

Abstract
Carbon monoxide (CO) and nitric oxide (NO) each have mechanistically unique roles in various inflammatory disorders. Although it is known that CO can induce production of NO and that NO can induce expression of the cytoprotective enzyme heme oxygenase 1 (HO-1), there is no information whether the protective effect of CO ever requires NO production or whether either gas must induce expression of HO-1 to exert its functional effects. Using in vitro and in vivo models of tumor necrosis factor α–induced hepatocyte cell death in mice, we find that activation of nuclear factor κB and increased expression of inducible NO are required for the protective effects of CO, whereas the protective effects of NO require up-regulation of HO-1 expression. When protection from cell death is initiated by CO, NO production and HO-1 activity are each required for the protective effect showing for the first time an essential synergy between these two molecules in tandem providing potent cytoprotection.
Keywords: carbon monoxide, nitric oxide, heme oxygenase, iNOS, hepatitis
Introduction
Inflammation and stress result in the induction of “protective genes” such as the inducible forms of heme oxygenase 1 (HO-1) and nitric oxide (NO) synthase II (inducible NO synthase [iNOS]) that generate the gaseous molecules carbon monoxide (CO) and NO, respectively. Both HO-1 and iNOS are cytoprotective enzymes and when expressed produce therapeutic benefits in a number of conditions/diseases. We and others have previously demonstrated that iNOS/NO is protective in models of fulminant hepatic failure (1, 2). HO-1 overexpression has been demonstrated to be cytoprotective in multiple models including endotoxemia (3), shock (4), ischemia/reperfusion (5, 6), and xenograft rejection (7). Recent findings suggest that one mechanism by which HO-1 protects is via generation of CO after the degradation of heme by HO-1 (8, 9).
These two gases are linked in that NO can up-regulate HO-1 expression leading to the formation of endogenous CO (10), and CO can bind to the heme group in the iNOS protein and influence the production of NO (11, 12). However, there is no information about any requisite functional interactions between the effects of CO and NO and the respective enzymes leading to their production. We hypothesized that in some cases CO requires up-regulation of iNOS and production of NO in mediating protective effects and that subsequent NO generation requires HO-1 induction to mediate its therapeutic effects. We have studied the role of iNOS and NO in a model of fulminant hepatic failure in which CO confers protective effects both in vitro and in vivo. Our findings demonstrate a system of interdependence: exogenous CO protects through a nuclear factor (NF)-κB–mediated, iNOS-dependent mechanism with NO subsequently protecting, at least in part, through an HO-1–dependent mechanism. Additionally, the protective effects initiated by CO are ultimately mediated, in part, via up-regulation of HO-1. Other roles NO plays in providing protection initiated by CO in this model are not clear. We show that each of these steps, iNOS induction/NO production and activation of HO-1 expression, is obligatory to obtain the therapeutic benefit initiated by CO.
Materials and Methods
Acute Hepatic Injury Models.
All experiments were approved by the institutional animal care and use committee of the University of Pittsburgh. Male C57BL/6J (Charles River Laboratories), 8–12-wk-old inos − / − mice and wild-type littermates (bred/maintained at the University of Pittsburgh) were used for in vivo experiments. Groups of mice were administered 0.3 μg TNF-α/8 mg D-galactosamine (D-gal)/mouse i.p. Depending on the experimental condition, some mice received 250 ppm CO, 10 mg/kg of the selective NO donor O2-vinyl 1-(pyrrolidin-1-yl) diazen-1-ium-1,2-diolate (V-PYRRO; Qbiogene) s.c., or 5 mg/kg cobalt protoporphyrin (CoPP; Frontier Scientific) i.p. Additionally, 5 mg/kg of the selective inhibitor of iNOS L-N6-(1-iminoethyl)-lysine-dihydrochloride (L-NIL; Qbiogene) or 50 μmol/kg of the HO-1 inhibitor tin protoporphyrin (SnPP; Frontier Scientific) was administered i.p. when specified. Where indicated, 500 mg/kg acetaminophen (APAP; Sigma-Aldrich) was administered i.p.
Hepatocyte Cell Culture.
Mouse primary hepatocytes were harvested from C57BL/6J, mkk3 − / −, inos − / − (in house breeding colony), or hmox-1 − / − (provided by E. Nabel, National Heart, Lung, and Blood Institute, Bethesda, MD, and S. Yet, Harvard Medical School, Boston, MA) mice as previously described (13). Hepatocytes were used on days 1–3 after the harvest.
Induction of Hepatocyte Death/Apoptosis.
Cells were treated with 10 ng/ml TNF-α and 200 ng/ml actinomycin-D (ActD; Sigma-Aldrich) to induce cell death. TNF-α/ActD treatment has previously been demonstrated to induce cell death, specifically apoptosis, in primary hepatocytes (13). Hepatocytes were treated with CO, 250–750 μM of the NO donor s-nitroso-N-acetyl-penicillamine (SNAP), and/or additional pharmacologic agents when specified. 12 h after TNF-α/ActD treatment, cells were washed and stained with crystal violet to determine viability as previously described (13). Where specified, 1–2 mM of the selective in vitro inhibitor of iNOS, L-N5-(1-iminoethyl)-ornithine-2HCl (L-NIO; Calbiochem), was used.
Gene Transfer/Plasmids.
In some experiments, gene transfer of an IκBα super repressor (provided by D. Brenner, University of North Carolina, Chapel Hill, NC; reference 14) or β-galactosidase using adenoviral vectors (10 PFU/cell) was performed 12 h before TNF-α/ActD treatment. Evaluation of NF-κB activation was performed using a luciferase reporter assay as previously described. (15) In brief, hepatocytes were cotransfected with NF-κB reporter constructs (100 ng/well; pGL3-κβ luciferase; provided by D. Golenbock, University of Massachusetts, Worcester, MA) and pIEP-Lac-z (0.5 μg/well) using Lipofectin (Invitrogen) according to the manufacturer's instructions. Evaluation of iNOS expression was performed using a luciferase reporter assay as previously described (16). In brief, hepatocytes were cotransfected with iNOS promoter reporter constructs (1 μg/well; pXP2; provided by C. Lowenstein, Johns Hopkins University, Baltimore, MD) and pIEP-LacZ (0.5 μg/well) as described above.
Luciferase Reporter Assays.
Hepatocytes were transfected with plasmids as described above. Hepatocytes were treated with various stimuli 24 h after transfection (see Results). Luciferase activity (reported as arbitrary units [A.U.]) was assayed 6 h after initiation of treatment using a luciferase assay kit (Promega) according to the manufacturer's instructions and measured on a Berthold Luminometer. Results were corrected for transfection efficiency and protein concentration.
Electrophoretic Mobility Shift Assay (EMSA).
Nuclei were extracted from hepatocytes after specified treatment. A double stranded DNA NF-κB consensus sequence (GGGGACTTTCCC; Santa Cruz Biotechnology, Inc.) was labeled with [γ-32P]-ATP and then incubated with 5 mg total nuclear protein. Some incubations were performed in the presence of antibodies against p65/RelA or p50 (Santa Cruz Biotechnology, Inc.) to evaluate for supershift. EMSAs were performed as previously described (17).
Immunoblot Analysis.
Western blot analysis was performed on primary hepatocytes in culture or from liver homogenates with antibodies to iNOS (1:1,000; Transduction Laboratories), HO-1 (1:2,000; Calbiochem), or β-actin (1:5,000; Sigma-Aldrich). 30 μg protein in cell culture experiments or 100 μg protein from liver homogenates was loaded per well for SDS-PAGE.
Histology/Immunohistochemistry.
For histology and immunohistochemistry, livers were fixed in 2% paraformaldehyde and then snap frozen in liquid nitrogen. Livers were then sectioned (7 microns thick) and stained with hematoxylin and eosin (H&E). Liver sections were also stained for TUNEL and activated caspase 3 using kits according to the manufacturer's instructions (Promega). Sections for iNOS immunocytochemistry were blocked with 5% goat serum containing 0.2% bovine serum albumin. Thereafter, sections were incubated for 1 h at room temperature with anti-iNOS antibody (1:300; Transduction Laboratories), and then washed and probed with a secondary antibody conjugated to Alexa 488 (Molecular Probes). Nuclei were stained with Hoechst dye. Images were acquired using an Olympus Provus microscope. Hepatocytes in culture were plated on gelatinized coverslips, stimulated as indicated, and then fixed in 2% paraformaldehyde containing 0.1% Triton X-100. Blocking and staining was similar to liver sections except that anti-p65/RelA antibody (1:350; Santa Cruz Biotechnology, Inc.) was used.
Results
HO-1 Protects against Liver Injury.
Pharmacological induction or gene transfer of HO-1 is hepatoprotective in models of ischemia/reperfusion (5), endotoxemia (3), hyperoxic lung injury (18), and hemorrhagic shock (19). Consistent with these findings, Sass et al. (20) showed that mice were more prone to TNF-α–induced hepatocyte cell death when HO-1 was inhibited. To study whether HO-1 is protective in a model of acute hepatic failure, we induced HO-1 expression in C57BL/6J mice by administering CoPP and then administering TNF-α/D-gal. Induction of HO-1 was able to prevent the injury as measured by serum alanine aminotransferase (ALT) levels (Fig. 1) .
Figure 1.

HO-1 protects against acute hepatitis. Male C57BL/6J mice were administered 5 mg/kg CoPP i.p. 24 h before 0.3 μg TNF-α/8 mg D-gal/mouse i.p. Serum ALT was determined 8 h later. Results are the mean ± SD of six to eight mice/group. *, P < 0.005.
CO Protects Hepatocytes.
Based on the protection conferred by HO-1 in vivo, we asked if exogenous CO would protect against hepatocyte cell death in vitro. Exposure of primary mouse, rat, and human hepatocytes to 250 ppm CO inhibited TNF-α–induced apoptosis (Fig. 2 , a and b), consistent with previous data showing inhibition of cell death in endothelial cells (21), β cells (22), and fibroblasts (23). As the effects of CO in other models were mediated by guanylyl cyclase and/or p38 MAPK (21, 24), the role of these signaling molecules was investigated in this model. Inhibition of hepatocyte apoptosis was independent of cGMP generation, as the selective guanylyl cyclase inhibitor 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one (Calbiochem) did not reverse the protection provided by CO (Fig. 2 a). Additionally, CO treatment inhibited cell death both in the presence of 3–30 μM SB203580 (Calbiochem), a selective inhibitor of p38 MAPK activation, and in hepatocytes from mkk3 − / − mice, the dominant upstream kinase for p38 (not depicted). Thus, the effects of CO were independent of the cGMP/p38 MAPK pathway. In all of these experiments, hepatocytes were pretreated with CO for 1 h before the addition of TNF-α/ActD to the medium. If the CO treatment was started after the addition of TNF-α, there was less protection (not depicted). We hypothesize that the CO pretreatment primes the molecular machinery in the cells such that upon stimulation with TNF-α, the antiapoptotic/survival pathways will predominate over the proapoptotic pathways.
Figure 2.


NF-κB activation is crucial in the ability of CO to provide cytoprotection against TNF-α/ActD-induced cell death. (a) Cells were preincubated with 250 ppm CO for 1 h (standard pretreatment time for all experiments) before the addition of 10 ng TNF-α/200 ng/ml ActD. Cells were maintained in CO for the duration of the experiment. 12 h thereafter, cell viability was determined as previously described (reference 13). Adenoviral experiments involved incubating hepatocytes overnight with 10 PFU/cell of the adenovirus before the addition of TNF-α/ActD. Hepatocytes were then assayed for viability by crystal violet. To evaluate the role of cGMP and confirm the role of NF-κB, hepatocytes were treated separately with 2–10μM of the soluble guanylate cyclase inhibitor 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one or 10 μM of the NF-κB inhibitor BAY 11-7082. Treatment with the inhibitors was for 1 h before the 1-h pretreatment with CO. TNF-α/ActD was then added and the cells were tested for viability 12 h later. Note that NF-κB activation was critical to the protection elicited by CO whereas cGMP was not involved. Exposure to CO led to suppression of cell death that was significantly lower (*, P < 0.01) than without CO. Results shown are the mean ± SE of triplicate wells from four independent experiments. (b) Human primary hepatocytes obtained from a donor liver resection (provided by S. Strom, University of Pittsburgh, Pittsburgh, PA) were treated with CO and TNF-α/ActD as described above. A similar protective effect of CO was observed. Results are mean ± SD of triplicate wells from three independent experiments. *, P < 0.05. (c) Evaluation of NF-κB activation was performed using a luciferase reporter assay as previously described (reference 15). In brief, hepatocytes were cotransfected with NF-κB reporter constructs and pIEP-Lac-z 24 h before the addition of 10 μM BAY 11-7082 or vehicle. Cells were incubated for 1 h before 250 ppm CO. Luciferase activity (reported as A.U.) was assayed 6 h after exposure to CO or a CM composed of 500 U/ml TNF-α, 100 U/ml IL-1β, and 100 U/ml IFNγ, which was used as a positive control for NF-κB activation. Results were corrected for transfection efficiency and protein concentration. Results shown are the mean ± SE of triplicate wells from three independent experiments. *, P < 0.001 versus Air. (d) NF-κB DNA binding evaluated by EMSA in hepatocytes treated with 250 ppm CO. Note the time-dependent increase in NF-κB binding (total) with expression peaking at 1 h (lanes 1, 4, and 7). Extracts were then supershifted to identify the different NF-κB dimers using antibodies against p50 (lanes 2, 5, and 8) and p65 (lanes 3, 6, and 9). Results are representative of two independent experiments. (e) Immunostaining for nuclear p65 localization in primary hepatocytes after 1 h exposure to 250 ppm CO. Images depict nuclear translocation of NF-κB (arrows pointing to green nuclei that depict the translocation of NF-κB) in both CM (used as a positive control) and CO-treated cells versus no localization in air-treated cells (arrows pointing to blue nuclei). Images are representative of six different fields. Bar, 10 μm.
CO-induced Protection Requires NF-κB.
The role of NF-κB in the liver as part of a prosurvival signaling pathway has been studied (25). We demonstrate here in mouse hepatocytes that CO induced an increase in NF-κB nuclear translocation and DNA binding as measured by EMSA, immunostaining for RelA/p65 nuclear translocation, and NF-κB luciferase reporter assay activity that peaked 1 h after placing cells in the CO atmosphere (Fig. 2, c–e). A cytokine mixture (CM) was included in the treatment groups as a positive signal as well as a standard for maximum reporter activity by which to evaluate the effects of CO. Transfection efficiency in primary hepatocytes is difficult, but even given this problem, our reporter activity was very significant (*, P < 0.001 vs. control). These data combined with our positive immunostaining and EMSA results support the notion that CO induces a moderate increase in NF-κB that in itself may in part result in selective gene expression. To evaluate whether NF-κB activity is needed for protection mediated by CO, we used adenoviral gene transfer of IκBα to prevent NF-κB translocation (14) or 1–10 mM BAY 11-7082 (Calbiochem) to inhibit NF-κB activation. The protective effects of CO were abrogated by inhibition of NF-κB activation (Fig. 2 a). NF-κB is known to be involved in the regulation of many antiapoptotic genes (21, 26, 27), some of which may play a role in the cytoprotection conferred by CO.
CO Requires NF-κB–dependent iNOS Expression for Protection of Hepatocytes.
Overexpression of iNOS and NO donors are protective in models of hepatocyte apoptosis similar to the one studied here (13, 28, 29). Because iNOS gene expression is regulated by NF-κB (17), we asked if protection mediated by CO requires expression of iNOS and generation of NO. Exposure of hepatocytes to CO produced a highly significant increase in activity in an iNOS luciferase reporter assay (Fig. 3 a). Again, a CM was used as both a positive control in these low efficiency transfections and as a standard by which to evaluate the effects of CO. Consistent with the NF-κB dependence of iNOS expression, we demonstrated decreased reporter activity in hepatocytes treated with BAY 11-7082 (Fig. 3 a). Additionally, iNOS protein was markedly increased in response to TNF-α in the presence of CO compared with TNF-α alone (Fig. 3 b). Using hepatocytes from iNOS knockout mice (inos − / −) as well as wild-type hepatocytes treated with 1 mM of the selective iNOS inhibitor L-NIO (Calbiochem), we tested whether CO could protect against TNF-α–induced death in the absence of iNOS activity. Hepatocytes lacking iNOS activity were not protected significantly by CO from TNF-α–induced cell death whereas wild-type hepatocytes were (Fig. 3 c). The slight protection observed, which did not reach statistical significance, in those groups treated with CO where iNOS/NO was inhibited suggests the potential existence of an additional protective mechanism by which CO exerts its effects. One possibility is that other protective genes that are activated by NF-κB exert protection. Taken together, these data show that CO requires NF-κB activation and iNOS expression to provide strong protection for hepatocytes from cell death in vitro, although we do not rule out an apparently weaker protective mechanism that does not require iNOS expression.
Figure 3.

The role of iNOS in CO-induced cytoprotection from TNF-α/ActD-induced cell death. (a) Evaluation of iNOS expression was performed using a luciferase reporter assay as previously described (16). In brief, hepatocytes were cotransfected with an iNOS promoter reporter construct and pIEP-LacZ 24 h before exposure to 10 μM BAY 11-7082 or vehicle. Cells were incubated with BAY 1 h before exposure to 250 ppm CO. Luciferase activity (reported as A.U.) was assayed as described above. CM (see above) was used as a positive control to induce iNOS expression and results were corrected for transfection efficiency and protein concentration. Results shown are the mean ± SE of triplicate wells from four independent experiments. *, P < 0.001 versus air- and air/BAY-treated cells. (b) Expression of iNOS protein was evaluated by immunoblotting. In brief, cell extracts from hepatocytes were treated with TNF-α/ActD for 6–8 h in the presence and absence of 250 ppm CO. Control cells received air or CO alone. Note that TNF-α induces iNOS expression minimally, whereas those cells treated with TNF-α in the presence of CO show a significantly greater induction in iNOS protein. The immunoblot is representative of three independent experiments. (c) Mouse hepatocytes were isolated from inos − / − or wild-type C57BL/6J mice that were then pretreated for 1 h with 1 mM L-NIO to inhibit iNOS before CO administration. Those groups exposed to CO received a 1-h pretreatment before the addition of TNF/ActD and were then returned to CO exposure. CO did not provide protection against cell death, as evaluated via crystal violet exclusion 12 h later in cells where iNOS expression was absent or inhibited. Results shown are the mean ± SE of triplicate wells from four independent experiments. *, P < 0.01 versus non-TNF/ActD and CO/TNF/ActD-treated cells.
Inhaled CO Prevents Liver Failure.
The above data guided the hypothesis that inhaled CO would prevent liver injury in the TNF-α/D-gal model of fulminant hepatic failure. To test this, mice were pretreated with 250 ppm CO for 1 h before administering 0.3 μg TNF-α/8 mg D-gal/mouse i.p. and then returned to the CO exposure chamber. Liver failure without CO occurred in 6–8 h, driven primarily by apoptosis of hepatocytes, as in the in vitro model described above (1, 13, 30). Serum ALT in mice treated with CO was 74% lower than in those that breathed air (Fig. 4 a; *, P < 0.001). CO treatment also decreased TUNEL+ cells (a marker of cell death) and staining of activated caspase 3, while preserving normal architecture by histological assessment (Fig. 4 b). Control mice kept in air that received TNF-α/D-gal showed marked hepatic inflammation, edema, hemorrhage, and loss of architecture (Fig. 4 b).
Figure 4.
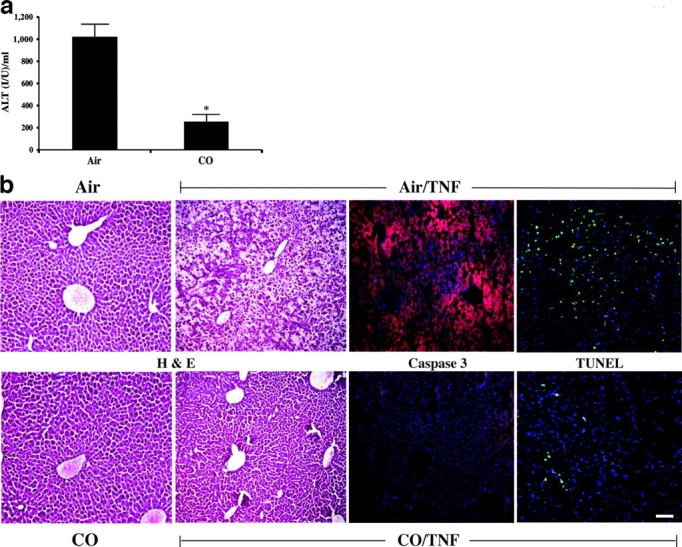
CO prevents TNF-α/D-gal–induced liver damage. (a) Male C57BL/6 mice were pretreated for 1 h with 250 ppm CO and then administered TNF-α/D-gal. Serum was analyzed for ALT levels 6–8 h later. Those mice exposed to CO had a >74% lower serum ALT level than mice receiving air instead of CO. Results presented as mean ± SD of 18–20 mice. *, P < 0.001 versus air treated. (b) Liver samples from mice treated with TNF-α/D-gal in the presence and absence of 250 ppm CO for 8 h were sectioned and stained for H&E, activated caspase 3 (as indicated by an increase in red intensity), and TUNEL+ cells (as demarcated by the increased green cellular staining). Nuclei are stained blue. Exposure to CO markedly reduced TNF-α/D-gal–induced liver damage as assessed by H&E staining. Both the increase in activated caspase 3 and TUNEL+ cells were significantly less in both staining intensity and the percentage of positively stained cells in mice treated with TNF-α/D-gal in the presence of CO. Images are representative sections from 15–20 sections/liver from three to four individual mice/group. Bar, 20 μm.
CO-mediated Protection against Liver Damage Requires iNOS.
Hepatic iNOS protein levels, determined by immunoblotting and immunohistochemistry, were increased in livers of mice 6 h after treatment with TNF-α/D-gal as compared with air-treated controls (Fig. 5 , a and b). To ascertain whether the expression of iNOS had a functional role, we tested whether CO would protect inos − / − mice or wild-type mice treated i.p. with 10 mg/kg of the selective iNOS inhibitor L-NIL (dosed every 2 h). CO failed to protect these animals as determined by serum ALT (Fig. 5 c) and histopathology (not depicted). Thus, we concluded that the protective effect of inhaled CO in TNF-α–induced liver failure is dependent on iNOS activity.
Figure 5.

Role of iNOS in CO-induced cytoprotection in vivo. (a) Male C57BL/6J mice were treated with air or 250 ppm CO 1 h before 0.3 μg TNF-α/8 mg D-gal/mouse i.p. administration. 6 h later, livers were harvested to evaluate iNOS expression by immunoblotting. Results show that iNOS expression was increased modestly in air/TNF-α/D-gal–treated mice, but was markedly increased in mice treated with TNF-α/D-gal and CO. As expected, inos − / − mice showed no expression of iNOS protein. (b) Immunostaining of iNOS expression in liver sections from mice treated with TNF-α/D-gal in the presence or absence of CO as well as from air and CO controls that received no TNF-α/D-gal. Results show a modest increase in iNOS expression in CO-treated mouse livers in the absence of TNF/D-gal, however a significantly greater increase in expression (indicated by the increase in green-stained cells) was observed in the liver of animals treated with TNF-α/D-gal in the presence of CO, which appeared to be localized around blood vessels. Images are representative of six separate animals and 6–10 different sections/liver sample. Bar, 20 μm. (c) The efficacy of CO-induced protection was tested in the absence of iNOS activity using inos − / − and wild-type mice that were treated i.p. with 5 mg/kg L-NIL, the selective inhibitor of iNOS, dosed every 2 h. L-NIL was administered 2 h before CO. CO-treated animals were then pretreated (250 ppm) for 1 h before TNF-α/D-gal. In the absence of iNOS function/expression, CO is unable to protect against liver damage as assessed by serum ALT levels. Results are mean ± SD of six to eight animals/group. *, P < 0.01 versus CO/TNF-α/D-gal and air and CO controls.
CO-induced Protection Is Ultimately Dependent on HO-1 Activity.
Based on our findings that CO-induced hepatoprotection is dependent on iNOS, plus the known hepatoprotective effects of NO and data showing that NO increases the expression of HO-1 (10), we hypothesized that CO and NO may protect through an HO-1–dependent mechanism. Immunoblotting of liver extracts from mice treated with CO in the presence or absence of TNF-α/D-gal showed up-regulation of HO-1 (Fig. 6 a). The addition of the iNOS inhibitor L-NIL to these above groups, which abrogated the protection (Fig. 5 c), also prevented the up-regulation of HO-1 (Fig. 6 b). To test the hypothesis that HO-1 was central to CO-elicited hepatoprotection, we used 50 μmol/kg SnPP (Frontier Scientific) s.c. as a selective inhibitor of HO-1 activity (31). SnPP very significantly diminished the protective effects of CO in this model (Fig. 6 c). SnPP administration in the absence of TNF-α/D-gal had no deleterious or protective effects (unpublished data). These results suggested that up-regulation of HO-1 was important for the protective effects of CO.
Figure 6.

Interrelationship between CO/HO-1 and NO/iNOS in providing protection against acute liver failure. (a) Immunoblot of HO-1 expression in the livers from mice administered TNF-α/D-gal in the presence and absence of 250 ppm CO. CO-treated mice showed a significant increase in HO-1 expression in both the presence and absence of TNF-α/D-gal. Blot is representative of two independent experiments. (b) To assess the role of iNOS on TNF/D-gal-induced HO-1 expression in the liver, mice were administered 5 mg/kg L-NIL i.p. 2 h before pretreatment with 250 ppm CO, and then every 2 h thereafter. Control mice received L-NIL and remained in room air. Note that CO increased HO-1 expression in vehicle-treated mice, but was unable to induce expression when iNOS was inhibited. L-NIL treatment alone had a minimal effect on HO-1 expression. Blot is representative of two independent experiments. (c) To test the protective role of CO-induced HO-1, mice were given 50 μmol/kg SnPP s.c., the selective inhibitor of HO-1, 5 h before CO. Alternatively, the mice were given 10 mg/kg V-PYRRO (VP) s.c., an NO donor. V-PYRRO was selectively designed to deliver NO directly to the liver. 1 h after the initial V-PYRRO dose the animals were exposed to CO for 1 h before the administration of TNF-α/D-Gal (see above). Serum ALT levels were determined 6–8 h later. Note that CO was not able to provide protection in animals where HO-1 activity was blocked. V-PYRRO, when administered 2 h before and then every 2 h thereafter, provided protection against injury as determined 8 h later by serum ALT measurements (reference 1). Results are expressed as mean ± SD of 8–10 mice/group. *, P < 0.05 versus CO/TNF/D-gal–treated mice. (d) Wild-type C57BL/6J mice were pretreated for 24 h with 4.5 mM L-NIL in the drinking water as previously described (reference 32). These mice and inos − / − mice were then administered CoPP. L-NIL was maintained in the water throughout the experiment. Control and inos − / − mice received normal drinking water. 24 h after CoPP, TNF-α/D-gal was administered and serum ALT was determined 6–8 h later. Note that induction of HO-1 provides protection regardless of the presence of iNOS. Results are mean ± SD of six to eight mice/group. *, P < 0.001 versus Air/TNF and L-NIL/TNF.
To determine if up-regulation of HO-1 would also be needed if protection was initiated by NO, mice were treated with the pharmacological NO donor V-PYRRO/NO. This agent is metabolized by the liver resulting in release of NO by hepatocytes (1). V-PYRRO/NO also provides protection after LPS/D-gal or TNF-α/D-gal administration (1). Mice were randomized and treated with TNF-α/D-gal with or without SnPP to evaluate the role of HO-1. As previously demonstrated, V-PYYRO/NO was protective as assayed by serum ALT. However, SnPP abrogated the ability of this NO donor to protect against liver damage (Fig. 6 c). Thus, we conclude that CO- or NO-initiated hepatoprotection are at least partially dependent on HO-1.
Because these data suggest that CO and NO require HO-1 activity to protect against TNF-α–induced hepatocyte death, we tested whether protection mediated by HO-1 required iNOS activity. Using inos − / − mice, we induced HO-1 via administration of CoPP. TNF-α/D-gal was injected 24 h thereafter, at the peak of HO-1 expression, and liver damage was assessed 6–8 h later. Results show that induction of HO-1 was able to significantly prevent liver injury independently of iNOS activity with a >50% reduction in serum ALT (Fig. 6 d). These results were confirmed using L-NIL. Mice were pretreated with drinking water containing 4.5 mM L-NIL for 24 h. This method effectively inhibits NO synthase activity (32). Control mice received normal water. Subsequently, CoPP was administered to induce HO-1 expression and 24 h thereafter mice were challenged with TNF-α/D-gal. L-NIL treatment alone did not change the severity of injury induced in this model. All animals receiving CoPP (with and without L-NIL) were protected from liver injury (Fig. 6 d).
To confirm the requirement for HO-1 expression in the observed protection by CO or NO, we isolated hepatocytes from HO-1–null mice (hmox-1 − / −) and wild-type (C57BL/6J) littermates and treated them with TNF-α/ActD in the presence and absence of 250 ppm CO or 500 μM of the NO donor SNAP. SNAP has previously been demonstrated to protect hepatocytes in this model (13). Air-treated wild-type and hmox-1 − / − cells exposed to TNF-α/ActD underwent cell death as expected whereas CO- or NO-treated wild-type cells were protected in the presence of TNF-α/ActD (Fig. 7 , a and b). The protection conferred by CO and NO was however lost in cells lacking functional HO-1 (Fig. 7, a and b). We conclude that HO-1 can provide protection in this model without the involvement of iNOS, suggesting that HO-1 or one or more of its catalytic products can in part exert cytoprotective effects in this model.
Figure 7.

HO-1 is required for CO- or NO-induced cytoprotection from TNF-α/ActD-induced hepatocyte cell death. (a) Mouse hepatocytes isolated from hmox-1 − / − or wild-type C57BL/6J mice were pretreated for 1 h with 250 ppm CO followed by treatment with TNF-α/ActD. Viability was assayed as described in Materials and Methods. CO significantly protected wild-type hepatocytes, but was unable to protect hepatocytes isolated from hmox-1 − / − mice. (b) 500 μM of the NO donor, SNAP, significantly protected against cell death in wild-type hepatocytes, but did not provide significant protection against cell death in hepatocytes isolated from hmox-1 − / − mice. *, P < 0.01 versus non–TNF-α/ActD-treated cells and versus TNF-α/ActD-treated cells that were also treated with SNAP or CO.
Inhaled CO Therapeutically Protects against APAP-induced Hepatitis.
The data above do not lend support to the use of either of these gases for potential treatment of hepatitis clinically. In fact, the TNFα/D-gal model does not lend itself to such an analysis given the rapidly fatal course of the disease. Therefore, we tested CO administration in another model of hepatitis in which the disease course is less violent: APAP-induced hepatitis. In Fig. 8 , the results of administering CO before and 4 h after administration of APAP to mice and the assessment of the results 16 h later (20 h after APAP) are shown. This protocol was designed to allow the hepatitis to begin to develop for 4 h before giving CO. CO administration still very significantly reduced damage to the liver as assessed by serum ALT (622 ± 44 vs. 175 ± 137, P < 0.01, as compared with controls; Fig 8). This protection was similar to that observed in a separate group of animals that had been pretreated with CO before APAP. These data support the potential therapeutic use of CO in a clinically relevant situation where treatment would begin after the initiation of the insult.
Figure 8.

Therapeutic effects of CO against APAP-induced hepatotoxicity. Male C57BL/6J mice were exposed to 250 ppm CO either 1 h before or 4 h after the i.p. administration of 500 mg/kg APAP. The mice were then maintained in CO for the duration of the experiment. Serum ALT levels were determined 20 h after APAP administration. Control mice received APAP and were maintained in air. *, P < 0.01 versus APAP-treated mice in air. Results are mean ± SD of four to eight mice/group.
Discussion
These results demonstrate that a low concentration of CO can protect against TNF-α/D-gal–induced fulminant hepatitis and illustrate a unique and previously unrecognized dependence on both HO-1 and iNOS in the CO-induced protection of livers from damage by TNF-α/D-gal. The model (Fig. 9) we propose is that CO-mediated protection operates by activating NF-κB, which in the presence of an inflammatory stimulus leads to the up-regulation of iNOS with the consequent production of NO. In addition to the induction of iNOS, other NF-κB–dependent antiapoptotic/protective genes might be induced (21, 26, 27). These protective genes may account for the relatively weak (and not significant) protection seen with exogenous CO even when iNOS was inhibited. During the 1-h pretreatment with CO and before the cells were treated with TNF-α, significant activation of NF-κB was present, which could be part of the priming of the cellular apparatus discussed above. The activation of NF-κB by CO may in part result from a mild increase in reactive oxygen species generation originating from the mitochondria (preliminary observations). 1 h might also permit time for expression of NF-κB–dependent antiapoptotic genes. The next step in our model would lead to NO production after the up-regulation of iNOS. NO leads to up-regulation of HO-1, the activity of which confers protective effects. The protective effect of HO-1 could be due to removal of heme or to any one or more of its three products: CO, biliverdin/bilirubin, or iron/ferritin (31). Given that we administer exogenous CO throughout the duration of these experiments, it seems unlikely that endogenously produced CO alone mediates HO-1 protection. However, the combination of CO with other products of HO-1 or these other products acting individually might be involved.
Figure 9.

CO signaling in the liver. The figure depicts the concept of a cycle where exogenous CO mediates its therapeutic effects through a series of steps, each of which is essential for hepatoprotection. We speculate in the text about the potential explanations for the need for HO-1 expression to mediate the protection provided by exogenous CO.
The primary goal of these studies was to examine the potential interdependence of CO and NO in the therapeutic effects these two gases have on hepatitis induced by TNF-α/D-gal, and the potential role of iNOS and HO-1 in the process. In these experiments CO or NO (or in the case of HO-1 induction, CoPP) was given as a pretreatment and thus these experiments do not address the hypothesis that CO may have therapeutic benefit even after the hepatitis process has started. How much CO is delivered to the liver by inhalation is unknown and well near impossible to measure with present technology. CO bound to hemoglobin has an off rate that presumably helps to determine the amount of CO that would reach the liver in addition to the high diffusion and nonreactivity of CO. The TNF-α/D-gal model is not a good one for testing such a hypothesis given the rapidity with which hepatic necrosis, circulatory collapse, and death occurs after TNF-α/D-gal administration. Therefore, we administered CO in a clinically relevant model of APAP-induced hepatitis that has a time course similar to the development of acute hepatitis in humans (33). Our data demonstrate that exposure to CO 4 h after i.p. administration of 500 mg/kg APAP resulted in a 62% reduction in liver injury (Fig. 8). In this model of APAP-induced liver injury, mice show signs of hepatitis as early as 2–4 h after APAP administration (34, 35) with hepatocyte cell death and lethality occurring by 24–48 h. Thus, CO was administered after the initiation of liver injury. Preliminary analyses of reduced glutathione levels in liver homogenates from these animals showed that CO prevented the APAP-induced decrease in glutathione levels, which points to CO maintaining the antioxidant potential of the tissue (not depicted). Consistent with the data in the APAP model are our results in a murine model of hemorrhagic shock where the therapeutic initiation of inhaled CO during resuscitation after a 2.5-h shock phase resulted in protection against liver injury (>65% reduction in serum ALT at 24 h, P < 0.01; n = 6–10/group). Further investigations into the potential therapeutic benefit of inhaled CO delivery as a therapeutic adjunct after an insult that leads to hepatitis are clearly needed.
In summary, using a model of liver injury driven principally by TNF-α–induced apoptosis, we show the following. First, inhaled CO can prevent hepatitis in this model. Second, protection by CO requires generation of a second gaseous molecule, NO. Third, NO exerts its beneficial effects, at least in part, via up-regulation of HO-1. Lastly, up-regulation of HO-1 is protective without a need for iNOS/NO activity, i.e., without an obligate continuation of the cycle. Taken together, these data demonstrate for the first time a functional relationship between HO-1/CO and iNOS/NO. We propose a novel protective cycle that relies on two gas molecules and their respective enzymes. Together, CO, NO, and HO-1 beneficially modulate the apoptotic/inflammatory state. Whether CO treatment can be used clinically was not the main question we probed in this paper. However, our limited but significant data in the APAP model suggests that CO exposure might prove beneficial as a therapy to ameliorate hepatic injury even if given shortly after the hepatic injury starts.
Acknowledgments
We would like to thank Emeka Ifedigbo for his technical engineering, Elisabeth Humphris for the hepatocyte harvests, Sean Alber for the immunohistochemistry, and Julieta Aranda for her graphic design work.
This work was supported by grants from the American Heart Association 0160332U and Atorvastatin Research Award (AHA) sponsored by Pfizer (awarded to L.E. Otterbein) and National Institutes of Health grants R01-GM-44100 (awarded to T.R. Billiar), HL 58688 (awarded to F.H. Bach), and HL60234 (awarded to A.M.K. Choi). B.S. Zuckerbraun is supported by the Ethicon-Society of University Surgeons Resident Research Award. F.H. Bach is the Lewis Thomas Professor at the Harvard Medical School and a paid consultant of Novartis Pharma.
Abbreviations used in this paper: ActD, actinomycin-D; ALT, alanine aminotransferase; APAP, acetaminophen; A.U., arbitrary units; CM, cytokine mixture; CO, carbon monoxide; CoPP, cobalt protoporphyrin; D-gal, D-galactosamine; EMSA, electrophoretic mobility shift assay; H&E, hematoxylin and eosin; HO-1, heme oxygenase 1; iNOS, inducible nitric oxide synthase; L-NIL, L-N6-(1-iminoethyl)-lysine-dihydrochloride; L-NIO, L-N5-(1-iminoethyl)-ornithine-2HCl; NF, nuclear factor; NO, nitric oxide; SNAP, s-nitroso-N-acetyl-penicillamine; SnPP, tin protoporphyrin; V-PYRRO, O2-vinyl 1-(pyrrolidin-1-yl) diazen-1-ium-1,2-diolate.
References
- 1.Saavedra, J.E., T.R. Billiar, D.L. Williams, Y.M. Kim, S.C. Watkins, and L.K. Keefer. 1997. Targeting nitric oxide (NO) delivery in vivo. Design of a liver-selective NO donor prodrug that blocks tumor necrosis factor-alpha-induced apoptosis and toxicity in the liver. J. Med. Chem. 40:1947–1954. [DOI] [PubMed] [Google Scholar]
- 2.Mojena, M., S. Hortelano, A. Castrillo, M.J. Diaz-Guerra, M.J. Garcia-Barchino, G.T. Saez, and L. Bosca. 2001. Protection by nitric oxide against liver inflammatory injury in animals carrying a nitric oxide synthase-2 transgene. FASEB J. 15:583–585. [DOI] [PubMed] [Google Scholar]
- 3.Otterbein, L., S.L. Sylvester, and A.M. Choi. 1995. Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am. J. Respir. Cell Mol. Biol. 13:595–601. [DOI] [PubMed] [Google Scholar]
- 4.Tamion, F., V. Richard, Y. Lacoume, and C. Thuillez. 2002. Intestinal preconditioning prevents systemic inflammatory response in hemorrhagic shock. Role of HO-1. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G408–G414. [DOI] [PubMed] [Google Scholar]
- 5.Amersi, F., R. Buelow, H. Kato, B. Ke, A.J. Coito, X.D. Shen, D. Zhao, J. Zaky, J. Melinek, and C.R. Lassman. 1999. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J. Clin. Invest. 104:1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogt, B.A., T.P. Shanley, A. Croatt, J. Alam, K.J. Johnson, and K.A. Nath. 1996. Glomerular inflammation induces resistance to tubular injury in the rat. A novel form of acquired, heme oxygenase-dependent resistance to renal injury. J. Clin. Invest. 98:2139–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soares, M.P., Y. Lin, J. Anrather, E. Csizmadia, K. Takigami, K. Sato, S.T. Grey, R.B. Colvin, A.M. Choi, K.D. Poss, et al. 1998. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat. Med. 4:1073–1077. [DOI] [PubMed] [Google Scholar]
- 8.Otterbein, L.E., F.H. Bach, J. Alam, M. Soares, L.H. Tao, M. Wysk, R.J. Davis, R.A. Flavell, and A.M. Choi. 2000. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 6:422–428. [DOI] [PubMed] [Google Scholar]
- 9.Fujita, T., K. Toda, A. Karimova, S.F. Yan, Y. Naka, S.F. Yet, and D.J. Pinsky. 2001. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat. Med. 7:598–604. [DOI] [PubMed] [Google Scholar]
- 10.Naughton, P., R. Foresti, S.K. Bains, M. Hoque, C.J. Green, and R. Motterlini. 2002. Induction of heme oxygenase 1 by nitrosative stress. A role for nitroxyl anion. J. Biol. Chem. 277:40666–40674. [DOI] [PubMed] [Google Scholar]
- 11.Kim, Y.M., H.A. Bergonia, C. Muller, B.R. Pitt, W.D. Watkins, and J.R. Lancaster, Jr. 1995. Loss and degradation of enzyme-bound heme induced by cellular nitric oxide synthesis. J. Biol. Chem. 270:5710–5713. [DOI] [PubMed] [Google Scholar]
- 12.Maulik, N., D.T. Engelman, M. Watanabe, R.M. Engelman, and D.K. Das. 1996. Nitric oxide–a retrograde messenger for carbon monoxide signalling in ischemic heart. Mol. Cell. Biochem. 157:75–86. [DOI] [PubMed] [Google Scholar]
- 13.Kim, Y.M., M.E. de Vera, S.C. Watkins, and T.R. Billiar. 1997. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-alpha-induced apoptosis by inducing heat shock protein 70 expression. J. Biol. Chem. 272:1402–1411. [DOI] [PubMed] [Google Scholar]
- 14.Hellerbrand, C., C. Jobin, Y. Iimuro, L. Licato, R.B. Sartor, and D.A. Brenner. 1998. Inhibition of NFkappaB in activated rat hepatic stellate cells by proteasome inhibitors and an IkappaB super-repressor. Hepatology. 27:1285–1295. [DOI] [PubMed] [Google Scholar]
- 15.Chow, J.C., D.W. Young, D.T. Golenbock, W.J. Christ, and F. Gusovsky. 1999. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 274:10689–10692. [DOI] [PubMed] [Google Scholar]
- 16.Lowenstein, C.J., E.W. Alley, P. Raval, A.M. Snowman, S.H. Snyder, S.W. Russell, and W.J. Murphy. 1993. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc. Natl. Acad. Sci. USA. 90:9730–9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor, B.S., M.E. de Vera, R.W. Ganster, Q. Wang, R.A. Shapiro, S.M. Morris, Jr., T.R. Billiar, and D.A. Geller. 1998. Multiple NF-kappaB enhancer elements regulate cytokine induction of the human inducible nitric oxide synthase gene. J. Biol. Chem. 273:15148–15156. [DOI] [PubMed] [Google Scholar]
- 18.Otterbein, L.E., J.K. Kolls, L.L. Mantell, J.L. Cook, J. Alam, and A.M.K. Choi. 1999. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung. J. Clin. Invest. 103:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pannen, B.H., N. Kohler, B. Hole, M. Bauer, M.G. Clemens, and K.K. Geiger. 1998. Protective role of endogenous carbon monoxide in hepatic microcirculatory dysfunction after hemorrhagic shock in rats. J. Clin. Invest. 102:1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sass, G., K. Koerber, and G. Tiegs. 2002. TNF tolerance and cytotoxicity in the liver: the role of interleukin-1beta, inducible nitric oxide-synthase and heme oxygenase-in D-galactosamine-sensitized mice. Inflamm. Res. 51:229–235. [DOI] [PubMed] [Google Scholar]
- 21.Brouard, S., L.E. Otterbein, J. Anrather, E. Tobiasch, F.H. Bach, A.M. Choi, and M.P. Soares. 2000. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 192:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunther, L., P.O. Berberat, M. Haga, S. Brouard, R.N. Smith, M.P. Soares, F.H. Bach, and E. Tobiasch. 2002. Carbon monoxide protects pancreatic beta-cells from apoptosis and improves islet function/survival after transplantation. Diabetes. 51:994–999. [DOI] [PubMed] [Google Scholar]
- 23.Petrache, I., L.E. Otterbein, J. Alam, G.W. Wiegand, and A.M. Choi. 2000. Heme oxygenase-1 inhibits TNF-alpha-induced apoptosis in cultured fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 278:L312–L319. [DOI] [PubMed] [Google Scholar]
- 24.Otterbein, L.E., B.S. Zuckerbraun, M. Haga, F. Liu, R. Song, A. Usheva, C. Stachulak, N. Bodyak, R.N. Smith, E. Csizmadia, et al. 2003. Carbon monoxide prevents intimal thickening associated with chronic graft rejection and with balloon injury. Nat. Med. 9:183–190. [DOI] [PubMed] [Google Scholar]
- 25.Beg, A.A., W.C. Sha, R.T. Bronson, S. Ghosh, and D. Baltimore. 1995. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 376:167–170. [DOI] [PubMed] [Google Scholar]
- 26.Beg, A.A., and D. Baltimore. 1996. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 274:782–784. [DOI] [PubMed] [Google Scholar]
- 27.Van Antwerp, D.J., S.J. Martin, T. Kafri, D.R. Green, and I.M. Verma. 1996. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 274:787–789. [DOI] [PubMed] [Google Scholar]
- 28.Kim, Y.M., T.H. Kim, D.W. Seol, R.V. Talanian, and T.R. Billiar. 1998. Nitric oxide suppression of apoptosis occurs in association with an inhibition of Bcl-2 cleavage and cytochrome c release. J. Biol. Chem. 273:31437–31441. [DOI] [PubMed] [Google Scholar]
- 29.Kim, Y.M., R.V. Talanian, and T.R. Billiar. 1997. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. 272:31138–31148. [DOI] [PubMed] [Google Scholar]
- 30.Sass, G., K. Koerber, R. Bang, H. Guehring, and G. Tiegs. 2001. Inducible nitric oxide synthase is critical for immune-mediated liver injury in mice. J. Clin. Invest. 107:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otterbein, L.E., M.P. Soares, K. Yamashita, and F.H. Bach. 2003. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 24:449–455. [DOI] [PubMed] [Google Scholar]
- 32.Stenger, S., N. Donhauser, H. Thuring, M. Rollinghoff, and C. Bogdan. 1996. Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J. Exp. Med. 183:1501–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinson, J.A., D.W. Roberts, R.W. Benson, K. Dalhoff, S. Loft, and H.E. Poulsen. 1990. Mechanism of paracetamol toxicity. Lancet. 335:732. [DOI] [PubMed] [Google Scholar]
- 34.Zhang, J., W. Huang, S.S. Chua, P. Wei, and D.D. Moore. 2002. Modulation of acetominophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science. 298:422–424. [DOI] [PubMed] [Google Scholar]
- 35.Henderson, C.J., C.R. Wolf, N. Kitteringham, H. Powell, D. Otto, and B.K. Park. 2000. Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proc. Natl. Acad. Sci. USA. 7:12741–12745. [DOI] [PMC free article] [PubMed] [Google Scholar]