

Abstract
Phosphoinositide 3-kinase (PI3K) is thought to contribute to the pathogenesis of asthma by effecting the recruitment, activation, and apoptosis of inflammatory cells. We examined the role of class IA PI3K in antigen-induced airway inflammation and hyperresponsiveness by i.p. administration into mice of Δp85 protein, a dominant negative form of the class IA PI3K regulatory subunit, p85α, which was fused to HIV-TAT (TAT-Δp85). Intraperitoneal administration of TAT-Δp85 caused time-dependent transduction into blood leukocytes, and inhibited activated phosphorylation of protein kinase B (PKB), a downstream target of PI3K, in lung tissues in mice receiving intranasal FMLP. Antigen challenge elicited pulmonary infiltration of lymphocytes, eosinophils and neutrophils, increase in mucus-containing epithelial cells, and airway hyperresponsiveness to methacholine. Except for modest airway neutrophilia, these effects all were blocked by treatment with 3–10 mg/kg of TAT-Δp85. There was also significant reduction in IL-5 and IL-4 secretion into the BAL. Intranasal administration of IL-5 caused eosinophil migration into the airway lumen, which was attenuated by systemic pretreatment with TAT-Δp85. We conclude that PI3K has a regulatory role in Th2-cell cytokine secretion, airway inflammation, and airway hyperresponsiveness in mice.
Keywords: allergy, cytokines, eosinophils, lung, inflammation
Introduction
Persistent inflammation of the airway and subsequent airway hyperresponsiveness are fundamental characteristics of bronchial asthma (1). Th2 lymphocytes mediate the inflammatory response in human asthma; mRNA expression for IL-4 and IL-5 and the presence of these cytokines in the airway are correlated to disease severity (2, 3). Th2 cytokine secretion is chemotactic for eosinophils, which release bronchoactive lipids, e.g., leukotriene C4 (LTC4) (4), platelet-activating factor (PAF) (5), and granular proteins (e.g., major basic protein) after migration into airways (6). The major basic protein of eosinophils causes bronchial hyperresponsiveness and sloughing of the airway epithelium (7).
Phosphoinositide 3-kinase (PI3K) is a signal transduction enzyme, which phosphorylates the D3 position of the inositol ring of phosphoinositide and its phosphorylated derivatives (8). The class IA PI3K is activated by tyrosine kinases and consists of a heterodimer composed of a 110-kD (p110α, β, δ) catalytic subunit and an adaptor protein (p85α, p85β, p55α, p55γ, p50α) (9). Recent studies suggest that PI3K may contribute to the pathogenesis of asthma by effecting the recruitment, activation, and apoptosis of inflammatory cells (10, 11). Class IA PI3K plays a key role in induction of the Th2 response (10–14). PI3K is also essential for IL-5–induced eosinophil release from bone marrow (12) and the migration of eosinophils caused by a number of chemoattractants (13). The catalytic subunits p110β and p110δ modulate FcɛR1-dependent mast cell activation (15). In addition to inflammatory cells, smooth-muscle cell mitogenesis and hyperplasia are also regulated by PI3K (14, 16). Enhanced basal activity of PI3K has been reported in eosinophils derived from allergic asthmatics (17). However, the determination of the precise role of class IA PI3K has been difficult because (a) knockout mice lacking p85α and its splicing variants die within a few weeks after birth (18) and (b) this deletion results in immunodeficiency caused by B cell dysfunction (19).
Recent investigations have demonstrated the usefulness of in vivo transduction of TAT-fusion proteins in mice (20). This allows for introduction of functional proteins directly in the intracellular compartment of all cells in rodents within 2 h. Transcellular administration (transduction) of dominant negative inhibitors accordingly can block enzyme systems otherwise requiring knockout and allow for immediate assessment, thus avoiding lethal mutations.
In these studies, we transduced dominant negative p85α (Δp85) (21) using an HIV-TAT protein transduction (PTD) domain into mice in vivo. In contrast to wild-type p85α (Wp85), Δp85 lacks the binding site for the catalytic p110 subunit (21). We constructed a Δp85 plasmid (pTAT-Δp85) containing 6 His residues, 11 amino acids of TAT, Δp85, and purified TAT-Δp85 fusion protein (Fig. 1 a). We tested the hypothesis that class IA PI3K mediates both antigen-induced airway inflammation and airway hyperresponsiveness to methacholine. We found that transduction of TAT-Δp85 into mice inhibited protein kinase B (PKB, also termed Akt) phosphorylation in lung tissues and that TAT-Δp85 blocked antigen-induced airway inflammation and hyperresponsiveness, and reduced Th2 cytokine level in BAL.
Figure 1.

Purified proteins used in this study. (a) Structure of TAT-Δp85 fusion protein. Six His residues and the 11–amino acid TAT peptide precede the NH2-terminal of the Δp85 protein. The 11 amino acids of TAT are the protein transduction domain (PTD). (b) Control Δp85 protein lacking the PTD. (c) TAT-GFP, a control protein lacking Δp85.
Materials and Methods
Generation of TAT-Δp85 and Its In Vivo Administration.
A cDNA fragment encoding dominant negative p85α was amplified by PCR from the Δp85 cDNA with the deletion of 35 amino acids from residues 478–513 with the insertion of two amino acids in pGEX (provided by Dr. M. Kasuga) with the following primers (sense: 5′-ACC GGT ATG AGT GCC GAG GGG TAC CAG TAC-3′; antisense: 5′-GAATTCTCATCGCCTCTGCTGCGCGTACACTGGGT-3′). The PCR products have AgeI and EcoRI cutting sites before and after Δp85 cDNA, respectively, and were inserted into a pCRII TOPO vector. The plasmids were digested with AgeI-EcoRI and ligated into an AgeI-EcoRI–digested pTAT vector (provided by Dr. S. Dowdy) using T4 ligase.
Purification of TAT fusion proteins was performed as described (22, 23). Briefly, TAT-protein was purified by sonication of high expressing BL21 cells in 10 ml of buffer Z (8 M urea, 20 mM Hepes, pH 8.0, 100 mM NaCl). Cellular lysates were resolved by centrifugation, loaded onto a 5-ml Ni-NTA column in buffer Z, washed, and eluted with imidazole in buffer Z sequentially in 100, 250, and 500 mM concentrations. Urea and imidazole were removed from the resultant protein solution using a low volume, 10,000 MWCO Slide-A-Lyzer dialysis cassette. Each fusion protein was flash-frozen at −80°C.
Mice.
Male C57BL/6 mice, 6–8-wk-old, were purchased from the Jackson Laboratory and housed in a pathogen-free biohazard level 2 facility maintained by The University of Chicago Animal Resources Center. The studies reported here conform to the principles outlined by the Animal Welfare and the National Health Services guidelines for the care and use of animals in biomedical research.
In protocols detailed below, animals receive i.p. TAT-Δp85 or control injection. The animals receiving injection of TAT-Δp85 or control proteins (His-Δp85 or TAT-GFP) were assigned randomly to the experimental groups consisting of six animals each. In preliminary studies, we demonstrated that TAT protein vector (administered as TAT-GFP) had no effect on OVA-induced airway inflammation and airway responsiveness versus saline buffer control.
Flow Cytometric Analysis of the TAT Fusion Protein Transduction into Blood Leukocytes.
Either TAT-Δp85 or control protein lacking the TAT PTD was labeled with FITC (Sigma-Aldrich) according to manufacturer's instructions. Mice were killed and blood was aspirated by cardiac puncture into a heparinized syringe for isolation of whole-blood leukocytes at specified times after i.p. injection of TAT-Δp85 or control Δp85 protein. Whole-blood leukocytes were isolated by hypotonic lysis of RBC. Isolated leukocytes were resuspended in calcium- and magnesium-free HBSS, pH 6.8, containing 1% BSA. The pH of the sheath fluid of the flow cytometer was adjusted to 6.8 to quench FITC fluorescence on the outside of the cell (24), and at least 10,000 cells were analyzed on a FACScan (Becton Dickinson).
Western Blot Analysis of the Uptake of TAT-Δp85 into Lung Tissue and the Efficacy of TAT-Δp85 Protein Transduction on FMLP-induced PKB Phosphorylation in Lung Tissues.
To examine the uptake of TAT-Δp85 into the lung, lungs were excised from mice at indicated times after i.p. administration of TAT-Δp85 (10 mg/kg). To examine the efficacy of TAT-Δp85 transduction, TAT-Δp85 (10 mg/kg) or control protein was administered i.p. at the indicated times before administration of FMLP. Animals were lightly anesthetized with 4.4 mg/kg i.p. xylazine and 65 mg/kg i.p. ketamine (25) and then challenged intranasally with 100 μl of 10−5 M FMLP dissolved in PBS through 24-g catheter. 15 min after FMLP instillation, mice were anesthetized with 30 mg/kg i.p. xylazine and 80 mg/kg i.p. ketamine, and lungs then were perfused through the right ventricle with 5 ml of PBS. The lungs were excised, rinsed in PBS, blotted dry, snap frozen in liquid nitrogen, and stored at −80°C. For preparation of whole-lung cell extracts, 10 mg of ground lung tissue was resuspended in 500 μl of lysis buffer (10 mM Tris, pH 7.4, 5 mM MgCl2, 50 U/ml DNase and RNase, 10 μg/ml each of leupeptin, aprotinin and pepstatin, 1 mM PMSF, and then homogenized for 30 s at a speed of 15,000 rpm in an Ultra-Turrax homogenizer). After a 30-min incubation on ice, homogenates were centrifuged at 16,000 g for 10 min. Protein concentrations were determined using the Bradford assay, the supernatants were mixed with Laemmli sample buffer, and boiled for 5 min. The samples were stored at −80°C.
Whole-lung cell extracts (30 μg) were subjected to SDS-PAGE, using acrylamide gels under reducing condition (15 mA/gel). Electrotransfer of proteins from the gels to polyvinylidene fluoride membrane was achieved using a semidry system (400 mA, 60 min). The membrane was blocked with 1% BSA for 60 min, then incubated with 1/1000 anti-phosphorylated PKB Ab, 1/1000 anti-6His tag (Cell Signaling, Beverly, MA), or 1/1000 anti-PKB Ab diluted in Tris buffered saline with Tween-20 (TBST) overnight. The membranes then were washed three times for 20 min with TBST. Donkey anti-rabbit IgG conjugated with HRP was diluted 1/3000 in TBST and incubated with polyvinylidene fluoride membrane for 60 min. The membrane was again washed three times with TBST and assayed by an ECL chemiluminescence system (Amersham).
Immunization and Airway Challenge with OVA.
Mice were sensitized and challenged as described previously (26). Briefly, mice were immunized with 10 μg of OVA and 1.125 mg of aluminum hydroxide (Imgect Alum; Pierce Chemical Co.) in 0.2 ml of sterile saline i.p. on days 0, 7, and 14. On days 21–24, mice were exposed to aerosolized OVA (1%) or saline for 40 min. To examine the effect of TAT-Δp85 on antigen-induced airway reactions, the animals received i.p. injection of TAT-Δp85 or control Δp85 protein lacking TAT PTD (Fig. 1 b) every 12 h from the first OVA challenge to the assessment of BAL, tissue, and airway responsiveness. For measurement of airway responsiveness, animals also received TAT vehicle lacking Δp85 (Fig. 1 c). In additional experiments, TAT fusion proteins were administered intranasally to mice to assess their effect on airway inflammation after antigen challenge. 2 h before OVA challenge, mice received intranasal administration of 50 μl of 0.3 or 1.0 mg/ml TAT-Δp85 through a 24 g catheter.
Measurement of Airway Responsiveness to Methacholine.
Methacholine challenge was performed 24 h after the final dose of OVA. Respiratory system resistance (Rrs) was measured through a computer-controlled small-animal ventilator (SAV) (Flexivent; SCIREQ) as described previously (27). Briefly, mice were anesthetized with 30 mg/kg xylazine and 80 mg/kg ketamine i.p., and the trachea was cannulated with an 18-gauge metal needle connected to the SAV. Mechanical ventilation was applied, and animals were ventilated quasisinusoidally at a frequency of 120 breaths/min at a tidal volume of 6 ml/kg. The expiratory valve of the SAV allowed the animal to empty passively through a water trap adjusted to maintain a positive end-expiratory pressure (PEEP) of 2.0 cmH2O. In preliminary experiments, this PEEP was shown to be optimal for the determination of methacholine-induced effects on Rrs (27). Increasing doses of methacholine (31.3 to 4,000 μg/ml) were infused through a jugular vein catheter at 5-min intervals.
Collection and Analysis of Bronchoalveolar Fluid Cells.
Airway inflammation was assessed 24 h after the final antigen challenge with OVA. BAL was performed by delivering 0.8 ml cold PBS into the airway through a trachea cannula and gently aspirating the fluid. The lavage was repeated three times to recover a total volume of 2–3 ml. The cells were stained with Trypan blue to determine viability and with Turk solution to obtain total nucleated cell counts using a hemocytometer. Cytospin (Cytospin 2; Shandon) slides were prepared from the BAL and were then fixed and stained using Diff-Quick (Dade Diagnostics). Differential cell counts were determined by counting a minimum of 300 cells/slide using standard morphological criteria in a single-blind method.
Measurement of Cytokine Levels in BAL.
The concentrations of IL-4, IL-5, and IFN-γ in BAL fluid were measured using a Mouse Th1/Th2 Cytokine CBA kit according to the manufacturer's protocol (BD Biosciences). The detection limits were 5 pg/ml for IL-4, 5 pg/ml for IL-5, and 2.5 pg/ml for IFN-γ.
Lung Histology.
Lungs removed from the chest cavity were fixed by injection of 4% buffered paraformaldehyde into the tracheal cannula at a pressure of 20 cm H2O and immersed in paraformaldehyde for 24 h. Lobes were sectioned sagitally, embedded in paraffin, cut into 5 μm sections, and stained with H&E for histological analysis. Additional sections were stained with alcian blue/PAS to identify mucus-containing cells. The severity of peribronchial inflammation was graded semiquantitatively for the following features: 0, normal; 1, few cells; 2, a ring of inflammatory cells 1 cell layer deep; 3, a ring of inflammatory cells 2–4 cells deep; 4, a ring of inflammatory cells of >4 cells deep. The numerical scores for the abundance of PAS-positive mucus-containing cell in each airway were determined as follows: 0, <0.5% PAS-positive cells; 1, 5–25%; 2, 25–50%; 3, 50–75%; 4, >75% (28).
IL-5–induced Airway Eosinophil Accumulation.
TAT-Δp85 (3 or 10 mg/kg) or a control Δp85 protein (excluding TAT PTD) was i.p. administered 2 h before challenge with IL-5. Animals were anesthetized with 4.4 mg/kg xylazine and 65 mg/kg i.p. ketamine (25) and then challenged intranasally with 100 μl of 100 ng/ml IL-5 dissolved in PBS through a 24-g catheter. BAL was performed 12 h after IL-5 instillation as described above.
Statistical Analysis.
Variation between three or more groups was analyzed using ANOVA followed by Fisher's least protected difference test. Statistical significance was claimed whenever P < 0.05.
Results
Kinetics and Efficacy of TAT-Δp85 Transduction.
The pharmacokinetics of in vivo TAT-Δp85 transduction were evaluated in mouse blood leukocytes. Mice were injected with 10 mg/kg fluorescein isothiocyanate (FITC)-labeled TAT-Δp85 i.p. or control Δp85 protein lacking TAT PTD. Flow cytometry of whole-blood leukocytes isolated 2–24 h after i.p. injection demonstrated the presence of TAT-Δp85. TAT-Δp85 was maximally expressed at 2–6 h, and disappeared by 24 h (Fig. 2 a). The T1/2 for TAT-Δp85 was 12 h. By contrast, control Δp85 protein lacking the TAT transduction domain was undetectable in blood leukocytes at any time (Fig. 2 b).
Figure 2.
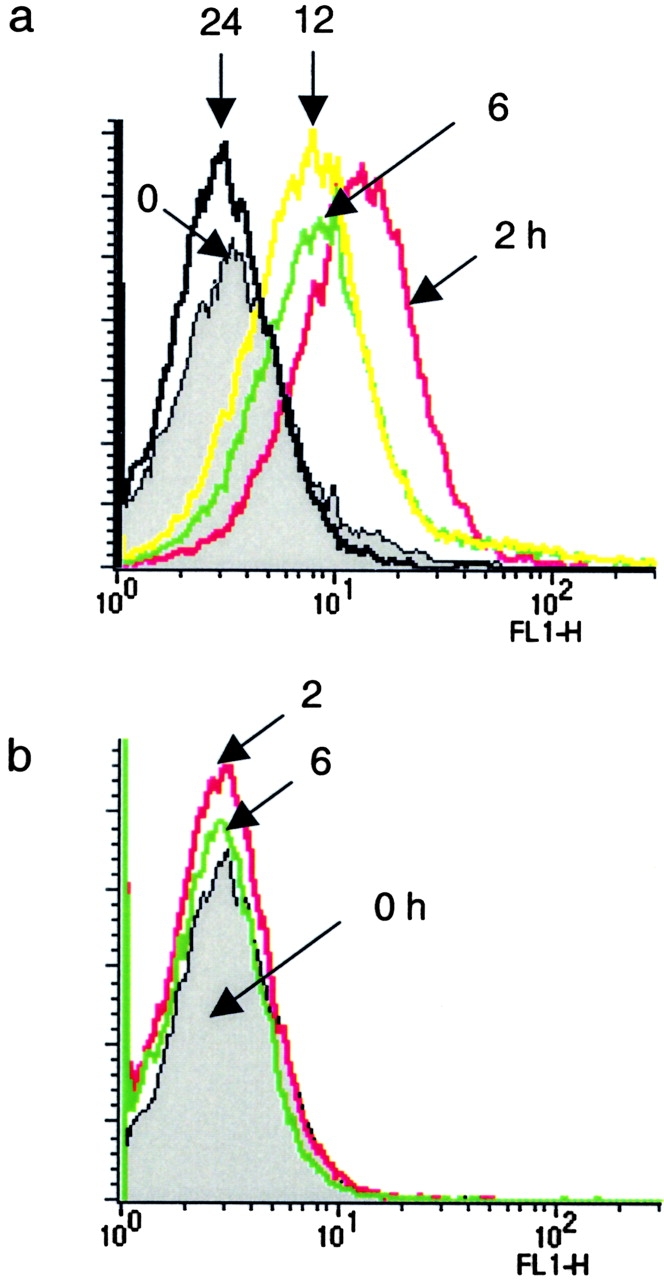
Kinetics of TAT-Δp85 transduction into whole-blood leukocytes in mice measured by flow cytometry. Whole-blood leukocytes were isolated at 0, 2, 6, 12, and 24 h after i.p. injection of FITC-labeled TAT-Δp85 to determine time-course and specificity of transmembrane uptake of TAT-Δp85. Nonspecific cell-surface fluorescence was quenched by resuspension in pH 6.8 HBSS. (a) Conjugated TAT-Δp85 demonstrates maximal transcellular uptake at 2 h and is progressively diminished over 24 h. (b) FITC-conjugated control Δp85 protein, which lacks the HIV-TAT domain, demonstrates no change in fluorescence versus after resuspension in pH 6.8 HBSS.
Western blot analysis of lung (Fig. 3 A), spleen, and liver (unpublished data) extracts demonstrated that TAT-Δp85 was maximally transduced at 2 h after i.p. administration of TAT-Δp85, and disappeared thereafter (Fig. 3 A). The inability of detecting of TAT-Δp85 at 4–12 h after i.p. injection may be due to the lower sensitivity of the Western blot than flow cytometry (see below). Intraperitoneal injection of TAT-Δp85 caused substantial inhibition of PKB phosphorylation in lung homogenates. Intranasal instillation of 10−5 M FMLP caused PKB phosphorylation in lung tissue extracts compared with vehicle PBS-stimulated controls; PKB phosphorylation was inhibited to baseline at 2–12 h after i.p. injection of 10 mg/kg TAT-Δp85 (Fig. 3 B). A control Δp85 protein absent the 11–amino acid TAT (Fig. 1 b) had no inhibitory effect on PKB phosphorylation (unpublished data). These results demonstrated that i.p. delivery of TAT-Δp85 fusion protein into mice was distributed into the lungs, and functionally suppressed PI3K-induced PKB phosphorylation. Based on the half-life of TAT-Δp85 in blood leukocytes, TAT-Δp85 was administered every 12 h in the subsequent experiments involving antigen challenge.
Figure 3.

Western blot analysis of the uptake of TAT-Δp85 into lung tissue and the efficacy of TAT-Δp85 protein transduction on FMLP-induced PKB phosphorylation in lung tissues. (A) Transduction of TAT-Δp85 into the lung. Lungs were excised from mice at indicated times after i.p. administration of TAT-Δp85 (10 mg/kg), and lung tissue extracts were mixed with loading buffer and separated by SDS-PAGE, and probed by anti-His antibody. (B) Western blot analysis of the effect of TAT-Δp85 on FMLP-induced PKB phosphorylation in whole-lung tissue. FMLP was instilled intranasally at 0, 2, 4, 6, 12, and 24 h after administration of TAT-Δp85. Whole-lung cell extracts were prepared and analyzed by Western blot with antiphosphorylation-specific PKB Ab (top). Equivalency of loading was established for each lane with anti-PKB Ab, which measures total PKB (phosphorylated and nonphosphorylated) (bottom). Inhibition of PKB phosphorylation is maximal at 2–6 h, corresponding to fluorescence data from Fig. 2. By 12 h, there is diminished inhibition of PKB phosphorylation by TAT-Δp85. The result shown is representative of three different experiments.
Effect of TAT-Δp85 on Antigen-induced Airway Inflammation.
OVA inhalation significantly increased eosinophil (P < 0.01), lymphocyte (P < 0.01), and neutrophil numbers (P < 0.05) in BAL fluid (Fig. 4) . Eosinophils were absent in BAL fluid collected from the saline challenged group. After OVA challenge, the number of eosinophils in the BAL fluid increased to 538 ± 60 × 103 cells (P < 0.01). Treatment of animals with 3–10 mg/kg TAT-Δp85 blocked the migration of eosinophils 24 h after OVA challenge in concentration-dependent manner. Eosinophil number decreased to 251 ± 5 × 103/ml with 3 mg/kg of TAT-Δp85 (P < 0.05) and further decreased to 82.6 ± 37.0 × 103/ml at 10 mg/kg (P < 0.01). By contrast, control Δp85 protein lacking the TAT PTD has no inhibitory effect on OVA-induced eosinophil count in BAL fluid.
Figure 4.

Effect of IP administration of TAT-Δp85 on airway inflammation after OVA sensitization and challenge. Analysis of inflammatory cells from BAL fluid 24 h after last OVA challenge. Positive (▪) and negative (□) controls are OVA and saline challenged animals, respectively. These animals received no other treatment. OVA challenge caused increased total cell count, which is largely composed of eosinophils. Increase in lymphocytes and neutrophils are recorded on a different scale (see ordinate). There was no effect on macrophage number. Migration of eosinophils and lymphocytes is blocked progressively with 3–10 mg/kg TAT-Δp85 but unaffected by 10 mg/kg Δp85 lacking the TAT domain. TAT-Δp85 has no effect on the migration of neutrophils, which is minimal. Each bar represents the mean ± SEM of 4–6 mice. *P < 0.05 and **P < 0.01 compared with positive control group. ND, not detectable.
BAL lymphocytes increased from 0.13 ± 0.06 × 103/ml for saline challenged control to 48.6 ± 7.2 × 103/ml 24 h after OVA challenge (P < 0.01, Fig. 4). TAT-Δp85 also blocked the increase in lymphocytes in the airway lumen after OVA challenge in concentration-dependent manner. Lymphocytes number decreased to 12.6 ± 5.3 × 103 at 3 mg/kg (P < 0.01) and 6.3 ± 1.9 × 103/ml at 10 mg/kg TAT-Δp85 (P < 0.01 vs. a Δp85 control protein treated group).
OVA challenge modestly increased neutrophil number in BAL to 13.1 ± 4.9 × 103/ml; neutrophil was not found in saline-challenged mice. Unlike eosinophils or lymphocytes, OVA-induced airway neutrophilia was not blocked by pretreatment with TAT-Δp85. Macrophage number in BAL was neither affected by OVA challenge nor by pretreatment with TAT-Δp85.
Effect of TAT-Δp85 on IL-5-induced Inflammation.
Further studies were performed to determine the role of PI3K in IL-5–induced eosinophil migration into airways. Eosinophils were not detected in BAL fluid in nonsensitized animals; 12 h after intranasal instillation of IL-5, the eosinophil count in BAL fluid was 5.0 ± 0.7 × 103/ml (P < 0.01, Fig. 5) . Pretreatment of animals with TAT-Δp85 concentration dependently blocked BAL eosinophilia to 2.4 ± 0.9 × 103 at 3 mg/kg TAT-Δp85 (P < 0.05) and 0.9 ± 0.103/ml at 10 mg/kg TAT-Δp85 (P < 0.01).
Figure 5.

Effects of TAT-Δp85 on eosinophil accumulation in BAL fluid 12 h after intranasal administration of IL-5. Cell counts were obtained from animals receiving IL-5 + either control Δp85 (lacking TAT domain) or TAT-Δp85. Eosinophils were not detectable in airways of animals before challenge with IL-5. TAT-Δp85 blocked progressively the eosinophil migration caused by IL-5. Δp85 lacking the TAT domain was not different from IL-5 alone. Accordingly, TAT-Δp85 blocks both IL-5 synthesis (Fig. 6) as well as the chemotactic effects of IL-5 on eosinophils. Each bar represents the mean ± SEM of 4–6 mice. *P < 0.05 and **P < 0.01 compared with positive control group. ND, not detectable.
Antigen-induced Th2 Cytokine Secretion in BAL Fluids.
To evaluate the role of Th2 cytokines in OVA-induced airway responses, we measured the concentrations of IL-4 and IL-5 in BAL fluid. OVA inhalation caused a significant increase in the concentration of IL-4 and IL-5, which was concentration dependently inhibited by TAT-Δp85. There was no reduction of these Th2 cell cytokines in control mice receiving Δp85 lacking the TAT PTD (Fig. 6) . OVA challenge also increased the barest concentration of Th1 cytokine, INF-γ, a Th-1 cell cytokine, in BAL fluid. TAT-Δp85 at 10 mg/kg had no inhibitory effect on INF-γ level in BAL fluid.
Figure 6.

Effect of TAT-Δp85 on Th1 and Th2 cytokine secretion after four OVA challenges on days 21, 22, 23, and 24. BAL fluids were collected 24 h after the last OVA challenge. Positive (▪) and negative (□) controls are OVA and saline challenged animals, respectively, without any pretreatment. TAT-Δp85 reduces the level of Th2 cytokines, IL-4 and IL-5, but has no effect on the Th1 cytokine IFNγ. Note difference in scale on the ordinate. Each bar represents the mean ± SEM of 4–6 mice. **P < 0.01 compared with positive control group. ND, not detectable.
Effect of TAT-Δp85 on Airway Histology.
OVA challenge induced inflammatory cell infiltration (largely eosinophils and mononuclear cells) in the lung interstitium around the airways and pulmonary blood vessels (Fig. 7 A, 1 and 2). Pretreatment with 3–10 mg/kg TAT-Δp85 blocked cell infiltration progressively at 24 h (Fig. 7, A 3 and B, left). Although many mucus-containing epithelial cells were apparent in OVA-challenged mice (Fig. 7 A, 4 and 5), very few of these cells were detected in mice treated with 10 mg/kg TAT-Δp85 (Fig. 7, A 6 and B, right).
Figure 7.

Histologic examination of lung tissues. (A) Representative sections of the lungs. The lung sections were obtained from saline- (1, 4) or OVA- (2, 3, 5, 6) challenged mice in the absence (1, 2, 4, 5) or presence (3, 6) of TAT-Δp85 (10 mg/kg). Sections are stained with haematoxylin and eosin (1, 2, 3) for morphological analysis of inflammation and with alcian blue/PAS (4, 5, 6) for analysis of mucin-containing cells. Tissue was examined by light microscopy (original magnification 400×). Substantial inflammation and mucin production caused by OVA (2, 5) are blocked by TAT-Δp85 (3, 6); compare with saline control (1, 4). (B) Semiquantitative analysis of the severity of peribronchial inflammation and the abundance of PAS-positive mucus-containing cells. Total lung inflammation and PAS-positive cells were defined as the average of the scores as described in Materials and Methods. *P < 0.05 and **P < 0.01 compared with positive control group.
Antigen-induced Airway Hyperresponsiveness.
Antigen-induced airway hyperresponsiveness to methacholine was attenuated by TAT-Δp85 (Fig. 8) . Rrs was comparable 24 h after the last OVA challenge in each group before methacholine challenge (time zero). Sensitization with OVA caused substantial increase in methacholine response compared with mice receiving saline inhalation. TAT-Δp85, but not Δp85 absent the TAT transduction domain, inhibited the augmented dose–response curve to methacholine in a dose-dependent manner. In a subsequent study, we demonstrated that TAT vehicle alone had no effect on the baseline airway responsiveness to methacholine. Hence, blockade of OVA-induced airway response to methacholine challenge was not caused by TAT protein in the absence of Δp85.
Figure 8.

Effect of TAT-Δp85 on antigen-induced airway hyperresponsiveness to methacholine in immunized mice following four OVA challenges on days 21, 22, 23 and 24. Airway responsiveness was measured 24 h after the last OVA challenge. Positive (□) and negative (Δ) controls are OVA and saline challenged animals, respectively, without any pretreatment. Δp85 lacking the TAT domain has no attenuating effect on antigen challenge in immune sensitized mice (□). By contrast, 10 mg/kg TAT-Δp85 (▪) blocked methacholine responsiveness in sensitized animals to control level (Δ), i.e., the response was comparable to that of sensitized, saline-challenged animals. Each bar represents the mean ± SEM of 4–6 mice. *P < 0.05 and **P < 0.01 compared with positive control group.
Discussion
The objective of this investigation was to determine the potential role of PI3K in antigen-induced airway inflammation, Th2 cytokine secretion, and airway hyperresponsiveness in mice. Our results show that TAT-Δp85 attenuates antigen-induced airway infiltration of eosinophils and lymphocytes in a concentration-dependent manner, reduces the number of mucus-containing cells in the airway lumen, reduces Th2 cytokine concentrations in BAL, and blocks airway hyperresponsiveness to methacholine. These findings suggest that PI3K signaling likely has a regulatory role in antigen-induced airway inflammation and hyperresponsiveness in mice.
The Th2 cytokine, IL-4, regulates allergic inflammation by promoting Th2 cell differentiation (29, 30), IgE synthesis and its receptor up-regulation (31, 32), VCAM-1 expression (33), and airway hypersecretion (34). Another Th2 cytokine, IL-5, promotes eosinophilic inflammation and infiltration into airways (35, 36). Our data demonstrate that blockade of PI3K signaling by i.p. administration of TAT-Δp85 reduces secretion of Th2 cytokines (IL-4 and IL-5), but has no effect on the secretion of the Th-1 cytokine, IFNγ, in the BAL fluid. These data suggest that PI3K regulates, in part, the balance between Th1 and Th2 lymphocytes (Fig. 6).
Although the mechanistic relationship between eosinophilic inflammation and airway hyperresponsiveness has not been established, eosinophils and their granular contents can cause airway smooth muscle contraction (7, 37, 38). Many inflammatory mediators attract and activate eosinophils through signal transduction pathways involving the enzyme, PI3K. In this study, TAT-Δp85 administration blocked antigen-induced both airway infiltration of eosinophils and airway hyperresponsiveness. Blockade of eosinophilic infiltration into airway can be caused by: (a) decreased release of eosinophils from bone marrow, (b) decreased eosinophil adhesion to endothelial cells and subsequent blockade of chemotaxis, or (c) decreased Th2 cytokine secretion. Palframan et al. (12) reported that wortmannin inhibited IL-5–induced release of eosinophils from perfused bone marrow, as well as selective eosinophil chemokinesis in vitro (12). We have demonstrated previously that the pharmacological inhibition of PI3K by LY294002 or wortmannin blocks eosinophil adhesion to ICAM-1 caused by IL-5 in vitro (39). In this study, TAT-Δp85 blocked Th2 cytokine secretion after antigen challenge (Fig. 6) and IL-5–induced eosinophil migration (Fig. 5), suggesting that PI3K is involved in cytokine-induced eosinophil migration. Our data are consistent with the results obtained in studies reported by others showing that PI3K inhibition blocks the increased number of eosinophils in OVA-challenged mice (40). Other investigations have shown that PI3K inhibition blocks eosinophil degranulation in vitro (41) and in vivo (40, 41). In our report, decreased airway hyperresponsiveness corresponded to blockade of infiltration of eosinophils into the conducting airways.
In contrast to eosinophils, TAT-Δp85 had no effect on neutrophil migration into the airway after antigen challenge. It has been reported that class IB PI3Kγ-deficient neutrophils have severe defects in migration and respiratory burst activity in response to heterotrimeric G protein–coupled receptor agonists and chemotactic agents (42). Thus, class IB PI3Kγ rather than class IA PI3K may regulate neutrophil migration.
The effects of transduction of TAT-Δp85 in decreasing airway hyperresponsiveness and inflammation appear to be fully specific to Δp85 domain of the TAT. Treatment with Δp85 lacking the TAT domain had no effect on airway responsiveness or inflammatory cell migration after allergen challenge. Likewise, TAT-GFP, which transmits the HIV-TAT domain, but not the Δp85 domain, had no effect on antigen-induced airway inflammation and methacholine-induced airway responsiveness versus saline controls.
A potential strategy for treatment asthma is to develop methods for blocking the signal transduction pathways causing inflammatory cell activation. A variety of PI3K effectors, such as PDK-1, PKB, p70 S6 kinase (p70S6K), and protein kinase C (PKC), and eliminators, such as PTEN (40), have been reported, and some of these were reported as critical signal transduction molecules in the pathogenesis of asthma. The p70S6K inhibitors, rapamycin and its analogue, were reported to inhibit antigen-induced airway inflammation and airway responsiveness in mice (43, 44). Rapamycin also blocked allergen-driven T cell proliferation and IL-5 production in PBMCs (43). Cellular PKC activity was increased in asthmatic patients (45), and increased expression and activation of PKCζ, which is phosphorylated in the activation loop site by PDK-1 (46, 47), correlated with antigen-induced late asthmatic responses (48). PTEN blocks the action of PI3K by dephosphorylating the signal lipid, PIP3. Intratracheal administration of adenoviruses carrying PTEN cDNA significantly reduced IL-4, IL-5, and eosinophil cationic protein levels in BAL fluids after OVA inhalation (40). Although the PI3K inhibitor, wortmannin, has been shown to inhibit antigen-induced airway hyperresponsiveness in mouse and guinea pigs (40, 41), wortmannin also exhibits an inhibitory effect on group IV-PLA2 (49), phospholipase D (50), or myosin light chain kinase (51). Accordingly, specific blockade of the PI3K signaling rather than a pharmacological inhibitor may be therapeutically more specific in airway inflammatory disease. In 1999, Dowdy et al. (20) reported the delivery of a biologically active protein into the mouse without signs of gross neurological problems or systemic distress. In this study, TAT-Δp85 had a marked effect on experimental antigen–induced asthmatic reactions, and there were no apparent adverse effects on BAL cell viability as assessed by trypan blue dye, histological change of the lung, or physiological airway smooth muscle response to methacholine in TAT-Δp85-treated mice. These results suggest possibilities for the development of protein therapies targeted to the specific pathways for bronchial asthma.
Our data, which link specific physiological events, i.e., airway hyperresponsiveness to inflammatory cell migration (i.e., eosinophils), also suggests that blockade of the latter during immune sensitization prevents the development of airway hyperresponsiveness. These data suggest that intervention with TAT-Δp85 protein could possibly be efficacious in preventing the development of airway hyperresponsiveness in humans. However, rodents do not develop the “human” asthma, and blockade of airway hyperresponsiveness in these studies was achieved in mice newly sensitized with antigen from the naïve state. Hence, it is not certain whether long established human asthma would respond comparably. It is important to acknowledge specific limitations of our findings. Our data were obtained in mice with experimentally induced antigen-mediated airway hyperresponsiveness. Although the histological changes in these airways resemble those of human asthma, these data cannot be extrapolated to the human state. Nonetheless, these data suggest both a possible pathogenetic mechanism and means for therapeutic intervention in human asthmatic states that is worthy of further consideration.
We conclude that blockade of PI3K in antigen sensitized animals reduces Th2 cytokine levels in the BAL, blocks eosinophil and lymphocyte migration into airways and consequent airway hyperresponsiveness to methacholine. Accordingly, PI3K may have a potential role in the immune regulation of Th2-mediated airway hyperresponsiveness.
Acknowledgments
We thank Dr. Steven Dowdy (University of California San Diego, La Jolla, CA) for providing TAT expression vector, Dr. Masato Kasuga (Kobe University Graduate School of Medicine, Kobe, Japan) for providing pGEX vectors containing Δp85 cDNA, and Dr. Julian Solway (The University of Chicago, Chicago, IL) for advice in measurement of airway responsiveness.
This work was supported by NHLBI grant HL-46368, by NHLBI SCOR grant HL-56399 (A.R. Leff), NIAID grant AI-52109 (X. Zhu), and by the Astra-Zeneca Traveling Fellowship (S. Myou).
Abbreviations used in this paper: FITC, fluorescein isothiocyanate; LTC4, leukotriene C4; PAF, platelet-activating factor; PEEP, positive end-expiratory pressure; PI3K, phosphoinositide 3-kinase; PKB, protein kinase B; PMSF, phenylmethylsulfonyl fluoride; PTD, protein transduction domain; Rrs, respiratory system resistance; SAV, small-animal ventilator; TBST, Tris-buffered saline with Tween-20; Wp85, wild-type p85α.
References
- 1.Bochner, B.S., B.J. Undem, and L.M. Lichtenstein. 1994. Immunological aspects of allergic asthma. Annu. Rev. Immunol. 12:295–335. [DOI] [PubMed] [Google Scholar]
- 2.Hamid, Q., M. Azzawi, S. Ying, R. Moqbel, A.J. Wardlaw, C.J. Corrigan, B. Bradley, S.R. Durham, J.V. Collins, P.K. Jeffery, et al. 1991. Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asthma. J. Clin. Invest. 87:1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson, D., Q. Hamid, S. Ying, A. Bentley, B. Assoufi, S. Durham, and A.B. Kay. 1993. Prednisolone treatment in asthma is associated with modulation of bronchoalveolar lavage cell interleukin-4, interleukin-5, and interferon-γ cytokine gene expression. Am. Rev. Respir. Dis. 148:401–406. [DOI] [PubMed] [Google Scholar]
- 4.Weiss, J.W., J.M. Drazen, N. Coles, E.R. McFadden, Jr., P.F. Weller, E.J. Corey, R.A. Lewis, and K.F. Austen. 1982. Bronchoconstrictor effects of leukotriene C in humans. Science. 216:196–198. [DOI] [PubMed] [Google Scholar]
- 5.Cuss, F.M., C.M. Dixon, and P.J. Barnes. 1986. Effects of inhaled platelet activating factor on pulmonary function and bronchial responsiveness in man. Lancet. 2:189–192. [DOI] [PubMed] [Google Scholar]
- 6.White, S.R., M.E. Strek, G.V. Kulp, S.M. Spaethe, R.A. Burch, S.P. Neeley, and A.R. Leff. 1993. Regulation of human eosinophil degranulation and activation by endogenous phospholipase A2. J. Clin. Invest. 91:2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefort, J., M.A. Nahori, C. Ruffie, B.B. Vargaftig, and M. Pretolani. 1996. In vivo neutralization of eosinophil-derived major basic protein inhibits antigen-induced bronchial hyperreactivity in sensitized guinea pigs. J. Clin. Invest. 97:1117–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitman, M., C.P. Downes, M. Keeler, T. Keller, and L. Cantley. 1988. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 332:644–646. [DOI] [PubMed] [Google Scholar]
- 9.Dhand, R., K. Hara, I. Hiles, B. Bax, I. Gout, G. Panayotou, M.J. Fry, K. Yonezawa, M. Kasuga, and M.D. Waterfield. 1994. PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 13:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukao, T., M. Tanabe, Y. Terauchi, T. Ota, S. Matsuda, T. Asano, T. Kadowaki, T. Takeuchi, and S. Koyasu. 2002. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3:875–881. [DOI] [PubMed] [Google Scholar]
- 11.Fukao, T., T. Yamada, M. Tanabe, Y. Terauchi, T. Ota, T. Takayama, T. Asano, T. Takeuchi, T. Kadowaki, J. Hata Ji, and S. Koyasu. 2002. Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat. Immunol. 3:295–304. [DOI] [PubMed] [Google Scholar]
- 12.Palframan, R.T., P.D. Collins, N.J. Severs, S. Rothery, T.J. Williams, and S.M. Rankin. 1998. Mechanisms of acute eosinophil mobilization from the bone marrow stimulated by interleukin 5: the role of specific adhesion molecules and phosphatidylinositol 3-kinase. J. Exp. Med. 188:1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunzendorfer, S., C. Meierhofer, and C.J. Wiedermann. 1998. Signaling in neuropeptide-induced migration of human eosinophils. J. Leukoc. Biol. 64:828–834. [DOI] [PubMed] [Google Scholar]
- 14.Krymskaya, V.P., R.B. Penn, M.J. Orsini, P.H. Scott, R.J. Plevin, T.R. Walker, A.J. Eszterhas, Y. Amrani, E.R. Chilvers, and R.A. Panettieri, Jr. 1999. Phosphatidylinositol 3-kinase mediates mitogen-induced human airway smooth muscle cell proliferation. Am. J. Physiol. 277:L65–L78. [DOI] [PubMed] [Google Scholar]
- 15.Smith, A.J., Z. Surviladze, E.A. Gaudet, J.M. Backer, C.A. Mitchell, and B.S. Wilson. 2001. p110β and p110delta phosphatidylinositol 3-kinases up-regulate Fc(epsilon)RI-activated Ca2+ influx by enhancing inositol 1,4,5- trisphosphate production. J. Biol. Chem. 276:17213–17220. [DOI] [PubMed] [Google Scholar]
- 16.Page, K., J. Li, J.A. Hodge, P.T. Liu, T.L. Vanden Hoek, L.B. Becker, R.G. Pestell, M.R. Rosner, and M.B. Hershenson. 1999. Characterization of a Rac1 signaling pathway to cyclin D(1) expression in airway smooth muscle cells. J. Biol. Chem. 274:22065–22071. [DOI] [PubMed] [Google Scholar]
- 17.Bracke, M., E. van de Graaf, J.W. Lammers, P.J. Coffer, and L. Koenderman. 2000. In vivo priming of FcαR functioning on eosinophils of allergic asthmatics. J. Leukoc. Biol. 68:655–661. [PubMed] [Google Scholar]
- 18.Fruman, D.A., F. Mauvais-Jarvis, D.A. Pollard, C.M. Yballe, D. Brazil, R.T. Bronson, C.R. Kahn, and L.C. Cantley. 2000. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 α. Nat. Genet. 26:379–382. [DOI] [PubMed] [Google Scholar]
- 19.Fruman, D.A., S.B. Snapper, C.M. Yballe, L. Davidson, J.Y. Yu, F.W. Alt, and L.C. Cantley. 1999. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science. 283:393–397. [DOI] [PubMed] [Google Scholar]
- 20.Schwarze, S.R., A. Ho, A. Vocero-Akbani, and S.F. Dowdy. 1999. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 285:1569–1572. [DOI] [PubMed] [Google Scholar]
- 21.Kotani, K., K. Yonezawa, K. Hara, H. Ueda, Y. Kitamura, H. Sakaue, A. Ando, A. Chavanieu, B. Calas, F. Grigorescu, et al. 1994. Involvement of phosphoinositide 3-kinase in insulin- or IGF-1-induced membrane ruffling. EMBO J. 13:2313–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagahara, H., A.M. Vocero-Akbani, E.L. Snyder, A. Ho, D.G. Latham, N.A. Lissy, M. Becker-Hapak, S.A. Ezhevsky, and S.F. Dowdy. 1998. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 4:1449–1452. [DOI] [PubMed] [Google Scholar]
- 23.Myou, S., X. Zhu, E. Boetticher, S. Myo, A. Meliton, A. Lambertino, N.M. Munoz, and A.R. Leff. 2002. Blockade of focal clustering and active conformation in β2-integrin-mediated adhesion of eosinophils to intercellular adhesion molecule-1 caused by transduction of HIV TAT-dominant negative Ras. J. Immunol. 169:2670–2676. [DOI] [PubMed] [Google Scholar]
- 24.Hall, D.J., J. Cui, M.E. Bates, B.A. Stout, L. Koenderman, P.J. Coffer, and P.J. Bertics. 2001. Transduction of a dominant-negative H-Ras into human eosinophils attenuates extracellular signal-regulated kinase activation and interleukin-5-mediated cell viability. Blood. 98:2014–2021. [DOI] [PubMed] [Google Scholar]
- 25.Mathur, M., K. Herrmann, X. Li, Y. Qin, J. Weinstock, D. Elliott, J. Monahan, and P. Padrid. 1999. TRFK-5 reverses established airway eosinophilia but not established hyperresponsiveness in a murine model of chronic asthma. Am. J. Respir. Crit. Care Med. 159:580–587. [DOI] [PubMed] [Google Scholar]
- 26.Humbles, A.A., B. Lu, C.A. Nilsson, C. Lilly, E. Israel, Y. Fujiwara, N.P. Gerard, and C. Gerard. 2000. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature. 406:998–1001. [DOI] [PubMed] [Google Scholar]
- 27.Duguet, A., K. Biyah, E. Minshall, R. Gomes, C.G. Wang, M. Taoudi-Benchekroun, J.H. Bates, and D.H. Eidelman. 2000. Bronchial responsiveness among inbred mouse strains. Role of airway smooth-muscle shortening velocity. Am. J. Respir. Crit. Care Med. 161:839–848. [DOI] [PubMed] [Google Scholar]
- 28.Ford, J.G., D. Rennick, D.D. Donaldson, R. Venkayya, C. McArthur, E. Hansell, V.P. Kurup, M. Warnock, and G. Grunig. 2001. Il-13 and IFN-γ: interactions in lung inflammation. J. Immunol. 167:1769–1777. [DOI] [PubMed] [Google Scholar]
- 29.Hsieh, C.S., A.B. Heimberger, J.S. Gold, A. O'Garra, and K.M. Murphy. 1992. Differential regulation of T helper phenotype development by interleukins 4 and 10 in an α β T-cell-receptor transgenic system. Proc. Natl. Acad. Sci. USA. 89:6065–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seder, R.A., W.E. Paul, M.M. Davis, and B. Fazekas de St Groth. 1992. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J. Exp. Med. 176:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coffman, R.L., J. Ohara, M.W. Bond, J. Carty, A. Zlotnik, and W.E. Paul. 1986. B cell stimulatory factor-1 enhances the IgE response of lipopolysaccharide-activated B cells. J. Immunol. 136:4538–4541. [PubMed] [Google Scholar]
- 32.Pawankar, R., M. Okuda, H. Yssel, K. Okumura, and C. Ra. 1997. Nasal mast cells in perennial allergic rhinitics exhibit increased expression of the Fc epsilonRI, CD40L, IL-4, and IL-13, and can induce IgE synthesis in B cells. J. Clin. Invest. 99:1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moser, R., J. Fehr, and P.L. Bruijnzeel. 1992. IL-4 controls the selective endothelium-driven transmigration of eosinophils from allergic individuals. J. Immunol. 149:1432–1438. [PubMed] [Google Scholar]
- 34.Dabbagh, K., K. Takeyama, H.M. Lee, I.F. Ueki, J.A. Lausier, and J.A. Nadel. 1999. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J. Immunol. 162:6233–6237. [PubMed] [Google Scholar]
- 35.Mauser, P.J., A.M. Pitman, X. Fernandez, S.K. Foran, G.K. Adams, III, W. Kreutner, R.W. Egan, and R.W. Chapman. 1995. Effects of an antibody to interleukin-5 in a monkey model of asthma. Am. J. Respir. Crit. Care Med. 152:467–472. [DOI] [PubMed] [Google Scholar]
- 36.Foster, P.S., S.P. Hogan, A.J. Ramsay, K.I. Matthaei, and I.G. Young. 1996. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med. 183:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabe, K.F., N.M. Munoz, A.J. Vita, B.E. Morton, H. Magnussen, and A.R. Leff. 1994. Contraction of human bronchial smooth muscle caused by activated human eosinophils. Am. J. Physiol. 267:L326–L334. [DOI] [PubMed] [Google Scholar]
- 38.Strek, M.E., F.S. Williams, G.J. Gleich, A.R. Leff, and S.R. White. 1996. Mechanisms of smooth muscle contraction elicited by cationic proteins in guinea pig trachealis. Am. J. Physiol. 270:L133–L140. [DOI] [PubMed] [Google Scholar]
- 39.Zhu, X., B. Jacobs, E. Boetticher, S. Myou, A. Meliton, H. Sano, A.T. Lambertino, N.M. Munoz, and A.R. Leff. 2002. IL-5-induced integrin adhesion of human eosinophils caused by ERK1/2- mediated activation of cPLA2. J. Leukoc. Biol. 72:1046–1053. [PubMed] [Google Scholar]
- 40.Kwak, Y.G., C.H. Song, H.K. Yi, P.H. Hwang, J.S. Kim, K.S. Lee, and Y.C. Lee. 2003. Involvement of PTEN in airway hyperresponsiveness and inflammation in bronchial asthma. J. Clin. Invest. 111:1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ezeamuzie, C.I., J. Sukumaran, and E. Philips. 2001. Effect of wortmannin on human eosinophil responses in vitro and on bronchial inflammation and airway hyperresponsiveness in Guinea pigs in vivo. Am. J. Respir. Crit. Care Med. 164:1633–1639. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki, T., J. Irie-Sasaki, R.G. Jones, A.J. Oliveira-dos-Santos, W.L. Stanford, B. Bolon, A. Wakeham, A. Itie, D. Bouchard, I. Kozieradzki, et al. 2000. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 287:1040–1046. [DOI] [PubMed] [Google Scholar]
- 43.Powell, N., S. Till, J. Bungre, and C. Corrigan. 2001. The immunomodulatory drugs cyclosporin A, mycophenolate mofetil, and sirolimus (rapamycin) inhibit allergen-induced proliferation and IL-5 production by PBMCs from atopic asthmatic patients. J. Allergy Clin. Immunol. 108:915–917. [DOI] [PubMed] [Google Scholar]
- 44.Fujitani, Y., and A. Trifilieff. 2003. In vivo and in vitro effects of SAR 943, a rapamycin analogue, on airway inflammation and remodeling. Am. J. Respir. Crit. Care Med. 167:193–198. [DOI] [PubMed] [Google Scholar]
- 45.Vachier, I., P. Chanez, T. Radeau, C. Le Doucen, C. Leger, and P. Godard. 1997. Cellular protein kinase C activity in asthma. Am. J. Respir. Crit. Care Med. 155:1211–1216. [DOI] [PubMed] [Google Scholar]
- 46.Le Good, J.A., W.H. Ziegler, D.B. Parekh, D.R. Alessi, P. Cohen, and P.J. Parker. 1998. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 281:2042–2045. [DOI] [PubMed] [Google Scholar]
- 47.Chou, M.M., W. Hou, J. Johnson, L.K. Graham, M.H. Lee, C.S. Chen, A.C. Newton, B.S. Schaffhausen, and A. Toker. 1998. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr. Biol. 8:1069–1077. [DOI] [PubMed] [Google Scholar]
- 48.Evans, D.J., M.A. Lindsay, B.L. Webb, H. Kankaanranta, M.A. Giembycz, B.J. O'Connor, and P.J. Barnes. 1999. Expression and activation of protein kinase C-zeta in eosinophils after allergen challenge. Am. J. Physiol. 277:L233–L239. [DOI] [PubMed] [Google Scholar]
- 49.Cross, M.J., A. Stewart, M.N. Hodgkin, D.J. Kerr, and M.J. Wakelam. 1995. Wortmannin and its structural analogue demethoxyviridin inhibit stimulated phospholipase A2 activity in Swiss 3T3 cells. Wortmannin is not a specific inhibitor of phosphatidylinositol 3-kinase. J. Biol. Chem. 270:25352–25355. [DOI] [PubMed] [Google Scholar]
- 50.Bonser, R.W., N.T. Thompson, R.W. Randall, J.E. Tateson, G.D. Spacey, H.F. Hodson, and L.G. Garland. 1991. Demethoxyviridin and wortmannin block phospholipase C and D activation in the human neutrophil. Br. J. Pharmacol. 103:1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arcaro, A., and M.P. Wymann. 1993. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 296:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]