

Abstract
The initiation of cell-mediated immunity to Epstein-Barr virus (EBV) has been analyzed with cells from EBV-seronegative blood donors in culture. The addition of dendritic cells (DCs) is essential to prime naive T cells that recognize EBV-latent antigens in enzyme-linked immunospot assays for interferon γ secretion and eradicate transformed B cells in regression assays. In contrast, DCs are not required to control the outgrowth of EBV-transformed B lymphocytes from seropositive donors. Enriched CD4+ and CD8+ T cells mediate regression of EBV-transformed cells in seronegative and seropositive donors, but the kinetics of T-dependent regression occurs with much greater speed with seropositives. EBV infection of DCs cannot be detected by reverse transcription–polymerase chain reaction with primers specific for mRNA for the EBNA1 U and K exons. Instead, DCs capture B cell debris and generate T cells specific for EBV latency antigens. We suggest that the cross-presentation of EBV-latent antigens from infected B cells by DCs is required for the initiation of EBV-specific immune control in vivo and that future EBV vaccine strategies should target viral antigens to DCs.
Keywords: herpesvirus, regression assay, cross-priming, T cell, B cell
Introduction
Epstein-Barr virus (EBV) is a human γ–herpesvirus that latently infects >90% of the adult population. Primary EBV infection in adolescents and adults can cause infectious mononucleosis (IM), which can be associated with a prolonged clinical course and significant morbidity. More importantly, EBV has growth-transforming capabilities and is associated with numerous malignancies. Virtually all cases of undifferentiated nasopharyngeal carcinoma, posttransplant lymphoproliferative disease (PTLD), and endemic Burkitt's lymphoma as well as half the cases of Hodgkin's disease (HD) are EBV positive (1). Passive immunotherapy with EBV-specific T cells has been used to treat EBV-associated lymphoproliferative disorders (2, 3), but it remains difficult to generate these T cells, especially in unprimed EBV-negative children who receive transplants from EBV-positive donors (4–6). Furthermore, there is no vaccine to prevent EBV infection.
An intact immune system is capable of containing the primary infection and preventing transformation of infected B cells. Transformation is seen in patients when cellular immunosurveillance is compromised pharmacologically, by disease or congenital defects (1) and in vitro with the expansion of lymphoblastoid cell lines (LCLs). In healthy carriers, the presence of a protective, cellular immune response to EBV correlates with in vitro regression of EBV-transformed B cells. The outgrowth of EBV-transformed cells is controlled and eliminated by virus-specific memory T cells in EBV-seropositive donors (7–13). However, in EBV-naive donors, the foci of transformed B cells proliferate due to a lack of primed EBV-specific T cells in culture (7–9, 14, 15). Interestingly, LCL outgrowth in these assays is also seen in patients with impaired EBV immune control leading to nasopharyngeal carcinoma and X-linked lymphoproliferative disease (16–19). Therefore, regression serves as an indicator of protective immunity in EBV carriers as well as a reliable in vitro model of primary EBV infection in seronegative donors.
The lack of T cell priming by EBV-infected B cells during the course of the regression assay implies the involvement of another antigen-presenting cell in the immune control of the primary phase of EBV infection. Recently, it has become evident that DCs are able to capture dying B cells and present EBV latency antigens to CD4+ (20) and CD8+ T cells (21). Furthermore, the strong CD4+ T cell response to the EBNA1 antigen is of the Th1 phenotype (22) and DCs are known to be strong stimulators of this protective form of cell-mediated immunity (23–25). Here, we show that addition of DCs to cocultures of EBV-infected B and T cells from EBV-seronegative donors leads to regression of LCL outgrowth. This effect is secondary to DC priming of T cells specific for EBV latency antigens. Although the DCs are not detectably infected with EBV by RT-PCR, they can capture B cells and directly expand EBV-reactive T cells, indicating a need for cross-presentation by DCs during the initiation of T cell–mediated antiviral responses in humans.
Materials and Methods
Dendritic, B, and T Cell Preparations.
PBMCs were obtained from leukocyte concentrates (New York Blood Center) and blood from lab donors (the latter with informed consent) by Ficoll-Hypaque centrifugation (Amersham Biosciences). DCs were prepared from blood monocytes as described previously (21). In brief, CD14+ monocytes were MACS purified and cultured in 500 U/ml rhuIL-4 (R&D Systems) and 1,000 U/ml rhuGM-CSF (Immunex) for 5–6 d. Where indicated, they were matured two more days with 10 ng/ml IL-1β, 1,000 U/ml IL-6, 10 ng/ml TNFα (all obtained from R&D Systems), and 1 μg/ml PGE2 (Sigma-Aldrich). For positive selection of CD19+, CD8+, and CD4+ cells, CD14− cells were treated with the appropriate monoclonal antibody conjugated to magnetic microbeads (Miltenyi Biotec) and MACS purified. Alternatively, B and CD4+ T cells were sorted on a FACSVantage™ SE cell sorter before coculture; these maintained 99.5% purity during the coculture. T cells, B cells, and DCs were used fresh or after cryopreservation in FCS and 5% DMSO.
Evaluation of EBV Serostatus.
ELISA kits for viral capsid antigen and Epstein-Barr nuclear antigen 1 (Sigma-Aldrich) were used on plasma. Donors were considered EBV seronegative if no reactivity was detected against both antigens.
Cell Lines.
We cultured the EBV+ B95-8 marmoset cell line (26), EBV− Ramos Burkitt's lymphomal cells (27), EBV− BJAB B cell lymphoma (28), and the EBV− HD-MY-Z Hodgkin's cell line (29) in RPMI 1640 fortified with 10% FCS, gentamicin, and Hepes buffer.
Preparation of EBV Viral Stock and Mock Inocula.
As described previously (8), supernatant from EBV+ B95-8 cells containing ∼105 transforming U/ml, and EBV− BJAB cells, were harvested and adjusted to 10 g NaCl and 8% (wt/vol) polyethylene glycol/liter (PEG-8000; Sigma-Aldrich). After overnight incubation, the precipitate was collected at 7,500 revolutions/min. Viral stocks were resuspended, aliquoted, and stored at −70°C. Alternatively, the EBV− Ramos cell line and the EBV+ B95-8 LCLs were seeded at 2 × 105/ml and cultured for 14 d without refeeding. The cultures were collected, centrifuged at 1,800 revolutions/min for 10 min, and the supernatant was passed through a 0.45-μm filter and frozen into aliquots.
T Cell Coculture.
CD14−/CD19− cells or PBMCs were plated at 2 × 106 cells/ml in RPMI 1640 fortified with 5% human serum, gentamicin, and Hepes buffer in 24-well plates in 1 ml/well. 1 ml of EBV or mock inoculum was added. B cells and DCs were plated at a density of 105 cells/ml. CD4+ or CD8+ T cells were positively selected and plated at a concentration of 2 × 106/well as indicated. To recapitulate the ratio found in peripheral blood, the CD4+ cells were resuspended at 1.33 × 106 cells/ml and the CD8+ cells were resuspended at 6.67 × 105 cells/ml to give a total of 2 × 106 cells/ml that were specified. Where indicated, cyclosporin A (CSA) was added at 1 μg/ml, whereas protein A–purified αHLA class I (W6/32) or αHLA class II (IVA12) antibodies were added every second day at a final concentration of 50 μg/ml. For cross-priming experiments, mature DCs from EBV-seropositive or -seronegative donors were generated from CD14+ cells. CD19+ cells were incubated for 24 h in 1 ml of supernatant from EBV+ B95-8 cells and were washed three times before addition to the coculture. DCs were incubated for 24 h either in B95-8 supernatant or 1:2 with EBV-exposed CD19+ cells. DCs were positively selected with αCD11c-PE antibody followed by αPE microbeads with subsequent magnetic positive selection. These were added 1:30 with 2 × 106 autologous CD14− CD19− cells for 12 d. EBV specificity was tested with IFNγ ELISPOT assays on 2 × 106 cultured cells with 30:1 DCs infected with indicated vaccinia vectors or 10:1 with autologous LCL or EBV− Hodgkin's cell line, HD-MY-Z.
DC Infection with Recombinant Vaccinia Viruses (vv).
Mature DCs were infected with recombinant vaccinia vectors (vvTK−) as a negative control or vaccinia vectors expressing EBV latency (EBNA1, 2, 3A, 3B, 3C, LMP1, and LMP2A) and lytic (BMLF1) antigens at a multiplicity of infection of 2 for 1 h at 37°C (provided by M. Kurilla, Dupont Pharmaceuticals Corp., Wilmington, NC). Vaccinia infection was verified at 6–12 h by intracellular staining (30) using VV1-6B6 antibody to a vaccinia early protein followed by FACS® analysis. Infection of DCs was 40–60%.
FACS® Staining for Transformed Cells and T Cell Subsets.
Cultures were harvested at the indicated time points and stained with αCD19-FITC (BD Biosciences), αCD21-allophycocyanin (BD Biosciences), αTCR-Vβ13.1 (clone IMMU 222; Immunotech), αTCR-Vβ3 (clone CH92; Immunotech) antibodies, and αCD23-PE antibody (BD Biosciences) for 30 min on ice. Cells were washed, fixed in 4% paraformaldehyde, and analyzed on a FACSort™ (BD Biosciences).
ELISPOT Assay for IFNγ- and IL-4–secreting Cells.
ELISPOT assays were performed on days 12–14 as described previously (22). 5 × 104 cells/well or 2 × 106 cells/well as indicated were stimulated in duplicate or triplicate with the effector/DCs ratio of 30:1 for 18 h. In some experiments, a ratio of 10:1 of cultured cells and autologous LCLs or the EBV− HD-MY-Z was used. A response was considered significant if it was 10 spots greater than the negative control and at least twice that of the negative control.
DC Phagocytosis of EBV-infected and Uninfected B Cells.
CD19+ cells were incubated overnight in B95-8 or Ramos supernatant, washed three times in RPMI 1640, stained with the fluorescent dye PKH26 (Sigma-Aldrich), and added at a ratio of 1:1 with mature DCs. Cocultures were stained for CD11c FITC (BD Biosciences) and examined on a FACScan™ as well as on a deconvolution microscope (Olympus). FACS® analysis was performed by gating on large cells by forward scatter and exclusion of autofluorescing cells in FL-3. Staining for deconvolution microscopy was performed on cocultures after a 60-h incubation as described previously (21).
RT-PCR for EBNA1.
Mature DCs and CD19+ cells were incubated for 14 d in either B95-8 or Ramos supernatant and washed three times with RPMI 1640. RNA was purified from 5 × 106 cells using the RNeasy kit (QIAGEN). Reverse transcription and PCR amplification were performed using One Step RT-PCR kit (QIAGEN) in a total volume of 50 μl. Primers used were as follows: EBNA1 (U exon), 5′-TTAGGAAGCGTTTCTTTGAGC-3′; EBNA1 (K exon), 5′-CATTTCCAGGTCCTGTACCT-3′ (31); and G3PDH positive control, 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′. Tubes were loaded in a Biometra T3 thermocycler. Reactions were incubated at 50°C for 30 min for reverse transcription reaction, followed by a 15-min incubation at 95°C. This was followed by denaturation at 95°C for 45 s, annealing at 62°C for 45 s, and extension at 72°C for 1 min, repeated for 35 cycles. At the conclusion of the PCR was a final incubation at 72°C for 10 min to complete the extension. PCR products were run on a 1% agarose gel with 1 μg/ml ethidium bromide. Bands were visualized using a gel doc (Bio-Rad Laboratories).
Online Supplemental Material.
Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20030646/DC1. Fig. S1 demonstrates that immature and mature DCs can prime EBV-specific T cells in culture and initiate regression. Fig. S2 depicts the kinetics of TCR-Vβ13+ and TCR-Vβ3+ CD4+ T cell subsets in regression assays of EBV-seropositive and -seronegative donors. Neither TCR-Vβ subset expands preferentially during regression assays or is correlated with regression.
Results
DCs Are Required for Priming Naive T Cells to EBV Antigens.
To evaluate the hypothesis that DCs, rather than EBV-infected B cells, are required to prime virus-specific T cells, we set up cocultures of T cells with EBV-infected or uninfected B cells (8) from four EBV-seronegative donors in the presence (B + DC + T) or absence (B + T) of mature DCs. After 12 d, the cultures were tested by ELISPOT for responses to a panel of EBV latency antigens (EBNA1, 2, 3A-C, LMP1, 2A) and the lytic antigen, BMLF1. Day 12 was chosen because keyhole limpet hemocyanin–pulsed DCs were capable of priming specific CD4+ T cell responses during this time period (32) and as shown in Fig. 4, this is the time when regression of EBV transformation is evident. To optimize ELISPOT assays, we used DCs infected with the respective recombinant vv as APCs. In all four EBV-seronegative donors, priming of T cells occurred only in cultures that contained DCs (Fig. 1, A–D) . Reactive T cells were of the Th1 type as demonstrated by secretion of IFNγ (Fig. 1, A–D) and no IL-4 (not depicted). The pattern of latency antigen recognition was similar to that seen in healthy carriers of EBV (i.e., reliable recognition of EBNA1 and EBNA3s but minimal recognition of LMP2A; reference 1). In all four donors, no reactivity to any EBV latency antigens was seen on day 0 or in the control cocultures of DCs with T cells and uninfected B cells (unpublished data). In the one seronegative donor from our laboratory, responses were similar in magnitude in four experiments (Fig. 1, A, G [donor 3], and H [donor 2]). The addition of CD14+ monocytes did not result in EBV-latent antigen-specific T cell priming (Fig. S1 B, available at http://www.jem.org/cgi/content/full/jem.20030646/DC1), but populations of immature DCs or mature DCs each could prime autologous LCL-specific T cells (Fig. S1 B). In contrast to the findings with cells from seronegative donors, DCs were not essential to expand EBV-specific T cells from seropositive donors beyond that seen with infected B and T cells (Fig. 1, E and F). However, DCs induced the expansion and/or survival of T cells for a broader panel of EBV latency antigens than B and T cell mixtures alone. The EBV-specific T cells again demonstrated a polarized Th1 phenotype with secretion of IFNγ and no IL-4 (unpublished data).
Figure 4.

Kinetics of EBV-mediated B cell transformation in culture. EBV-infected B cells were cultured with autologous T cells from EBV-seropositive (top two rows) or -seronegative donors (bottom two rows) without (B + T) or with DCs (B + DC + T). Aliquots of the cultures were taken at the indicated time points, and CD19+CD23+ cells were quantified by FACS®.
Figure 1.

DCs are required for priming naive T cells to EBV antigens. T cells and EBV-infected B cells from EBV-seronegative (A–D) and -seropositive (E and F) donors were cultured in the presence (B + DC + T) or the absence (B + T) of mature DCs for 12–14 d. Results shown have the vector control, vvTK−, subtracted from the specific response, where vvTK− responses were always <20 spots. A response was considered meaningful if it was at least twice that of the negative control (vvTK−) as well as 10 spots greater than the vvTK− background and is indicated with an asterisk. (G) The seronegative B + DC + T cultures were also tested against autologous LCLs and the EBV-negative HD cell line HD-MY-Z by IFNγ ELISPOT. (H) Two sources of EBNA1 were compared for loading of DCs, recombinant vaccinia viruses (vv) leading to the expression of Gly-Ala–deficient EBNA1 (vvE1) or recombinant EBNA1 protein (rE1). DCs loaded with rPCNA (rP) or infected with vvTK− were used as controls. The responses in A, G (donor 3), and H (donor 2) are representative of four experiments with the same seronegative donor.
The recognition of EBV latency antigens by primed T cells was confirmed by restimulating EBV-seronegative B + DC + T cultures with autologous LCLs and the EBV-negative HD cell line HD-MY-Z as a control in ELISPOT assays (Fig. 1 G, left). Autologous EBV-transformed cell lines from three EBV-seronegative donors elicited IFNγ secretion far above recognition of the EBV− HD control. In the EBV-infected DCs, B and T cell cocultures of seronegative donors, the responses of the sum of EBV antigen–specific ELISPOTS, or the LCL-specific T cells, amounted on average to 0.4% of the cells. We also verified that the EBNA1 specific response with recombinant vv as the antigenic formulation could also be detected with a different source of this EBV-latent antigen (Fig. 1 H). We compared IFNγ secretion by DC-primed, EBV-seronegative cultures in response to DCs loaded with the immunogenic COOH-terminal domain of recombinant EBNA1 protein (from Escherichia coli; reference 22) and DCs infected with vvEBNA1ΔGA. Comparable T cell responses were observed, and these were exclusively present in B + DC + T cultures from EBV-seronegative donors. A recombinant control protein (rPCNA) purified by the same method as rEBNA1 did not elicit any detectable responses (Fig. 1 H). Therefore, the presence of DCs is sufficient to prime T cells specific for several EBV latency antigens in freshly transformed B cells and LCLs. In contrast, EBV-specific memory T cells from healthy carriers can be expanded in vitro by EBV-infected B cells, at least for some latency antigens.
T Cells Primed by DCs In Vitro Control the Outgrowth of LCLs in Cocultures.
Next, we investigated whether EBV-naive T cells from seronegative donors could mediate regression of transformed B cells if the cultures were supplemented with autologous DCs. Activated B cells, including those undergoing EBV-associated transformation, express the activation marker, CD23, and consequently, the percentage of transformed cells can be easily monitored by FACS® (14, 15, 33, 34). We confirmed that CD23+-activated B cells expressed EBV-latent antigens by immunofluorescence staining for EBNA1 (unpublished data).
When we analyzed our regression assays for the presence of CD19+/CD23+ B cells by flow cytometry in EBV-seronegative donors, there was outgrowth of 20–38% EBV-transformed B cells in the B + DC and B + T cocultures, but not in the B + DC + T cocultures (Fig. 2, A–C) . In contrast, B + T cocultures from EBV-seropositive donors had regressed without DCs (Fig. 2, D and E, center). Immature and mature DCs induced similar regression of EBV-transformed B cells in B + T cultures of EBV-seronegative donors, but monocytes could not mediate regression (Fig. S1 C). DCs could not induce regression in the absence of T cells in cultures of both EBV-seropositive and -seronegative donors (Fig. 2, A–E, left). No CD19+CD23+ B cell transformation was seen using EBV mock-infected cells (unpublished data). The data in Fig. 2 are representative of six EBV-seronegative and three EBV-seropositive donors.
Figure 2.
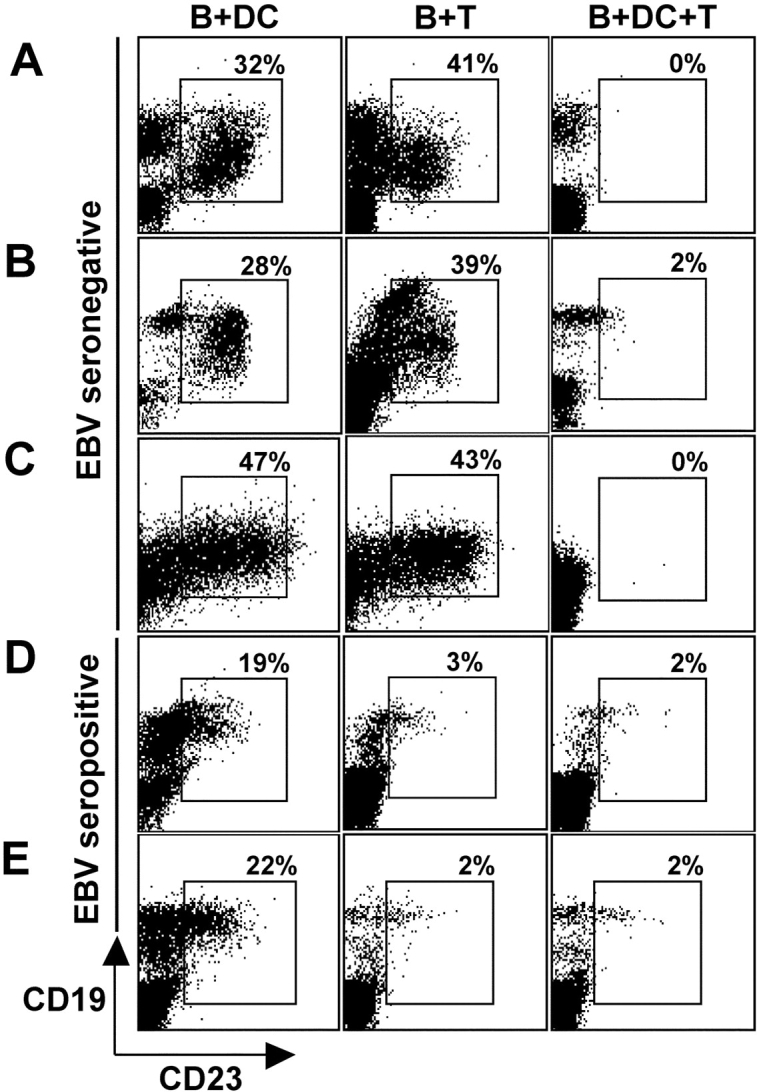
T cells primed by DCs in vitro control LCL outgrowth. T cells and EBV-infected B cells from EBV-seronegative (A–C) and -seropositive (D and E) donors were cocultured in the presence (B + DC + T) or absence (B + T) of mature DCs. After 18 d, cultures were collected and examined for outgrowth of LCLs as demonstrated by CD23+ and CD19+ double-positive cells quantified by FACS® analysis. Cocultures of B cells and DCs alone (B + DC) were used as a control for B cell transformation. Gates were set on transformed B cells by forward and side scatter and to exclude autofluorescing cells in FL-3.
To provide evidence that the observed regression was mediated by T cells, we first used CSA to inhibit T cell reactivity (35, 36). In B + DC + T cultures from both EBV-seropositive and -negative donors, the addition of CSA led to outgrowth of ∼30% of EBV-transformed B cells, whereas without T cell inhibition, <1% CD19+ CD23+ cells could be observed (Fig. 3) . To the extent that CSA specifically inhibits T cell function, these data indicate that primed T cells mediate regression in B + DC + T cultures from EBV-seronegative donors.
Figure 3.

EBV-transformed B cell regression is sensitive to cyclosporin A (CSA). EBV-infected B cells were cultured with autologous T cells from EBV-seropositive (top row) or -seronegative donors (bottom row) without (B + T) or with DCs (B + DC + T). To some of the cultures CSA was added to suppress T cell reactivity. CD19+CD23+ cells were evaluated by FACS® staining 21 d after EBV infection and start of the coculture.
Different Kinetics of Regression in Cultures with Primary and Secondary EBV-specific Immune Responses.
Regression of EBV-transformed B cells in B + DC + T cultures from EBV-seronegative donors coincided with the time point (days 12–14) at which we had assessed the T cell responses in IFNγ ELISPOT assays (Fig. 4) . With seropositive donors, CD19+CD23+ B cells were detectable at day 3 after infection in both B + T and B + DC + T cultures, but their numbers were already markedly decreased compared with cultures of seronegative donors. By day 6, EBV-transformed B cells were further reduced and most had vanished by day 11 (Fig. 4, top rows). In contrast, CD19+CD23+ B cells accumulated without obvious control in B + T cultures from EBV-seronegative donors (Fig. 4, bottom rows). In B + DC + T cultures of EBV-seronegative donors, the first indication for regression was at day 11 (average of 24 ± 4% in B + T vs. 7.2 ± 3.8% in B + DC + T), and the numbers decreased more markedly by day 14 (average of 29 ± 11% in B + T vs. 4.5 ± 3.5% in B + DC + T). These experiments are representative for two EBV-seronegative and three EBV-seropositive donors. Therefore, T cell reactivity reached protective levels around days 11–14 to control EBV transformation in cultures of EBV-seronegative donors.
Both CD4+ and CD8+ Lymphocytes Contribute to Regression of LCL Outgrowth.
Once clinical manifestations of IM are present, patients demonstrate a brisk cell-mediated immune response, characterized by numerous atypical lymphocytes in the peripheral blood (1). A majority of these T cells are CD8+ with much smaller expansions in CD4+ T cells (37). To evaluate the contribution of CD4+ and CD8+ T lymphocytes in the control of LCL outgrowth, we performed regression assays with lymphocytes from three EBV-seronegative donors positively selected for CD4 or CD8 expression. CD4+, CD8+, or a mixture of CD4+ and CD8+ cells were cultured with EBV-infected B cells and DCs for 18 d. After 18 d, CD4+ cultures had <1% contaminating CD8+ T cells and CD8+ cultures had <6% contaminating CD4+ cells as analyzed by FACS® (unpublished data). Cultures were evaluated for LCL outgrowth by CD19+ CD23+ double staining. FACS® data in Fig. 5 from three different EBV-seronegative donors show LCL regression in cultures with highly purified CD4+ or CD8+ T cells alone or in the mixed CD4+/CD8+ T cell cultures. To further verify the contribution of CD4+ and CD8+ T cells, we did additional studies by adding antibodies to MHC class I or class II every 2 d to cultures of EBV-infected PBMCs. Both the αHLA class I antibody W6/32 and the αHLA class II antibody IVA12 increased the yield of CD19+CD23+ B cells in EBV-infected PBMC cultures of seropositive donors (unpublished data). The generation of a protective response in both the CD8+ and CD4+ T cell compartment reflects the conditions seen in patients recovering from IM. Furthermore, the priming of both EBV-specific CD4+ as well as CD8+ T cells most likely ensures comprehensive and efficient immune control of latent EBV infection.
Figure 5.

CD4+ and CD8+ T cells contribute to LCL regression. EBV-infected B cells and DCs were cultured with either CD4+ T cells (B + DC + CD4), CD8+ T cells (B + DC + CD8), or a mixture of both (B + DC + CD4 + CD8) from three EBV-seronegative donors (A–C). Cultures were analyzed by FACS® after 18 d to identify LCLs as CD19+/CD23+ double-positive cells representing LCL outgrowth. Gates were set on transformed B cells by forward and side scatter and to exclude autofluorescing cells in FL-3.
Because transactivation of the human endogenous retrovirus HERV-K18 by lytic EBV infection had been reported recently and HERV-K18 encodes a superantigen that stimulates TCR-Vβ13 expressing CD4+ T cells (38, 39), we were concerned that low levels of lytic EBV infection in our regression cultures could trigger TCR-Vβ13–carrying T cells and their expansion could contribute to the regression of EBV-transformed B cells. However, when we followed the kinetics of TCR-Vβ13 and control TCR-Vβ3–expressing CD4+ T cells in EBV-seropositive and -seronegative regression cultures at days 0, 4, 11, 14, and 20 (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20030646/DC1), we found no selective TCR-Vβ13 T cell expansion over TCR-Vβ3 T cell expansion at any of these time points. Moreover, we found no correlation between TCR-Vβ13 T cell numbers and regression of EBV-transformed B cells in our cultures (Fig. 2). Namely, we did not observe any Vβ13 T cell expansion at day 4 in seropositive regression cultures, nor specifically in regressing EBV-infected DCs, B, and T cell cocultures of seronegative donors at day 11 and 14 in comparison to unregressing B and T cell cocultures. Although we cannot rule out that a low level of T cell stimulation by the HERV-K18 encoded superantigen was present in our regression cultures, we conclude from these data that it probably contributes minimally to the observed protection against EBV transformation.
DCs Phagocytize B Cell Fragments in the Presence or Absence of EBV Infection.
Uptake of cellular debris with subsequent processing and presentation of antigens by DCs has been described for many infectious and tumor-derived antigens (for reviews see references 40, 41). Therefore, we investigated uptake of EBV-infected B cells by DCs in our cultures. After an overnight incubation in supernatants from the B95-8 EBV-producing and Ramos EBV-negative cell lines, B cells were stained with PKH26, a membrane stain. These cells were cultured with DCs at 37°C and collected after 30 min, 6 h, and 18 h. As a negative control, DCs and B cells were incubated together at 4°C for 18 h; DCs were stained for CD11c and analyzed by FACS® (Fig. 6 A) and deconvolution microscopy (Fig. 6 B). The DCs took up B cell fragments after EBV or mock infection equally well at each of the specified time points of coculture (Fig. 6 A). Deconvolution microscopy visualized many intracellular vesicles with PKH26 stained B cell fragments (Fig. 6 B), thereby excluding extracellular adherence as a possible explanation for the double-positive cells seen in Fig. 6 A. These observations provide evidence that DCs obtain EBV latency antigens to prime naive T cells by taking up and processing EBV-infected B cells, as shown previously for continuous EBV-transformed cell lines (21, 42, 43).
Figure 6.

DCs phagocytize B cell fragments in the presence or absence of EBV infection. CD19+ cells were incubated overnight in the supernatant from the B95-8 EBV+ (+ virus) and Ramos EBV− (− virus) cell lines. CD19+ cells were stained with PKH26 and added to autologous mature DCs at a ratio of 1:1. After the specified time periods at the indicated temperatures, cultures were collected and stained for CD11c and analyzed by FACS® (A) and deconvolution microscopy (B). For FACS® analysis, gates were set on large cells (forward scatter) and there was exclusion of autofluorescing cells in FL-3.
DCs Exposed to EBV Do Not Express Detectable Levels of EBV Latency Antigens.
To further investigate the mechanism of T cell priming by DCs, we examined the possibility of latent EBV infection of DCs. Though B cells were the only cells directly exposed to the virus during the 24 h incubation period, the possibilities of residual EBV virions adhering to the surface of infected B cells or productive infection with release of infectious virions in the coculture could not be excluded. Using RT-PCR, we examined B cells and DCs after 14 d of incubation in B95-8 supernatant for the presence of EBNA1, using primers detecting the BKRF1 mRNA transcript encoding the U and K exons (31). EBNA1 mRNA is present in all forms of EBV latency. In infected B cells, this mRNA transcript was present at levels comparable to that seen in the LCL-positive control but was not present in the DCs (Fig. 7 A). No EBNA1 mRNA transcript was detected in B cells or DCs incubated in Ramos supernatant. Therefore, direct infection of DCs with subsequent production of EBV latency antigens most likely is not the mechanism for DC priming of naive T cells.
Figure 7.

DCs exposed to EBV do not express detectable levels of EBV latency antigens. CD19+ cells and mature DCs were cultured for 14 d in supernatant from the EBV− cell line, Ramos (Mock), or in B95-8 supernatant. RT-PCR was performed on mRNA derived from the cells using primers spanning the U–K exon splice site of the mRNA for the EBNA1 latency protein (A). Primer pairs for G3PDH were used as a positive control for the reaction. PCR products were run on a 1% agarose gel. (B) Mature DCs were incubated for 24 h in media containing concentrated EBV viral stock (DC) or 1:2 with B cells infected with EBV for 24 h before DC/B coculture (B + DC). DCs were positively selected using anti-CD11c antibodies and cultured for 12 d with T cells from an EBV-seronegative (B, left) or -seropositive donor (B, right). DCs, infected with vaccinia vectors specific for the indicated EBV latency antigens, or EBV− Ramos cells (negative control) and autologous LCLs were used to restimulate antigen specific cells in IFNγ ELISPOT assays. Results shown are representative of two EBV-seronegative donors (left) and four seropositive donors (right). Results shown have the vector control, vvTK−, subtracted from the specific response, and positive responses (*) were determined as in Fig. 1.
To further address the possibility of low level or intermittent infection of DCs by EBV, we used a functional assay in which T cells were cocultured with DCs that had either been directly exposed to EBV (Fig. 7 B, DCs alone, white bar) or to EBV-infected B cells (Fig. 7 B, B + DC, black bars). B cells were incubated with EBV virus for 24 h and cocultured with DCs for an additional 24 h. CD11c+ cells were positively selected and cocultured with autologous T cells. If EBV infection of DCs was responsible for T cell priming, or if sufficient EBV antigens were contained in the virus preparation, one would expect to see EBV-specific T cells in cultures containing DCs exposed to viral particles. However, in agreement with our RT-PCR results, T cell priming was seen only in those cultures in which DCs were incubated with EBV-infected B cells, but not in the cultures containing EBV-exposed DCs (Fig. 7 B, left) in two EBV-seronegative donors tested. These results were further reinforced by experiments using blood from EBV-seropositive donors. Again EBV-exposed DCs alone did not expand EBV-specific memory T cells (Fig. 7 B, right). We conclude that for significant expansion or priming of EBV-specific T cells to occur, DCs need to present antigen derived from EBV-infected B cells.
Discussion
The importance of the T cell–mediated immune response in the resolution of IM and control of EBV-latent infection is highlighted in patients with T cell immunodeficiencies (e.g., patients after bone marrow transplantation, in AIDS, and in hereditary T cell disorders; references 44, 45). Not only has functional T cell depletion been demonstrated to be responsible for the development of EBV-associated malignancies, particularly PTLD, but adoptive transfer of EBV-specific cytotoxic T cells is now used in the clinical setting as treatment and prophylaxis against these tumors in immunosuppressed patients (3). In this paper, we study cells from EBV-seronegative donors to show that DCs can prime T cells specific for a panel of EBV latency antigens (Fig. 1) and are essential for the T-dependent regression of EBV transformation in culture (Fig. 2). Although antigen-bearing B cells are widely regarded as “professional” antigen presenting cells, EBV-infected B cells fail to prime T cells and fail to elicit regression when cells from seronegative individuals are studied. DC cross-presentation of EBV antigens from exogenous B cells (Fig. 6) seems critical for their function in inducing resistance to EBV transformation, because direct infection of DCs is undetectable by both molecular and immunologic assays (Fig. 7).
Our findings that DCs initiate protective EBV immunity in culture have important implications clinically and immunologically. EBV-seronegative recipients of EBV+ solid organs have an increased likelihood of developing PTLD (4, 6, 46). In EBV-seropositive patients with PTLD, EBV-specific cytotoxic T lymphocytes for adoptive immunotherapy are easily expanded in vitro using autologous LCLs (2, 3). However, in EBV-seronegative patients, current protocols using autologous LCLs fail to efficiently prime protective EBV-specific cytotoxic T cells without repeated restimulations and the addition of rhIL-2 and rhIL-12 (5, 47). In this paper, we have shown that DCs within 12 d are able to prime T cells that recognize a wide array of EBV latency antigens (Fig. 1) and control the proliferation of EBV-transformed B cells (Fig. 2). In contrast, a previous work used DC preparations loaded with necrotic/apoptotic LCLs as well as LCLs fused with DCs, but the DCs were inferior to LCLs in priming of EBV-specific T cell responses from EBV-seronegative children (48). The differences in our work may lie in the antigenic formulation (cocultured freshly EBV-infected B cells), the responder T cell population (adult vs. pediatric EBV-seronegative donors), and the DC preparation. For example, we routinely detect the CD83 maturation marker on 100% of our monocyte-derived DCs, whereas Savoldo et al. reported 15–40% of their DCs were CD83+ (48). We believe that these differences might account for the higher efficiency of our DC preparations in EBV-specific T cell priming.
We observed that on average 0.4% of cells in our regression cultures responded on day 12–14 to EBV-latent antigens with IFNγ secretion (Fig. 1; sum of individual EBV specificities and reactivity against autologous LCLs). This time point correlated with time of regression of EBV-transformed B cells in B + DC + T cultures of EBV-seronegative donors (Fig. 4). In cultures of EBV-seropositive donors, regression of EBV-transformed B cells could be detected much more quickly. Already at day 3, the cultures contained fewer CD19+ CD23+ B cells than cultures of EBV seronegative donors. This indicates that the frequency of EBV-specific T cells in peripheral blood of EBV-seropositive donors is sufficient to control EBV transformation rapidly in vitro. Most EBV specificities can be found at frequencies of 0.01–0.1% in whole PBMCs (49–52). In total, this corresponds roughly to the 0.4% we induced in our cultures, and in both instances, regression was observed. The yields of newly primed EBV-specific T cells in our cultures probably underestimate the potential of DCs to expand specific T cells after EBV infection in vivo, because in lymph nodes, a large proportion of T cell repertoire circulates by DCs and is able to be selected for priming.
We have not investigated the mechanisms required by highly enriched CD4+ and CD8+ T cells to protect against EBV transformation in culture, and regression in these assays may require additional events than the known cytolytic capacities of these T cells. The effector mechanisms of the primed CD4+ and CD8+ T cell populations were not analyzed in this paper. However, both CD4+ as well as CD8+ T cell clones have been reported to be efficient in mediating EBV-transformed B cell regression (53–55). The literature lists several effector functions of EBV-specific T cells that could contribute to the regression of EBV-transformed B cells. These include perforin–granzyme-mediated lysis by CD8+ T cells (56–58), Fas–FasL-mediated lysis by CD4+ T cells (59), and IFNγ secretion by CD8+ (55) and probably also CD4+ T cells.
To prime to EBV latency antigens, DCs must either take up exogenous EBV antigens or endogenously generate these proteins after productive infection with the virus. The lack of detectable EBNA1 mRNA in EBV-exposed DCs by RT-PCR (Fig. 7 A) coupled with the inability of EBV-exposed DCs to prime or even expand EBV-specific T cells (Fig. 7 B) argues against an immunologically relevant DC infection by EBV. Therefore, we suggest that the most likely mechanism of priming is via uptake of cellular debris from EBV-infected B cells for processing and presentation to naive T cells. Immature and mature DCs were both able to mediate regression and T cell priming in EBV-seronegative cultures. This suggests to us that there is sufficient DC maturation in the regression cultures to render immature DCs immunogenic for T cell priming and that our preparations of cytokine-matured DCs retain sufficient endocytosis capacity (as demonstrated in Fig. 6) to take up antigens for T cell priming.
There is additional compelling in vivo evidence that supports cross-presentation of EBV antigens to naive T cells during primary infection. The evidence involves the EBV latency antigen, EBNA1, which is protected from endogenous processing onto MHC class I molecules for presentation to CD8+ T cells by its Gly-Ala repeat sequence (60, 61). Because of this, EBV-infected B cells cannot stimulate, much less prime, EBNA1-specific CD8+ T cells. Nevertheless, up to 5% of peripheral blood CD8+ T cells in HLA-B*3501 patients with IM can be EBNA1 specific (62). The inability of these CTLs to recognize autologous LCLs confirms that EBV-transformed B cells do not load EBNA1 epitopes onto MHC class I after endogenous processing (62, 63). In contrast, DCs loaded with EBNA1 protein are lysed by these CTLs, indicating that EBNA1 peptides can be exogenously processed and loaded onto MHC class I molecules by DCs (62). Furthermore, DCs have been shown to expand CD8+ T cells specific for other EBV latency antigens after cross-presentation of dying LCLs (21). Therefore, the generation of EBV immunity in culture and probably in infected individuals utilizes the capacity of DCs to process or cross-present other cells that die during infection (64), in this case, B cells, killed by lytic EBV infection, might induce both CD4+ and CD8+ T cell responses. The in vitro regression assays that we have used here are currently the most direct ex vivo manifestation of protection against transformation. To the extent that these findings are indicative of protection in vivo, EBV is an example of a human pathogen that would require cross-presentation by DCs to be efficiently controlled by the immune system.
Understanding the normal sequence of events necessary to control primary and latent EBV infections should benefit the development of novel immune-based therapies for patients with EBV-associated malignancies. In EBV-seronegative patients, especially in the pediatric population, EBV-latent antigen-bearing DCs can be used in vivo or ex vivo to prime naive T cells. Additionally in tumor-bearing adults, active immunization with DCs might be used to augment waning EBV immune responses or to realign responses to the more protective Th1 type. Our results suggest that future EBV vaccine strategies target EBV antigens to mature DCs, thereby exploiting their specialized roles in initiating protective immunity.
Acknowledgments
We thank K. Velinzon and S. Mazel for cell sorting.
This work was supported by grants from National Institute of Allergy and Infectious Diseases (AI40045 to R.M. Steinman and A149958 to K. Bickham). C. Münz is a recipient of a Special Fellowship from the Leukemia and Lymphoma Society and a grant from the Speaker's Fund for Public Health Research by the City of New York.
The online version of this article includes supplemental material.
Abbreviations used in this paper: CSA, cyclosporin A; EBV, Epstein-Barr virus; HD, Hodgkin's disease; IM, infectious mononucleosis; LCL, lymphoblastoid cell line; PTLD, posttransplant lymphoproliferative disease; vv, vaccinia viruses.
References
- 1.Rickinson, A.B., and E. Kieff. 2001. Epstein-Barr virus. Fields Virology. P.M. Knipe and P.M. Howley, editors. Lippincott-Raven, Philadelphia. 2575–2627.
- 2.Gahn, B., G. Hunt, C.M. Rooney, and H.E. Heslop. 2002. Immunotherapy to reconstitute immunity to DNA viruses. Semin. Hematol. 39:41–47. [DOI] [PubMed] [Google Scholar]
- 3.Heslop, H.E., C.Y. Ng, C. Li, C.A. Smith, S.K. Loftin, R.A. Krance, M.K. Brenner, and C.M. Rooney. 1996. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat. Med. 2:551–555. [DOI] [PubMed] [Google Scholar]
- 4.Boyle, G.J., M.G. Michaels, S.A. Webber, A.S. Knisely, G. Kurland, L.A. Cipriani, B.P. Griffith, and F.J. Fricker. 1997. Posttransplantation lymphoproliferative disorders in pediatric thoracic organ recipients. J. Pediatr. 131:309–313. [DOI] [PubMed] [Google Scholar]
- 5.Metes, D., W. Storkus, A. Zeevi, K. Patterson, A. Logar, D. Rowe, M.A. Nalesnik, J.J. Fung, and A.S. Rao. 2000. Ex vivo generation of effective Epstein-Barr virus (EBV)-specific CD8+ cytotoxic T lymphocytes from the peripheral blood of immunocompetent Epstein Barr virus-seronegative individuals. Transplantation. 70:1507–1515. [DOI] [PubMed] [Google Scholar]
- 6.Sokal, E.M., H. Antunes, C. Beguin, M. Bodeus, P. Wallemacq, J. de Ville de Goyet, R. Reding, M. Janssen, J.P. Buts, and J.B. Otte. 1997. Early signs and risk factors for the increased incidence of Epstein-Barr virus-related posttransplant lymphoproliferative diseases in pediatric liver transplant recipients treated with tacrolimus. Transplantation. 64:1438–1442. [DOI] [PubMed] [Google Scholar]
- 7.Konttinen, Y.T., H.G. Bluestein, and N.J. Zvaifler. 1985. Regulation of the growth of Epstein-Barr virus-infected B cells. I. Growth regression by E rosetting cells from VCA-positive donors is a combined effect of autologous mixed leukocyte reaction and activation of T8+ memory cells. J. Immunol. 134:2287–2293. [PubMed] [Google Scholar]
- 8.Nikiforow, S., K. Bottomly, and G. Miller. 2001. CD4+ T-cell effectors inhibit Epstein-Barr virus-induced B-cell proliferation. J. Virol. 75:3740–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okano, M., and D.T. Purtilo. 1995. Simple assay for evaluation of Epstein-Barr virus specific cytotoxic T lymphocytes. J. Immunol. Methods. 184:149–152. [DOI] [PubMed] [Google Scholar]
- 10.Rickinson, A.B., D.J. Moss, J.H. Pope, and N. Ahlberg. 1980. Long-term T-cell-mediated immunity to Epstein-Barr virus in man. IV. Development of T-cell memory in convalescent infectious mononucleosis patients. Int. J. Cancer. 25:59–65. [DOI] [PubMed] [Google Scholar]
- 11.Moss, D.J., A.B. Rickinson, and J.H. Pope. 1979. Long-term T-cell-mediated immunity to Epstein-Barr virus in man. III. Activation of cytotoxic T cells in virus-infected leukocyte cultures. Int. J. Cancer. 23:618–625. [DOI] [PubMed] [Google Scholar]
- 12.Rickinson, A.B., D.J. Moss, and J.H. Pope. 1979. Long-term C-cell-mediated immunity to Epstein-Barr virus in man. II. Components necessary for regression in virus-infected leukocyte cultures. Int. J. Cancer. 23:610–617. [DOI] [PubMed] [Google Scholar]
- 13.Moss, D.J., A.B. Rickinson, and J.H. Pope. 1978. Long-term T-cell-mediated immunity to Epstein-Barr virus in man. I. Complete regression of virus-induced transformation in cultures of seropositive donor leukocytes. Int. J. Cancer. 22:662–668. [DOI] [PubMed] [Google Scholar]
- 14.Calender, A., M. Billaud, J.P. Aubry, J. Banchereau, M. Vuillaume, and G.M. Lenoir. 1987. Epstein-Barr virus (EBV) induces expression of B-cell activation markers on in vitro infection of EBV-negative B-lymphoma cells. Proc. Natl. Acad. Sci. USA. 84:8060–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurley, E.A., and D.A. Thorley-Lawson. 1988. B cell activation and the establishment of Epstein-Barr virus latency. J. Exp. Med. 168:2059–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan, S.H., and T.S. Chew. 1981. Lack of regression in Epstein-Barr virus infected leucocyte cultures of nasopharyngeal carcinoma patients. Lancet. 2:1353. [DOI] [PubMed] [Google Scholar]
- 17.Harada, S., K. Sakamoto, J.K. Seeley, T. Lindsten, T. Bechtold, J. Yetz, G. Rogers, G. Pearson, and D.T. Purtilo. 1982. Immune deficiency in the X-linked lymphoproliferative syndrome. I. Epstein-Barr virus-specific defects. J. Immunol. 129:2532–2535. [PubMed] [Google Scholar]
- 18.Moss, D.J., S.H. Chan, S.R. Burrows, T.S. Chew, R.G. Kane, J.A. Staples, and N. Kunaratnam. 1983. Epstein-Barr virus specific T-cell response in nasopharyngeal carcinoma patients. Int. J. Cancer. 32:301–305. [DOI] [PubMed] [Google Scholar]
- 19.Tamura, S., A. Yamazaki, M. Kunimoto, K. Takemura, T. Tabata, Y. Hinuma, and O. Yoshie. 1992. Impaired long-term T cell immunity to Epstein-Barr virus in patients with nasopharyngeal carcinoma. Jpn. J. Cancer Res. 83:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Münz, C., K.L. Bickham, M. Subklewe, M.L. Tsang, A. Chahroudi, M.G. Kurilla, D. Zhang, M. O'Donnell, and R.M. Steinman. 2000. Human CD4+ T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J. Exp. Med. 191:1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subklewe, M., C. Paludan, M.L. Tsang, K. Mahnke, R.M. Steinman, and C. Münz. 2001. Dendritic cells cross-present latency gene products from Epstein-Barr virus-transformed B cells and expand tumor-reactive CD8+ killer T cells. J. Exp. Med. 193:405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bickham, K., C. Münz, M.L. Tsang, M. Larsson, J.F. Fonteneau, N. Bhardwaj, and R. Steinman. 2001. EBNA1-specific CD4+ T cells in healthy carriers of Epstein-Barr virus are primarily Th1 in function. J. Clin. Invest. 107:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macatonia, S.E., N.A. Hosken, M. Litton, P. Vieira, C.S. Hsieh, J.A. Culpepper, M. Wysocka, G. Trinchieri, K.M. Murphy, and A. O'Garra. 1995. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J. Immunol. 154:5071–5079. [PubMed] [Google Scholar]
- 24.Koch, F., U. Stanzl, P. Jennewein, K. Janke, C. Heufler, E. Kampgen, N. Romani, and G. Schuler. 1996. High level IL-12 production by murine dendritic cells: up-regulation via MHC class II and CD40 molecules and down-regulation by IL-4 and IL-10. J. Exp. Med. 184:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cella, M., D. Scheidegger, K. Palmer-Lehmann, P. Lane, A. Lanzavecchia, and G. Alber. 1996. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin 12 and enhances T cell stimulatory capacity: T–T help via APC activation. J. Exp. Med. 184:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller, G., J. Robinson, L. Heston, and M. Lipman. 1975. Differences between laboratory strains of Epstein-Barr virus based on immortalization, abortive infection and interference. IARC Sci. Publ. 11:395–408. [PubMed] [Google Scholar]
- 27.Klein, G., B. Giovanella, A. Westman, J.S. Stehlin, and D. Mumford. 1975. An EBV-genome-negative cell line established from an American Burkitt lymphoma; receptor characteristics. EBV infectibility and permanent conversion into EBV-positive sublines by in vitro infection. Intervirology. 5:319–334. [DOI] [PubMed] [Google Scholar]
- 28.Steinitz, M., and G. Klein. 1975. Comparison between growth characteristics of an Epstein–Barr virus (EBV)-genome-negative lymphoma line and its EBV-converted subline in vitro. Proc. Natl. Acad. Sci. USA. 72:3518–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bargou, R.C., M.Y. Mapara, C. Zugck, P.T. Daniel, M. Pawlita, H. Dohner, and B. Dorken. 1993. Characterization of a novel Hodgkin cell line, HD-MyZ, with myelomonocytic features mimicking Hodgkin's disease in severe combined immunodeficient mice. J. Exp. Med. 177:1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subklewe, M., A. Chahroudi, A. Schmaljohn, M.G. Kurilla, N. Bhardwaj, and R.M. Steinman. 1999. Induction of Epstein-Barr virus-specific cytotoxic T-lymphocyte responses using dendritic cells pulsed with EBNA-3A peptides or UV-inactivated, recombinant EBNA-3A vaccinia virus. Blood. 94:1372–1381. [PubMed] [Google Scholar]
- 31.Savard, M., C. Belanger, M. Tardif, P. Gourde, L. Flamand, and J. Gosselin. 2000. Infection of primary human monocytes by Epstein-Barr virus. J. Virol. 74:2612–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee, A.W., T. Truong, K. Bickham, J.F. Fonteneau, M. Larsson, I. Da Silva, S. Somersan, E.K. Thomas, and N. Bhardwaj. 2002. A clinical grade cocktail of cytokines and PGE(2) results in uniform maturation of human monocyte-derived dendritic cells: implications for immunotherapy. Vaccine. 20:A8–A22. [DOI] [PubMed] [Google Scholar]
- 33.Azim, T., and D.H. Crawford. 1988. Lymphocytes activated by the Epstein-Barr virus to produce immunoglobulin do not express CD23 or become immortalized. Int. J. Cancer. 42:23–28. [DOI] [PubMed] [Google Scholar]
- 34.Swendeman, S., and D.A. Thorley-Lawson. 1987. The activation antigen BLAST-2, when shed, is an autocrine BCGF for normal and transformed B cells. EMBO J. 6:1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borel, J.F., C. Feurer, H.U. Gubler, and H. Stahelin. 1976. Biological effects of cyclosporin A: a new antilymphocytic agent. Agents Actions. 6:468–475. [DOI] [PubMed] [Google Scholar]
- 36.Ruegger, A., M. Kuhn, H. Lichti, H.R. Loosli, R. Huguenin, C. Quiquerez, and A. von Wartburg. 1976. (Cyclosporin A, a Peptide Metabolite from Trichoderma polysporum (Link ex Pers.) Rifai, with a remarkable immunosuppressive activity) [in German]. Helv. Chim. Acta. 59:1075–1092. [DOI] [PubMed] [Google Scholar]
- 37.Maini, M.K., N. Gudgeon, L.R. Wedderburn, A.B. Rickinson, and P.C. Beverley. 2000. Clonal expansions in acute EBV infection are detectable in the CD8 and not the CD4 subset and persist with a variable CD45 phenotype. J. Immunol. 165:5729–5737. [DOI] [PubMed] [Google Scholar]
- 38.Sutkowski, N., B. Conrad, D.A. Thorley-Lawson, and B.T. Huber. 2001. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity. 15:579–589. [DOI] [PubMed] [Google Scholar]
- 39.Sutkowski, N., T. Palkama, C. Ciurli, R.P. Sekaly, D.A. Thorley-Lawson, and B.T. Huber. 1996. An Epstein-Barr virus–associated superantigen. J. Exp. Med. 184:971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.den Haan, J.M., and M.J. Bevan. 2001. Antigen presentation to CD8+ T cells: cross-priming in infectious diseases. Curr. Opin. Immunol. 13:437–441. [DOI] [PubMed] [Google Scholar]
- 41.Bickham, K., and C. Münz. 2003. Contrasting roles of dendritic cells and B cells in the immune control of Epstein-Barr virus. Curr. Top. Microbiol. Immunol. 276:55–76. [DOI] [PubMed] [Google Scholar]
- 42.Herr, W., E. Ranieri, W. Olson, H. Zarour, L. Gesualdo, and W.J. Storkus. 2000. Mature dendritic cells pulsed with freeze-thaw cell lysates define an effective in vitro vaccine designed to elicit EBV-specific CD4+ and CD8+ T lymphocyte responses. Blood. 96:1857–1864. [PubMed] [Google Scholar]
- 43.Ferlazzo, G., C. Semino, G.M. Spaggiari, M. Meta, M.C. Mingari, and G. Melioli. 2000. Dendritic cells efficiently cross-prime HLA class I-restricted cytolytic T lymphocytes when pulsed with both apoptotic and necrotic cells but not with soluble cell-derived lysates. Int. Immunol. 12:1741–1747. [DOI] [PubMed] [Google Scholar]
- 44.Boshoff, C., and R. Weiss. 2002. AIDS-related malignancies. Nat. Rev. Immunol. 2:373–382. [DOI] [PubMed] [Google Scholar]
- 45.O'Reilly, R.J., T.N. Small, E. Papadopoulos, K. Lucas, J. Lacerda, and L. Koulova. 1997. Biology and adoptive cell therapy of Epstein-Barr virus-associated lymphoproliferative disorders in recipients of marrow allografts. Immunol. Rev. 157:195–216. [DOI] [PubMed] [Google Scholar]
- 46.Finn, L., J. Reyes, J. Bueno, and E. Yunis. 1998. Epstein-Barr virus infections in children after transplantation of the small intestine. Am. J. Surg. Pathol. 22:299–309. [DOI] [PubMed] [Google Scholar]
- 47.Misko, I.S., T.B. Sculley, C. Schmidt, D.J. Moss, T. Soszynski, and K. Burman. 1991. Composite response of naive T cells to stimulation with the autologous lymphoblastoid cell line is mediated by CD4 cytotoxic T cell clones and includes an Epstein-Barr virus-specific component. Cell. Immunol. 132:295–307. [DOI] [PubMed] [Google Scholar]
- 48.Savoldo, B., M.L. Cubbage, A.G. Durett, J. Goss, M.H. Huls, Z. Liu, L. Teresita, A.P. Gee, P.D. Ling, M.K. Brenner, et al. 2002. Generation of EBV-specific CD4+ cytotoxic T cells from virus naive individuals. J. Immunol. 168:909–918. [DOI] [PubMed] [Google Scholar]
- 49.Leen, A., P. Meij, I. Redchenko, J. Middeldorp, E. Bloemena, A. Rickinson, and N. Blake. 2001. Differential immunogenicity of Epstein-Barr virus latent-cycle proteins for human CD4+ T-helper 1 responses. J. Virol. 75:8649–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan, L.C., N. Gudgeon, N.E. Annels, P. Hansasuta, C.A. O'Callaghan, S. Rowland-Jones, A.J. McMichael, A.B. Rickinson, and M.F. Callan. 1999. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J. Immunol. 162:1827–1835. [PubMed] [Google Scholar]
- 51.Hislop, A.D., N.H. Gudgeon, M.F. Callan, C. Fazou, H. Hasegawa, M. Salmon, and A.B. Rickinson. 2001. EBV-specific CD8+ T cell memory: relationships between epitope specificity, cell phenotype, and immediate effector function. J. Immunol. 167:2019–2029. [DOI] [PubMed] [Google Scholar]
- 52.Chakrabarti, S., D.W. Milligan, D. Pillay, S. Mackinnon, K. Holder, N. Kaur, D. McDonald, C.D. Fegan, H. Waldmann, G. Hale, et al. 2003. Reconstitution of the Epstein-Barr virus-specific cytotoxic T-lymphocyte response following T-cell-depleted myeloablative and nonmyeloablative allogeneic stem cell transplantation. Blood. 102:839–842. [DOI] [PubMed] [Google Scholar]
- 53.Nikiforow, S., K. Bottomly, G. Miller, and C. Münz. 2003. Cytolytic CD4+ T-cell clones reactive to EBNA1 inhibit Epstein-Barr virus-induced B-cell proliferation. J. Virol. 77:12088–12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Omiya, R., C. Buteau, H. Kobayashi, C.V. Paya, and E. Celis. 2002. Inhibition of EBV-induced lymphoproliferation by CD4+ T cells specific for an MHC class II promiscuous epitope. J. Immunol. 169:2172–2179. [DOI] [PubMed] [Google Scholar]
- 55.Shi, Y., and C.T. Lutz. 2002. Interferon-γ control of EBV-transformed B cells: a role for CD8+ T cells that poorly kill EBV-infected cells. Viral Immunol. 15:213–225. [DOI] [PubMed] [Google Scholar]
- 56.Khanna, R., S.R. Burrows, M.G. Kurilla, C.A. Jacob, I.S. Misko, T.B. Sculley, E. Kieff, and D.J. Moss. 1992. Localization of Epstein-Barr virus cytotoxic T cell epitopes using recombinant vaccinia: implications for vaccine development. J. Exp. Med. 176:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murray, R.J., M.G. Kurilla, J.M. Brooks, W.A. Thomas, M. Rowe, E. Kieff, and A.B. Rickinson. 1992. Identification of target antigens for the human cytotoxic T cell response to Epstein-Barr virus (EBV): implications for the immune control of EBV-positive malignancies. J. Exp. Med. 176:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steven, N.M., A.M. Leese, N.E. Annels, S.P. Lee, and A.B. Rickinson. 1996. Epitope focusing in the primary cytotoxic T cell response to Epstein-Barr virus and its relationship to T cell memory. J. Exp. Med. 184:1801–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paludan, C., K. Bickham, S. Nikiforow, M.L. Tsang, K. Goodman, W.A. Hanekom, J.F. Fonteneau, S. Stevanovic, and C. Münz. 2002. EBNA1 specific CD4+ Th1 cells kill Burkitt's lymphoma cells. J. Immunol. 169:1593–1603. [DOI] [PubMed] [Google Scholar]
- 60.Levitskaya, J., M. Coram, V. Levitsky, S. Imreh, P.M. Steigerwald-Mullen, G. Klein, M.G. Kurilla, and M.G. Masucci. 1995. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature. 375:685–688. [DOI] [PubMed] [Google Scholar]
- 61.Levitskaya, J., A. Sharipo, A. Leonchiks, A. Ciechanover, and M.G. Masucci. 1997. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA. 94:12616–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blake, N., T. Haigh, G. Shaka'a, D. Croom-Carter, and A. Rickinson. 2000. The importance of exogenous antigen in priming the human CD8+ T cell response: lessons from the EBV nuclear antigen EBNA1. J. Immunol. 165:7078–7087. [DOI] [PubMed] [Google Scholar]
- 63.Blake, N., S. Lee, I. Redchenko, W. Thomas, N. Steven, A. Leese, P. Steigerwald-Mullen, M.G. Kurilla, L. Frappier, and A. Rickinson. 1997. Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity. 7:791–802. [DOI] [PubMed] [Google Scholar]
- 64.den Haan, J.M., and M.J. Bevan. 2002. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8+ and CD8− dendritic cells in vivo. J. Exp. Med. 196:817–827. [DOI] [PMC free article] [PubMed] [Google Scholar]