group showed a similar trend, but was not significant due to the small sample size (OR = 1.93; 95% CI = 0.74–5.03). The OR for the association of allele 1775G with LD was 1.60 (95% CI = 1.04–2.45; P = 0.03) compared with the entire control group and 1.83 (95% CI = 1.02–3.28; P = 0.04) for the matched cohort. There was no association of SNP T1846C with LD. To determine if coinheritance of 1174T and 1775G further increased the risk of pneumonia, we examined the association of TLR5 haplotypes with susceptibility to LD (22, 23). However, no haplotypes containing both 1174T and 1775G were detected in either cases or controls (Table III, labeled 11). In the analysis by haplotypes, the association of 1174T-1775A (haplotype 10; OR = 1.90; P = 0.03) and 1174C-1775G (haplotype 01; OR = 1.68; P = 0.02) with susceptibility to LD was also present (23). Next, we considered whether any variables might be confounders in the analysis, an unlikely possibility given that there is no overt biologic relationship of TLR5 to any of the variables in Table I. We repeated the analysis with an adjustment for sex and age (stratified into <40, 40–49, 50–60, and >60) and found a similar trend in the association of TLR5 with LD (SNP 1174T; OR = 1.56 [95% CI = 0.82, 2.98], SNP 1775G, OR = 1.71 [95% CI = 1.07, 2.72]). In addition, there was no statistical association between any of the TLR5 SNPs and diabetes (unpublished data).
Table II.
TLR5 SNP Genotype Frequencies in Cases and Controls
| Cases n = 108 |
All controls n = 89 |
Matched controls n = 89 |
Total n = 616 |
||
|---|---|---|---|---|---|
| Genotype | Amino Acid |
n (freq) | n (freq) | n (freq) | n (freq) |
| 1174 CC | 392RR | 90 (0.833) | 457 (0.905) | 82 (0.921) | 547 (0.892) |
| 1174 CT | 392R* | 18 (0.167) | 46 (0.091) | 6 (0.067) | 64 (0.104) |
| 1174 TT | 392* | 0 (0) | 2 (0.004) | 1 (0.011) | 2 (0.003) |
| 1775 AA | 592NN | 72 (0.692) | 383 (0.777) | 71 (0.798) | 455 (0.762) |
| 1775 AG | 592NS | 28 (0.269) | 106 (0.215) | 18 (0.202) | 134 (0.224) |
| 1775 GG | 592SS | 4 (0.038) | 4 (0.008) | 0 (0) | 8 (0.010) |
| 1846 TT | 616FF | 28 (0.277) | 126 (0.260) | 21 (0.239) | 154 (0.263) |
| 1846 TC | 616FL | 49 (0.485) | 252 (0.520) | 53 (0.602) | 301 (0.514) |
| 1846 CC | 616LL | 24 (0.238) | 107 (0.221) | 14 (0.159) | 131 (0.224) |
TLR5 genotypes were determined in cases and controls.
, stop codon.
Table III.
TLR5 SNP Allele and Haplotype Frequencies in Cases and Controls
| SNP
|
Cases n = 108 |
All controls n = 508 |
Matched controls n = 89 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | BP | Allele | freq | freq | OR | (95% CI) | P | freq | OR | (95% CI) | P |
| 1. | 1174C | 0 | 0.917 | 0.950 | 0.955 | ||||||
| 1174T | 1 | 0.083 | 0.050 | 1.75 | (1.00, 3.05) | 0.05 | 0.045 | 1.93 | (0.74, 5.03) | 0.18 | |
| 2. | 1775A | 0 | 0.827 | 0.884 | 0.899 | ||||||
| 1775G | 1 | 0.173 | 0.116 | 1.60 | (1.04, 2.45) | 0.03 | 0.101 | 1.83 | (1.02, 3.28) | 0.04 | |
| 3. | 1846T | 0 | 0.520 | 0.520 | 0.540 | ||||||
| 1846C | 1 | 0.480 | 0.480 | 1.00 | (0.72, 1.38) | 0.98 | 0.460 | 1.09 | (0.71, 1.68) | 0.70 | |
| Haplotype 1174-1775 | |||||||||||
| CA | 00 | 0.744 | 0.835 | 1 | 0.861 | 1 | |||||
| CG | 01 | 0.173 | 0.115 | 1.68 | (1.10, 2.59) | 0.02 | 0.094 | 2.11 | (1.16, 3.85) | 0.02 | |
| TA | 10 | 0.083 | 0.049 | 1.90 | (1.07, 3.35) | 0.03 | 0.038 | 2.53 | (0.86, 7.49) | 0.09 | |
| TG | 11 | 0 | 0 | 0 | |||||||
0 represents the common allele at each locus; 1 is the uncommon allele. Haplotype alleles for SNP1 (1174) and SNP2 (1775) are listed in order. Odds ratio (OR) and 95% confidence intervals represent a comparison of the cases with the two respective control groups with an unadjusted analysis.
Because smoking has previously been shown to be significantly associated with LD, and this association was confirmed here, we investigated whether there was an interaction between smoking and TLR5 SNPs (Table IV; references 19, 30). The analysis revealed that the association of 1174T-1775A and 1174C-1775G haplotypes with LD was only found in the nonsmokers (ORs = 2.43 [CI = 1.00, 5.89; P = 0.05] and 1.99 [CI = 1.04, 3.80; P = 0.04], respectively), whereas the association became marginal among smokers. This result implies that smoking may be a dominant risk factor for LD and, thus, may mask genetic associations. Together, these results suggest that TLR5 SNPs are associated with LD in nonsmokers.
Table IV.
Multivariate Analysis of Association of TLR5 SNPs with Legionnaires' Disease in Smokers and Nonsmokers
| SNP
|
Smokers
|
Nonsmokers
|
||||||
|---|---|---|---|---|---|---|---|---|
| No. | BP | Allele | OR | (95% CI) | P | OR | (95% CI) | P |
| 1. | 1174C | 0 | 1 | 1 | ||||
| 1174T | 1 | 0.72 | (0.18, 2.86) | 0.64 | 2.16 | (0.89, 5.23) | 0.09 | |
| 2. | 1775A | 0 | 1 | 1 | ||||
| 1775G | 1 | 1.33 | (0.54, 3.26) | 0.54 | 1.84 | (0.97, 3.48) | 0.06 | |
| Haplotype 1174-1775 | ||||||||
| CA | 00 | 1 | 1 | |||||
| CG | 01 | 1.33 | (0.55, 3.25) | 0.53 | 1.99 | (1.04, 3.80) | 0.04 | |
| TA | 10 | 0.76 | (0.19, 3.05) | 0.70 | 2.43 | (1.00, 5.89) | 0.05 | |
Comparison is between cases (n = 108) and the entire control group (= 508), excluding those with no information on smoking status. Odds ratio (OR) and 95% confidence intervals were calculated by a multivariate logistic regression analysis. All analyses are adjusted for gender and age (stratified at <40, 40–49, 50–60, and 60+), and then ORs for smokers and nonsmokers were calculated separately through an interaction term.
TLR5 Is the Predominant Mediator of IL-8 Expression in Lung Epithelia.
These genetic findings raise the question of why a deficiency in TLR5 would be deleterious to the host because there are other PAMP receptors, including TLR4, which could presumably still recognize L. pneumophila and mediate an inflammatory response. To understand TLR5 regulation of signaling in pulmonary epithelia, we stimulated A549 lung cell lines and measured culture supernatant IL-8 levels. As seen in previous analyses, LPS was unable to stimulate IL-8 production, even at doses of 1,000 ng ml−1 (Fig. 3 A, not depicted) (31). In contrast, flagellin potently stimulated IL-8 in a dose-dependent fashion, starting at concentrations of 1 ng ml−1. Furthermore, although heat-killed wild-type L. pneumophila induced IL-8 production, a flagellin mutant strain (FlaA−) was nonstimulatory (Fig. 3 B). Similar results were obtained for IL-6 and IL-8 production in Calu-3 lung epithelial cells (Fig. 3, C and D; and not depicted). In contrast to epithelial cells, both wild-type and FlaA− strains were similarly able to induce TNF-α production in a whole blood cytokine assay (Fig. 3 E). This control assay indicates that the FlaA− mutant did not have a generalized loss of PAMPs that compromised its overall stimulatory capacity. Overall, these results suggest a dominant role for flagellin and TLR5 in regulating the pulmonary epithelial innate immune response to L. pneumophila.
Figure 3.
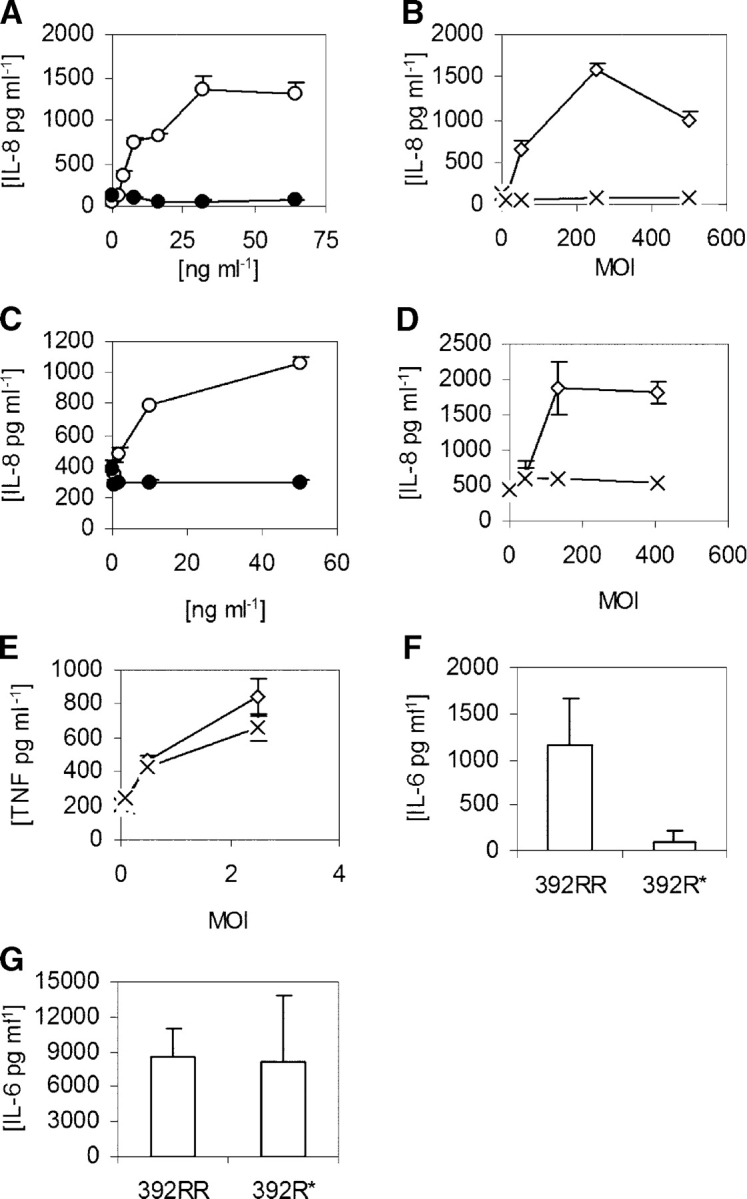
FliC stimulation of cytokine production. (A–E) Cells were stimulated with LPS (•), purified S. typhimurium FliC (○), heat-killed wild-type L. pneumophila (⋄), or FlaA− (FliC mutant) L. pneumophila (×). (A and B) A549 cells; (C and D) Calu-3 cells; and (E) whole blood. (F–G) PBMCs were harvested from individuals with homozygous wild-type TLR5 (392RR; n = 6) or stop codon TLR5 heterozygotes (392R*; n = 4). Cells were stimulated with FliC at 30 ng/ml−1 (F) or LPS at 10 ng/ml−1 (G). (A–G) Cells were stimulated for 18 h and supernatants were assayed for cytokine production by ELISA. Data are representative of experiments performed at least twice in triplicate with standard deviations indicated. (F and G) The mean level and standard deviation of IL-6 were derived from averaging the responses of different individual's cells stimulated in triplicate.
TLR5392STOP Has a Dominant Effect.
Because individuals who are heterozygous for the TLR5 stop codon are more susceptible to LD, we examined whether TLR5392STOP acts in a dominant fashion. We stimulated PBMCs from individuals with flagellin or LPS and measured IL-6 cytokine production by ELISA. Heterozygous stop codon individuals (392R*) had significantly decreased IL-6 production in comparison to those with the wild-type arginine at amino acid 392 (Fig. 3 F, 392RR; P < 0.05). In comparison, there was no difference in LPS-induced IL-6 production (Fig. 3 G). This result indicates that TLR5392STOP exerts a dominant effect and suggests that heterozygous TLR5 stop codon individuals are more susceptible to LD due to impaired production of proinflammatory cytokines. We reanalyzed the genetic association of TLR5 C1174T with susceptibility to LD with the assumption that TLR5392STOP exhibits a dominant effect. The genotype frequency of 1174TC and TT (combined) was 16.7% in the cases compared with 9.5% in the entire control group and 7.9% in the matched control group. The OR for the association of genotype 1174T (C,T) with developing LD was 1.90 (95% CI = 1.06–3.42; P = 0.03) when compared with the entire group and 2.34 (95% CI = 0.93–5.90; P = 0.07) in comparison to the matched control group. This analysis confirmed and further strengthened the association of TLR5392STOP with susceptibility to LD.
Discussion
In this paper, we show for the first time that a common TLR5 stop codon polymorphism acts in a dominant fashion and is associated with susceptibility to infection with a flagellated organism in humans. We further show that TLR5–flagellin interactions control IL-8 production in lung epithelial cell lines stimulated by L. pneumophila.
Although these results suggest an association of TLR5 with LD, we cannot exclude that these SNPs are in linkage disequilibrium with a nearby causative gene or that there is a confounding effect from population admixture. The latter possibility is unlikely given that the entire cohort was Dutch with >95% of both cases and controls being Caucasian Dutch. In addition, the matched control group showed similar trends of association. This control group included place of residence as a matching variable as a strategy to control for more subtle influences of population stratification. The association of the stop codon SNP with LD is more striking given that this is almost exclusively a heterozygous population. Furthermore, no DNA was available from the 18 individuals with LD who died, a group with an increased probability of having a susceptible genetic profile.
Previous works have found a number of risk factors for acquiring LD, including age, smoking, chronic lung disease, cancer, and immunosuppression (19, 27, 30, 32). This is the first study with evidence that supports a genetic basis for susceptibility to LD. In fact, association of SNPs with susceptibility to bacterial pneumonia has only been linked to a small number of genes, including mannose binding lectin and FcRIIa (33, 34). Our analytic approach highlights the importance of separating the effects of genetic and environmental variables on disease outcome because the genetic association of TLR5 with LD was exclusive to the nonsmoking population.
Our finding that flagellin is the major L. pneumophila stimulant of IL-8 production in lung epithelial cell lines has important implications for models of innate immunity. Previous investigators have demonstrated that the A549 lung epithelial cell line does not respond to LPS, even at doses of 1 μg ml−1 (31, 35). Normal lung epithelia may down-regulate LPS recognition through TLR4 as a protective mechanism to avoid an overly exuberant inflammatory response (36). A previous paper found that Pseudomonas flagellin, but not LPS, stimulated IL-8 production in tracheal epithelial cells (37). Several recent works with intestinal and lung epithelial cells also demonstrate that flagellin is a major stimulus of proinflammatory cytokine production (38–44). Together, these results suggest that PAMP recognition in epithelial cells is more constrained than macrophages and may explain why a selective TLR5 deficiency can predispose an individual to an increased risk of pneumonia due to a flagellated organism.
If there is redundancy in the innate immune system, there may be no detriment to having a stop codon in TLR5 if an individual has a wild-type version of TLR4. However, there are several reasons why TLR4 is unlikely to easily replace the function of TLR5. First, lung epithelial cell lines recognize LPS poorly and suggest that there are cell and organ-specific differences in TLR regulation and function. Second, L. pneumophila LPS is ∼1000-fold less stimulatory than S. typhimurium LPS (45–47). With such low stimulatory properties, L. pneumophila LPS may not be a major target of innate immune system recognition. Third, we have shown previously that TLR4-defective mice are not more susceptible to aerosolized infection with L. pneumophila (48). In addition, we found that TLR4 is not involved with recognition of heat-killed L. pneumophila by murine macrophages. These studies suggest that receptors other than TLR4 mediate recognition of L. pneumophila during an in vivo infection. Finally, even if TLR4 mediates L. pneumophila recognition, there may be qualitative or quantitative differences in the TLR4 induced gene expression profiles in comparison to TLR5 stimulation.
Although the importance of TLRs in regulating the innate immune response has been clearly demonstrated in animal models, the clinical significance of TLR polymorphisms in human disease susceptibility is only beginning to be elucidated. Recent analyses with TLR4 SNPs suggest a PAMP-specific influence of this gene on infectious disease susceptibility because TLR4 is associated with gram-negative bacterial infections (7, 8, 10, 11). The finding that TLR5 SNPs are associated with susceptibility to a flagellated bacterium supports the hypothesis that susceptibility profiles will be PAMP-dependent and vary between infections. The high frequency of the TLR5 stop codon mutation in apparently healthy individuals raises the question of whether there is an evolutionary advantage to having this genetic variant. Linkage papers have mapped a major susceptibility locus for systemic lupus erythematosus in humans and mice to chromosome 1q41 where TLR5 resides (49, 50). Given TLR5's role in mediating inflammatory signaling pathways, it will be of interest to examine its possible association with an autoimmune disease such as systemic lupus erythematosus.
The dominant effect of the stop codon mutation implies that ∼10% of individuals have severely impaired ability to recognize flagellated bacteria and may be more susceptible to infection. The presence of the stop codon in the extracellular ligand-binding domain, in contrast to the intracellular signaling domain, suggests that the dominant effect occurs through a unique mechanism. Because TLR5 acts as a homodimer, the stop codon variant may bind to the normal variant and inhibit its proper assembly or localization. Understanding this unusual mechanism may lead to novel insights for immunomodulatory treatment of inflammatory diseases.
Acknowledgments
We thank H.C. Boshuizen for establishing the database for controls, and A. Siegel and N. Khalid for statistical insights.
This work was supported by the National Institutes of Health grants to T.R. Hawn, K. Smith, L.P. Zhao, and A. Aderem. A Postdoctoral Physician Fellowship from the Howard Hughes Medical Institute supported T.R. Hawn.
Abbreviations used in this paper: CHO, Chinese hamster ovary; CI, confidence interval; ELAM, endothelial leukocyte adhesion molecule; LD, Legionnaires' disease; OR, odds ratio; PAMP, pathogen-associated molecular pattern; SNP, single nucleotide polymorphism; TLR, Toll-like receptor.
References
- 1.Bartlett, J.G., and L.M. Mundy. 1995. Community-acquired pneumonia. N. Engl. J. Med. 333:1618–1624. [DOI] [PubMed] [Google Scholar]
- 2.Cooke, G.S., and A.V. Hill. 2001. Genetics of susceptibility to human infectious disease. Nat. Rev. Genet. 2:967–977. [DOI] [PubMed] [Google Scholar]
- 3.Aderem, A., and R.J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature. 406:782–787. [DOI] [PubMed] [Google Scholar]
- 4.Janeway, C.A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. [DOI] [PubMed] [Google Scholar]
- 5.Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C.V. Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 6.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. [DOI] [PubMed] [Google Scholar]
- 7.Lorenz, E., J.P. Mira, K.L. Frees, and D.A. Schwartz. 2002. Relevance of mutations in the TLR4 receptor in patients with gram-negative septic shock. Arch. Intern. Med. 162:1028–1032. [DOI] [PubMed] [Google Scholar]
- 8.Agnese, D.M., J.E. Calvano, S.J. Hahm, S.M. Coyle, S.A. Corbett, S.E. Calvano, and S.F. Lowry. 2002. Human Toll-like receptor 4 mutations but not CD14 polymorphisms are associated with an increased risk of gram-negative infections. J. Infect. Dis. 186:1522–1525. [DOI] [PubMed] [Google Scholar]
- 9.Arbour, N., E. Lorenz, B. Schutte, J. Zabner, J. Kline, M. Jones, K. Frees, J. Watt, and D. Schwartz. 2000. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Gen. 25:187–191. [DOI] [PubMed] [Google Scholar]
- 10.Kiechl, S., E. Lorenz, M. Reindl, C.J. Wiedermann, F. Oberhollenzer, E. Bonora, J. Willeit, and D.A. Schwartz. 2002. Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 347:185–192. [DOI] [PubMed] [Google Scholar]
- 11.Smirnova, I., N. Mann, A. Dols, H.H. Derkx, M.L. Hibberd, M. Levin, and B. Beutler. 2003. Assay of locus-specific genetic load implicates rare Toll-like receptor 4 mutations in meningococcal susceptibility. Proc. Natl. Acad. Sci. USA. 100:6075–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi, F., K.D. Smith, A. Ozinsky, T.R. Hawn, E.C. Yi, D.R. Goodlett, J.K. Eng, S. Akira, D.M. Underhill, and A. Aderem. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 410:1099–1103. [DOI] [PubMed] [Google Scholar]
- 13.Smith, K.D., and A. Ozinsky. 2002. Toll-like receptor-5 and the innate immune response to bacterial flagellin. Curr. Top. Microbiol. Immunol. 270:93–108. [DOI] [PubMed] [Google Scholar]
- 14.Ibrahim, G.F., G.H. Fleet, M.J. Lyons, and R.A. Walker. 1985. Method for the isolation of highly purified Salmonella flagellins. J. Clin. Microbiol. 22:1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of Myd88-deficient mice to endotoxin. Immunity. 11:115–122. [DOI] [PubMed] [Google Scholar]
- 16.Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, and S. Akira. 1999. Cutting edge: toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749–3752. [PubMed] [Google Scholar]
- 17.Heuner, K., L. Bender-Beck, B.C. Brand, P.C. Luck, K.H. Mann, R. Marre, M. Ott, and J. Hacker. 1995. Cloning and genetic characterization of the flagellum subunit gene (flaA) of Legionella pneumophila serogroup 1. Infect. Immun. 63:2499–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadosky, A.B., L.A. Wiater, and H.A. Shuman. 1993. Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect. Immun. 61:5361–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Den Boer, J.W., E.P. Yzerman, J. Schellekens, K.D. Lettinga, H.C. Boshuizen, J.E. Van Steenbergen, A. Bosman, S. Van den Hof, H.A. Van Vliet, M.F. Peeters, et al. 2002. A large outbreak of Legionnaires' disease at a flower show, the Netherlands, 1999. Emerg. Infect. Dis. 8:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boshuizen, H.C., S.E. Neppelenbroek, H. van Vliet, J.F. Schellekens, J.W. den Boer, M.F. Peeters, and M.A. Conyn-van Spaendonck. 2001. Subclinical Legionella infection in workers near the source of a large outbreak of Legionnaires' disease. J. Infect. Dis. 184:515–518. [DOI] [PubMed] [Google Scholar]
- 21.Lettinga, K.D., A. Verbon, G.J. Weverling, J.F. Schellekens, J.W. Den Boer, E.P. Yzerman, J. Prins, W.G. Boersma, R.J. Van Ketel, J.M. Prins, and P. Speelman. 2002. Legionnaires' disease at a dutch flower show: prognostic factors and impact of therapy. Emerg. Infect. Dis. 8:1448–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, S.S., N. Khalid, C. Carlson, and L.P. Zhao. 2003. Estimating haplotype frequencies and standard errors for multiple single nucleotide polymorphisms. Biostatistics. 4:513–522. [DOI] [PubMed] [Google Scholar]
- 23.Zhao, L.P., S.S. Li, and N. Khalid. 2003. A method for the assessment of disease associations with single-nucleotide polymorphism haplotypes and environmental variables in case-control studies. Am. J. Hum. Gen. 72:1231–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon, D., C. Abajian, and P. Green. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8:195–202. [DOI] [PubMed] [Google Scholar]
- 25.Hajjar, A.M., D.S. O'Mahony, A. Ozinsky, D.M. Underhill, A. Aderem, S.J. Klebanoff, and C.B. Wilson. 2001. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J. Immunol. 166:15–19. [DOI] [PubMed] [Google Scholar]
- 26.Swanson, M.S., and B.K. Hammer. 2000. Legionella pneumophila pathogesesis: a fateful journey from amoebae to macrophages. Annu. Rev. Microbiol. 54:567–613. [DOI] [PubMed] [Google Scholar]
- 27.Stout, J.E., and V.L. Yu. 1997. Legionellosis. N. Engl. J. Med. 337:682–687. [DOI] [PubMed] [Google Scholar]
- 28.Roig, J., C. Domingo, and J. Morera. 1994. Legionnaires' disease. Chest. 105:1817–1825. [DOI] [PubMed] [Google Scholar]
- 29.Reingold, A.L. 1988. Role of legionellae in acute infections of the lower respiratory tract. Rev. Infect. Dis. 10:1018–1028. [DOI] [PubMed] [Google Scholar]
- 30.Fraser, D.W., T.R. Tsai, W. Orenstein, W.E. Parkin, H.J. Beecham, R.G. Sharrar, J. Harris, G.F. Mallison, S.M. Martin, J.E. McDade, et al. 1977. Legionnaires' disease: description of an epidemic of pneumonia. N. Engl. J. Med. 297:1189–1197. [DOI] [PubMed] [Google Scholar]
- 31.Pugin, J., C.C. Schurer-Maly, D. Leturcq, A. Moriarty, R.J. Ulevitch, and P.S. Tobias. 1993. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc. Natl. Acad. Sci. USA. 90:2744–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marston, B.J., H.B. Lipman, and R.F. Breiman. 1994. Surveillance for Legionnaires' disease. Risk factors for morbidity and mortality. Arch. Intern. Med. 154:2417–2422. [PubMed] [Google Scholar]
- 33.Yee, A.M., H.M. Phan, R. Zuniga, J.E. Salmon, and D.M. Musher. 2000. Association between FcgammaRIIa-R131 allotype and bacteremic pneumococcal pneumonia. Clin. Infect. Dis. 30:25–28. [DOI] [PubMed] [Google Scholar]
- 34.Roy, S., K. Knox, S. Segal, D. Griffiths, C.E. Moore, K.I. Welsh, A. Smarason, N.P. Day, W.L. McPheat, D.W. Crook, and A.V. Hill. 2002. MBL genotype and risk of invasive pneumococcal disease: a case-control study. Lancet. 359:1569–1573. [DOI] [PubMed] [Google Scholar]
- 35.Standiford, T.J., S.L. Kunkel, M.A. Basha, S.W. Chensue, J.P. Lynch, III, G.B. Toews, J. Westwick, and R.M. Strieter. 1990. Interleukin-8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. J. Clin. Invest. 86:1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin, T.R. 2000. Recognition of bacterial endotoxin in the lungs. Am. J. Respir. Cell Mol. Biol. 23:128–132. [DOI] [PubMed] [Google Scholar]
- 37.DiMango, E., H.J. Zar, R. Bryan, and A. Prince. 1995. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J. Clin. Invest. 96:2204–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steiner, T.S., J.P. Nataro, C.E. Poteet-Smith, J.A. Smith, and R.L. Guerrant. 2000. Enteroaggregative Escherichia coli expresses a novel flagellin that causes IL-8 release from intestinal epithelial cells. J. Clin. Invest. 105:1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reed, K.A., M.E. Hobert, C.E. Kolenda, K.A. Sands, M. Rathman, M. O'Connor, S. Lyons, A.T. Gewirtz, P.J. Sansonetti, and J.L. Madara. 2002. The Salmonella typhimurium flagellar basal body protein FliE is required for flagellin production and to induce a proinflammatory response in epithelial cells. J. Biol. Chem. 277:13346–13353. [DOI] [PubMed] [Google Scholar]
- 40.Sierro, F., B. Dubois, A. Coste, D. Kaiserlian, J.P. Kraehenbuhl, and J.C. Sirard. 2001. Flagellin stimulation of intestinal epithelial cells triggers CCL20-mediated migration of dendritic cells. Proc. Natl. Acad. Sci. USA. 98:13722–13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liaudet, L., C. Szabo, O.V. Evgenov, K.G. Murthy, P. Pacher, L. Virag, J.G. Mabley, A. Marton, F.G. Soriano, M.Y. Kirov, et al. 2003. Flagellin from gram-negative bacteria is a potent mediator of acute pulmonary inflammation in sepsis. Shock. 19:131–137. [DOI] [PubMed] [Google Scholar]
- 42.Gewirtz, A.T., P.O. Simon, Jr., C.K. Schmitt, L.J. Taylor, C.H. Hagedorn, A.D. O'Brien, A.S. Neish, and J.L. Madara. 2001. Salmonella typhimurium translocates flagellin across intestinal epithelia, inducing a proinflammatory response. J. Clin. Invest. 107:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gewirtz, A.T., T.A. Navas, S. Lyons, P.J. Godowski, and J.L. Madara. 2001. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 167:1882–1885. [DOI] [PubMed] [Google Scholar]
- 44.Eaves-Pyles, T., K. Murthy, L. Liaudet, L. Virag, G. Ross, F.G. Soriano, C. Szabo, and A.L. Salzman. 2001. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: I kappa B alpha degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J. Immunol. 166:1248–1260. [DOI] [PubMed] [Google Scholar]
- 45.Zahringer, U., Y.A. Knirel, B. Lindner, J.H. Helbig, A. Sonesson, R. Marre, and E.T. Rietschel. 1995. The lipopolysaccharide of Legionella pneumophila serogroup 1 (strain Philadelphia 1): chemical structure and biological significance. Prog. Clin. Biol. Res. 392:113–139. [PubMed] [Google Scholar]
- 46.Wong, K.H., C.W. Moss, D.H. Hochstein, R.J. Arko, and W.O. Schalla. 1979. “Endotoxicity” of the Legionnaires' disease bacterium. Ann. Intern. Med. 90:624–627. [DOI] [PubMed] [Google Scholar]
- 47.Neumeister, B., M. Faigle, M. Sommer, U. Zahringer, F. Stelter, R. Menzel, C. Schutt, and H. Northoff. 1998. Low endotoxic potential of Legionella pneumophila lipopolysaccharide due to failure of interaction with the monocyte lipopolysaccharide receptor CD14. Infect. Immun. 66:4151–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lettinga, K.D., S. Florquin, P. Speelman, R. van Ketel, T. van der Poll, and A. Verbon. 2002. Toll-like receptor 4 is not involved in host defense against pulmonary Legionella pneumophila infection in a mouse model. J. Infect. Dis. 186:570–573. [DOI] [PubMed] [Google Scholar]
- 49.Tsao, B.P., R.M. Cantor, K.C. Kalunian, C.J. Chen, H. Badsha, R. Singh, D.J. Wallace, R.C. Kitridou, S.L. Chen, N. Shen, et al. 1997. Evidence for linkage of a candidate chromosome 1 region to human systemic lupus erythematosus. J. Clin. Invest. 99:725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wakeland, E.K., K. Liu, R.R. Graham, and T.W. Behrens. 2001. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 15:397–408. [DOI] [PubMed] [Google Scholar]