

Abstract
The Leishmania major LACK antigen is a key target of the immune response in susceptible BALB/c mice and remains a viable vaccine candidate for human leishmaniasis. We describe the genomic organization of the four lack genes in the L. major diploid genome together with results of selected lack gene targeting. Parasites containing a single lack gene in either the upstream or downstream locus grew comparably to wild-type promastigotes in vitro, but failed to parasitize BALB/c mice efficiently, even in a T cell–deficient environment. The replication of single copy lack mutants as amastigotes was attenuated in macrophages in vitro, and parasites failed to increase in numbers in immunodeficient mice, despite their persistence over months. Complementation with an additional lack copy was sufficient to induce robust lesion development, which also occurred using parasites with two lack genes. Conversely, attempts to generate lack-null parasites failed, suggesting that LACK is required for parasite viability. These data suggest that LACK is critical for effective mammalian parasitization and thus represents a potential drug target for leishmaniasis.
Keywords: Leishmania major, LACK, protozoa, gene targeting, WD repeat proteins
Introduction
Leishmania major is a protozoan pathogen transmitted by phlebotomine sandflies and thrives in phagolysosomal compartments within macrophages of vertebrate hosts. The L. major LACK (Leishmania homologue of mammalian RACKs, the receptors for activated C kinase) antigen was identified by screening a parasite expression library as the antigen recognized by a protective murine CD4 T cell clone (1). Additional studies suggested that experimental infection of susceptible BALB/c mice with L. major nucleated a dominant population of LACK-specific Vβ4+ Th2 cells necessary for progressive infection (2, 3). Antagonism of the LACK-specific CD4 T cell response by altered peptide ligands (4) or deletion of LACK-reactive T cells by expression of a LACK transgene in the mouse thymus (5) resulted in a Th1 response and control of disease in BALB/c mice.
The immunologic response to LACK has been exquisitely characterized in BALB/c mice. Within the first 3–4 d after inoculation of parasites, a small number of naive CD4 T cells with appropriate specificity for a dominant I-Ad LACK peptide expand over 120-fold and begin to express IL-4 transcripts that characterize Th2 immune responses (6). Intriguingly, the preinfection repertoire and the postinfection expansion and acquisition of IL-4 transcripts among LACK-reactive CD4 T cells were not different in comparing susceptible and resistant inbred mouse strains. The factors that underlie the immunodominance of the LACK epitope in the I-Ad Th response remain unknown, although these are unlikely to reflect the overall abundance of the LACK protein because other L. major surface proteins such as GP63 are more highly expressed (4, 7). Whether the focus of the immune response on LACK is detrimental to the parasite remains unknown, but immunization with LACK is highly efficacious in protection against subsequent infection (8–10).
L. major LACK and its mammalian homologue, RACK1, belong to the WD repeat protein family. These macromolecules are evolutionarily conserved tryptophan aspartate motif proteins and have diverse but critical functions in eukaryotes, including signal transduction, RNA processing, and cell cycle control (11). In most cases, however, little is known about their molecular functions. The crystal structure has been best determined for one eukaryote WD repeat protein, the Gβ subunit of transducin, a heterotrimeric G protein in the visual signaling pathway of rod cells (12). The Gβ subunit adopts a flat, seven-bladed, propeller-like structure, suggesting that WD repeat proteins may function by providing a stable interaction surface upon which to scaffold a variety of multiprotein signaling complexes (13). The RACK1 protein was identified (14) based on its ability to bind activated protein kinase Cβ (βIIPKC) and modulate its subcellular localization (15, 16). Consistent with a more generalized role as an adaptor molecule, RACK1 has subsequently been found to interact with additional proteins, such as dynamin-1 and Src, and protein complexes (17–21).
Few studies have addressed the physiologic role of the LACK family of proteins in kinetoplastids. Trypanosomes up-regulate the homologous WD repeat protein, designated TRACK, in parasites undergoing apoptosis and in terminally differentiated bloodstream forms (22). In Leishmania infantum, LACK was localized near the kinetoplast, and peptide display suggested an interaction with sequences from proteins involved in DNA replication and RNA synthesis (23). In contrast, the homologue in Crithidia fasciculata, designated CACK, localized to the plasma membrane, consistent with a role in protein kinase C (PKC) localization similar to RACK1 (24). It remains possible that kinetoplastid LACK homologues interfere with their mammalian counterparts during parasitization. RACK1 has been implicated in negative regulation of microbicidal oxygen radical generation in HL60 cells (25) and the Epstein Barr virus RACK-binding protein, ZEBRA, was implicated in interfering with PKC-mediated phagocytosis and inflammatory cytokine production by infected macrophages (26). Macrophages infected with L. major demonstrated a delay in the dissociation of activated PKCβ that correlated with down-regulation of TNFα expression (27). Clearly, further genetic and biochemical studies are required to understand the biology of the LACK family proteins in kinetoplastids.
We used gene-targeting strategies to create LACK-deficient strains of L. major to determine the role of LACK in the parasite life cycle. The lack genes of L. major consist of a tandem duplicate family. lack-null parasites could not be generated, suggesting an essential role for LACK in viability. Although haploid parasites containing a single allele with two genes were indistinguishable from wild-type L. major, parasites containing single copy lack1 or lack2 genes were highly attenuated when used to infect susceptible BALB/c mice. Indeed, single copy mutants were unable to productively infect T- or T- and B cell–deficient mice, suggesting a necessary role for LACK in vertebrate parasitization.
Materials and Methods
Parasites.
L. major strain WHOM/IR/-/173 was cultured in medium 199 (M199; Sigma-Aldrich) supplemented with 10% heat-inactivated FCS (Atlanta Biologicals) as previously described (28). For growth assays, designated parasites from stationary phase cultures were diluted 1 in 50 and 1 in 15 in fresh medium and incubated at 27 and 37°C, respectively. Parasites were enumerated daily by counting in a hemocytometer after fixation in 0.4% formaldehyde in PBS. Metacyclogenesis of designated L. major mutants was assessed by staining parasites from 2- and 8-d cultures with 12 μg/ml Alexa 488 conjugated to peanut agglutinin (Ax-PNA; Molecular Probes) in PBS for 30 min at 4°C. Parasites were washed and analyzed by flow cytometry (FACSCalibur™; Becton Dickinson).
Constructs for lack Gene Targeting and Complementation.
A series of targeting constructs was assembled in plasmid pGEM-9Zf(−) (Promega) using regions flanking the tandem lack genes, designated lack1 and lack2, coupled to various drug selection cassettes (see Fig. 1, A and B). The entire lack1/lack2 locus, including the 3.3-kb intergenic region, was targeted using the 1.3-kb sequence immediately upstream of the lack1 start codon, the hygromycin B phosphotransferase coding region (hyg) ligated to the 3′ UTR of the L. major dhfr-ts gene (28, 29), and a 0.9-kb fragment beginning 190 bp 3′ of the lack2 stop codon. The final targeting vector, designated pLHDL, was verified by direct sequencing, as were each of the subsequent targeting constructs (see Fig. 1 B). A comparable targeting construct, designated pLSDL, was identical to pLHDL except that the hyg coding region was replaced by the 0.5-kb region coding for the streptothrycin acetyltransferase gene (sat) that confers nourseothricin resistance (28, 29).
Figure 1.

Targeting the L. major lack genes. (A) Physical map showing organization of the L. major lack genes. Restriction enzyme cut sites are indicated: B, BamHI; H, HindIII; K, KpnI; N, NsiI; No, NotI; S, StuI; Sp, SphI; Spe, SpeI; X, XbaI. Gray boxes denote lack1 and lack2 open reading frames. Fragments g and h (triple lines) were used as probes for Southern hybridization analysis. Fragments a–f (heavy lines) were used to make targeting constructs (B, 1–4) and episomal GFP-LACK and GFP expression constructs (B, 5 and 6). (B) Cartoon of the targeting constructs used for generating organisms used in these studies (refer to Materials and Methods). (C) Southern hybridization of L. major transfectant genomic DNAs. Genomic DNAs (10 μg/lane) from untransfected L. major (lane 1) or LHDL transfectants (L. major lack++/−−; lane 2) were digested with KpnI. Genomic DNAs from L. major lack++/−− (lane 3), L. major lack++/−− transfected with L2PDL2 (L. major lack+−/−−; lane 4), and L. major lack++/−− transfected with L1SDL1 (L. major lack−+/−−; lane 5) were double digested with StuI and BamHI. DNAs from L. major lack++/−− transfected with L2PD-LKΔ-L2 (L. major lack++Δ/−) were digested with StuI (lane 6). Blots were either hybridized with fragment h (lanes 1 and 2) or fragment g (lanes 3–6).
The lack1 gene was targeted using vector pL1SDL1, containing a sat drug selection cassette flanked by 1.3 kb upstream lack1 sequence and the 3.3-kb intergenic sequence. The lack2 gene was targeted using vector pL2PDL2, containing a puromycin acetyltransferase (pac) selection cassette inserted downstream of a 1.5-kb sequence immediately upstream of the lack2 start codon and upstream of a 0.6-kb sequence beginning 202 bp downstream of the lack2 stop codon (see Fig. 1, A and B).
Genetic complementation of LACK-deficient parasites was performed by inserting an XhoI-tagged lack wild-type coding region between the puromycin selection cassette and the 0.6-kb lack2 3′ flanking sequence in pL2PDL2. The resulting targeting vector was designated pL2PD-LKΔ-L2 (see Fig. 1 B).
To determine the cellular location of LACK in L. major, the green fluorescent protein (GFP) coding sequence (30) was fused using PCR to the 5′ end of the lack gene and inserted downstream of the DHFR sequence in construct pL1SDGLKL1 (see Fig. 1 B, 5). A control construct, pL1SDGL1, was made identical to pL1SDGLK1 except the gfp coding sequence was used alone in place of the fused gfp-lack sequence (see Fig. 1 B, 6).
Transfection and Selection of lack-targeted L. major.
The first lack allele was targeted by electroporation of 20 μg Qiaex II–purified insert (QIAGEN) from pLHDL double digested with NotI/XbaI (see Fig. 1 B). Deficient clones, designated lack++/−−, were selected after plating on semisolid M199 containing 32 μg/ml hygromycin B as previously described (28, 29). Deletion of the remaining lack allele was attempted after electroporation of 20 μg purified insert from pLSDL and selection on plates containing 60 μg/ml nourseothricin followed by culture in liquid media containing 30 μg/ml nourseothricin plus 16 μg/ml hygromycin B, as previously described (29).
To target the individual lack1 and lack2 genes, lack++/−− parasites were transfected with 20 μg Qiaex II–purified insert excised from pL1SDL1 and pL2PDL2, respectively. Transfectants were selected in the presence of nourseothricin (60 μg/ml semisolid media and 30 μg/ml liquid media) and puromycin (22 μg/ml semisolid media and 11 μg/ml liquid media), respectively. Effective complementation of L. major lack+−/−− parasites was achieved by transfection of L. major lack++/−− with 20 μg purified targeting fragment L2PD-LKΔ-L2 containing the XhoI-modified LACK2 construct.
For analysis of the cellular localization of LACK, lack++/−− parasites were transfected with 30 μg pL1SDGLKL1 or pL1SDGL1 and selected on plates and in liquid media as described for the pLSDL insert.
Southern Blotting to Confirm Gene Targeting.
Genomic DNAs were isolated from wild-type and L. major transfectant clones as previously described (31), digested with the designated restriction enzymes and size fractionated by agarose gel electrophoresis. The nucleic acids were blotted onto nylon membranes (Hybond N+; Amersham Biosciences) and hybridized using Rapid-Hyb (Amersham Biosciences) with DNA fragments g or h corresponding to the lack1 coding region and the most 3′ region of the lack allele, respectively (see Fig. 1 A), according to the manufacturer's instructions. Blots were washed at 65°C with 2X SSC-0.2X SSC/0.5% SDS.
Western Blotting Analysis.
Lysates from 2 × 106 or 2 × 107 parasites per lane were run on 4–20% gradient SDS-PAGE gels (Invitrogen) and blotted onto Hybond-C (Amersham Biosciences) according to the manufacturer's instructions. After overnight blocking in 5% milk powder (32) in TBS with 0.05% Tween 20 (TBS/T) at 4°C, blots were incubated with 1:4,000 rabbit anti–LACK antiserum (1) for 1 h at room temperature. After washing in TBS/T, blots were incubated with 1:3,000 goat anti–rabbit-Ig conjugated with horseradish peroxidase for 30 min. The blots were washed and developed using ECL chemiluminescence reagent (Amersham Biosciences) according to the manufacturer's instructions. Loading was verified by stripping the blots (62.5 mM Tris HCl, pH 6.8, 2% SDS, and 100 mM β-mercaptoethanol at 65°C for 30 min) and reprobing with polyclonal antisera (1:300) against the conserved 40S ribosomal protein, S6 (Santa Cruz Biotechnology, Inc.).
Fluorescence Microscopy.
Parasites transfected with pL1SDGLKL1 and pL1SDGL1 were diluted 1/10 in PBS/0.02% formaldehyde. Images of fluorescent parasites were acquired using a Nikon ECLIPSE E800 microscope (Nikon) in the FITC channel and viewed at ×1,000.
Mice and Infections.
BALB/c and BALB/c × RAG-2-deficient mice were purchased from The Jackson Laboratory. TCR-Cα–deficient mice were crossed 10 generations to BALB/c (33, 34). Animals were maintained in the University of California San Francisco specific pathogen-free barrier facility according to institutional protocols.
Cohorts of designated mice were inoculated subcutaneously in the left hind footpad with 2 × 106 metacyclic promastigotes, prepared as previously described (4). The relatively high inoculum was selected as a rigorous test of virulence based on preliminary experiments determining the capacity of resistant C57BL/6 and IL-4Rα–deficient BALB/c mice to control these inocula (unpublished data). Lesions were measured weekly using a vernier caliper. At the end of the infection, popliteal lymph nodes were collected from infected mice, dispersed into 5 ml M199 with 10% FCS, and diluted into microtiter wells in duplicate. After 7 d of culture at 27°C, parasite numbers were quantitated by microscopic determination of the last well containing viable organisms.
Analysis of the LACK-specific CD4 T Cell Response.
BALB/c mice were inoculated in the footpad with 2 × 106 designated metacyclic parasites. After 96 h, the draining popliteal lymph nodes were harvested and the CD4 T cells were incubated with I-Ad/LACK peptide MHC class II tetramers as previously described (6). Tetramer+ T cells were analyzed using a MoFlo® high speed cell sorter (DakoCytomation) and FlowJo® software (Tree Star) as previously described (6).
Macrophage Infections.
Bone marrow–derived macrophages from BALB/c mice were cultured on glass chamber slides (Lab-Tek; Nunc) as previously described (35). Promastigotes, at a ratio of approximately three per macrophage, were coincubated with the macrophages for 16 h at 35°C and washed in warm PBS to remove extracellular organisms. Monolayers were stained immediately after 96 h of incubation at 35°C using Diff-Quik (DADE Behring) and the number of amastigotes per cell was determined by light microscopy.
Results
The lack Gene Locus in L. major.
The organization of the lack genes was determined by Southern blot analysis using the lack coding region as a hybridization probe. The genes are arrayed in two tandemly linked, head to tail copies on a 10-kb StuI fragment on parasite chromosome 29 (Fig. 1 A and unpublished data). The cloned genomic fragment was sequenced and the two predicted lack protein-coding regions were identical. Comparisons of the 5′ and 3′ noncoding sequences showed divergence of the two genes 244 nucleotides upstream of the start codons and 190 nucleotides downstream of the stop codons. The 5′ mini-exon consensus capping sequence (36) is spliced onto the LACK mRNA at an AG dinucleotide 33 bp upstream of the start codon. Northern blots using gene-specific probes generated from the unique flanking regions showed the 5′ and 3′ lack genes, designated lack1 and lack2, encode 1.8 and 1.5 kb mRNAs, respectively (unpublished data).
Generation of Double and Single Copy lack Parasites.
Gene targeting constructs were generated from the cloned and sequenced 5′ and 3′ regions flanking the lack open reading frames as described in Materials and Methods (Fig. 1 B). After selection in drug-containing media, parasites deficient in one lack allele, designated lack++/−−, were expanded and characterized by Southern blotting. Consistent with deletion of one of the lack alleles, a 13-kb KpnI fragment containing both lack genes in the wild-type parasites was reduced to a 10-kb fragment in lack++/−− parasites when hybridized with probe h (Fig. 1 C, lanes 1 and 2).
Targeting the remaining two lack genes in lack++/−− parasites was attempted using the same targeting construct except that the hygromycin B phosphotransferase selection cassette was replaced with the streptothrycin acetyltransferase cassette, thus allowing selection in nourseothricin (refer to Materials and Methods). Repeated transfections resulted in failure to grow parasites in double drug selection media, suggesting that lack and/or its associated flanking regions are necessary for promastigote viability. This was not due to failure of selection using the nourseothricin cassette, as lack++/−− parasites could be readily generated using this targeting construct to delete one of the lack alleles (unpublished data).
Single copy lack parasites were generated by targeting the remaining lack1 or lack2 genes in lack++/−− organisms, using further selection in nourseothricin or puromycin, respectively (refer to Materials and Methods; Fig. 1 B). Southern blotting using the lack coding region as a hybridization probe showed loss of the 3-kb StuI/BamHI (lack1) and 7-kb StuI/BamHI (lack2) fragments in the respective lines, thus confirming generation of mutants with a single haploid copy of either lack1+−/−− or lack2−+/−− (Fig. 1 C, lanes 4 and 5, respectively). Attempts to delete the remaining copy of lack1 in lack+−/−− parasites were unsuccessful, consistent with an essential role for the lack locus in L. major.
To effectively determine the effects of complementation with the lack2 coding region, the lack2 gene in lack++/−− parasites was replaced in cis using a lack2 mutant (+D; Fig. 1 B, 4) containing an XhoI-tagged lack coding region to allow discrimination from endogenous lack. Southern blotting of the resulting parasite's DNA after StuI digestion revealed lack-hybridizing fragments of 4.9 and 5.4 kb, representing the endogenous lack1 gene and the reconstituted XhoI-marked lack2 copy, respectively (Fig. 1 C, lane 6). The reconstituted double copy mutants were designated lack++Δ/–.
Levels of LACK protein were assessed in the various lack mutants using Western blotting with anti-LACK antiserum. In multiple experiments, LACK protein levels were comparable between wild-type and lack++/−− L. major (unpublished data). After adjusting for differences in loading based on the intensity of eukaryote ribosomal protein S6 expression (Fig. 2 A, bottom), densitometric analysis of the bands showed the level of LACK protein was approximately threefold reduced in L. major lack+−/−− and lack++Δ/–, with relative band densities of 1.0 and 0.8, respectively, as compared with L. major lack++/−−, which showed a relative band density of 2.7 (Fig. 2 A, top).
Figure 2.
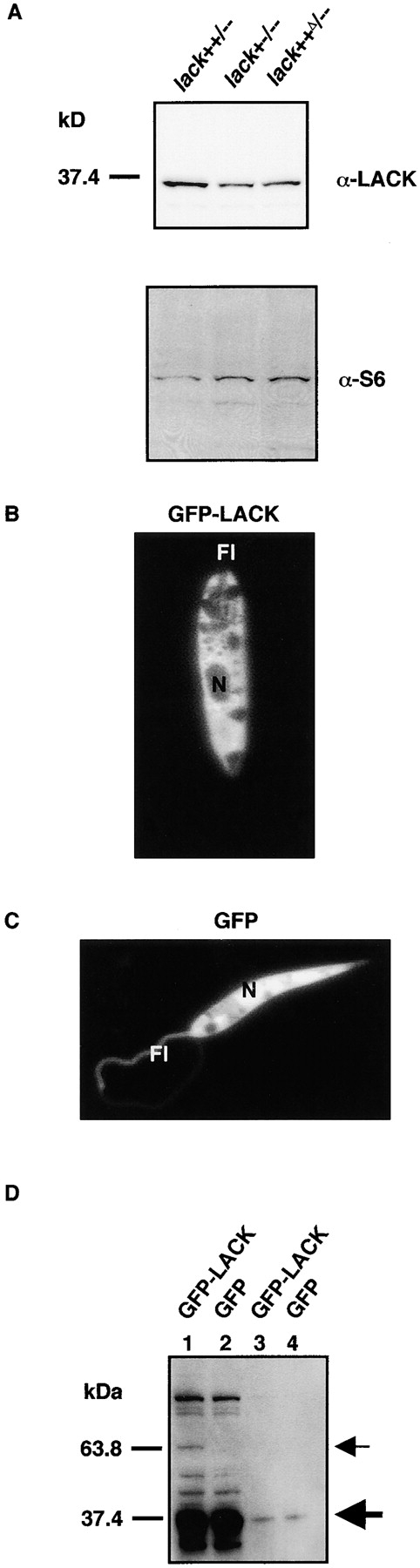
LACK protein determinations and subcellular localization. (A) LACK protein expression in lack-targeted L. major lines. Lysates from 2 × 107 lack-targeted L. major promastigotes were loaded, blotted, and probed with rabbit polyclonal antiserum raised against recombinant LACK protein (top). The blot was stripped and reprobed with goat polyclonal antiserum against eukaryotic 40S ribosomal protein S6 (bottom). Size markers (kD) are indicated. (B) Localization of GFP-LACK in L. major promastigotes. Stationary phase pL1SDGLKL1 transfectants were diluted 1 in 10 in PBS/0.02% formaldehyde. The parasites were viewed at ×1,000 with fluorescent excitation at 460–500 nm. Promastigote nucleus (N) and flagellum (Fl) are marked. (C) Localization of GFP in L. major promastigotes. Stationary phase pL1SDGL1 transfectants were treated and analyzed as described in B. (D) GFP-LACK protein expression in L. major lack++/−− transfectants. Parasites expressing GFP-LACK or GFP are indicated. Lanes 1 and 2, 2 × 107 organisms; lanes 3 and 4, 2 × 106 organisms. Location of endogenous LACK and GFP-LACK are indicated by large and small arrows, respectively.
Localization of LACK by immunofluorescence microscopy of pL1SDGLKL1 transfectants indicated that the GFP-LACK fusion protein localized to the cytosol of the promastigote cell body but was excluded from organelles such as the nucleus and other vacuolar structures and was also absent from the flagellum (Fig. 2 B). In contrast, GFP from pL1SDGL1 transfectants, although excluded from certain vacuolar structures, was localized throughout the cytosol, nucleus, and flagellum (Fig. 2 C). Comparable fluorescence patterns were observed in both strongly and weakly fluorescent transfectants, containing high and low copy numbers of the construct, respectively, suggesting that this distribution was not an effect of LACK overexpression. Further, and consistent with the relatively low level expression of the GFP-LACK fusion protein, Western blotting of lysates from pL1SDGLKL1 transfectants using anti-LACK antiserum revealed a 63-kD protein that was substantially less abundant than the endogenous LACK, even in lack++/−− organisms that contained a single allele (Fig. 2 D). Immunolocalization of LACK in promastigotes using anti-LACK antiserum showed a pattern of staining similar to the fluorescence observed in the pL1SDGLKL1 transfectants (unpublished data).
Attenuation of Virulence in Mice Using Single Copy lack Mutant Parasites.
An epitope from the LACK protein of L. major displayed in I-Ad constitutes a crucial recognition element for susceptibility-promoting Th2 cells in BALB/c mice (4). To ascertain whether moderation of LACK expression would affect the capacity to cause disease, the various mutant organisms were used to infect BALB/c mice. We first established that each of the parasites grew comparably as promastigotes at 27°C in vitro and that metacyclogenesis remained unimpaired. Growth of wild-type parasites in vitro and in the sandfly in vivo is associated with differentiation of promastigotes from noninfectious, rapidly dividing forms, to highly infectious, nondividing forms, termed metacyclics. Modifications of parasite surface carbohydrate moieties that accompany this transition allow metacyclic parasites to be distinguished from noninfectious stages based on their reduced ability to bind the lectin peanut agglutinin (PNA; reference 37). Parasites were collected during rapid growth on day 2 and during stationary phase on day 8, and assessed for PNA binding using flow cytometry. Both the percentages of organisms binding PNA and the intensity of staining were similar among all lack-targeted promastigotes when compared with wild-type L. major (Fig. 3 and unpublished data). These data suggest that metacyclogenesis is not impaired in the various LACK mutant promastigotes.
Figure 3.

Analysis of metacyclogenesis in lack-targeted L. major. Promastigotes from 2- or 8-d cultures were stained with Alexa 488 conjugated to PNA and analyzed by flow cytometry. Dashed line, L. major lack++/−−; solid line, L. major lack+−/−−; dotted line, unstained control.
Cohorts of BALB/c mice were infected in the footpads with selected, metacyclic parasites, and the course of disease was followed over time. In multiple experiments, double copy lack++/−− parasites produced lesions in mice that were indistinguishable from wild-type L. major that had been comparably passaged (unpublished data). However, in comparison to lack++/−− metacyclics, lack+−/−− and lack−+/−− metacyclic promastigotes showed significantly impaired lesion development (Fig. 4 A) and diminished recovery of viable parasites from infected mice (Fig. 4 E and unpublished data). Replacement of the endogenous lack2 coding region of lack++/−− parasites with the XhoI-tagged lack2 coding region (+Δ) preserved the capacity to produce lesions and recover parasites in recipient mice, suggesting that virulence had been attenuated by LACK deficiency (Fig. 4, D and E). Thus, despite comparable growth in vitro as promastigotes, the single copy lack parasites demonstrated marked attenuation of growth in vivo in susceptible mice.
Figure 4.
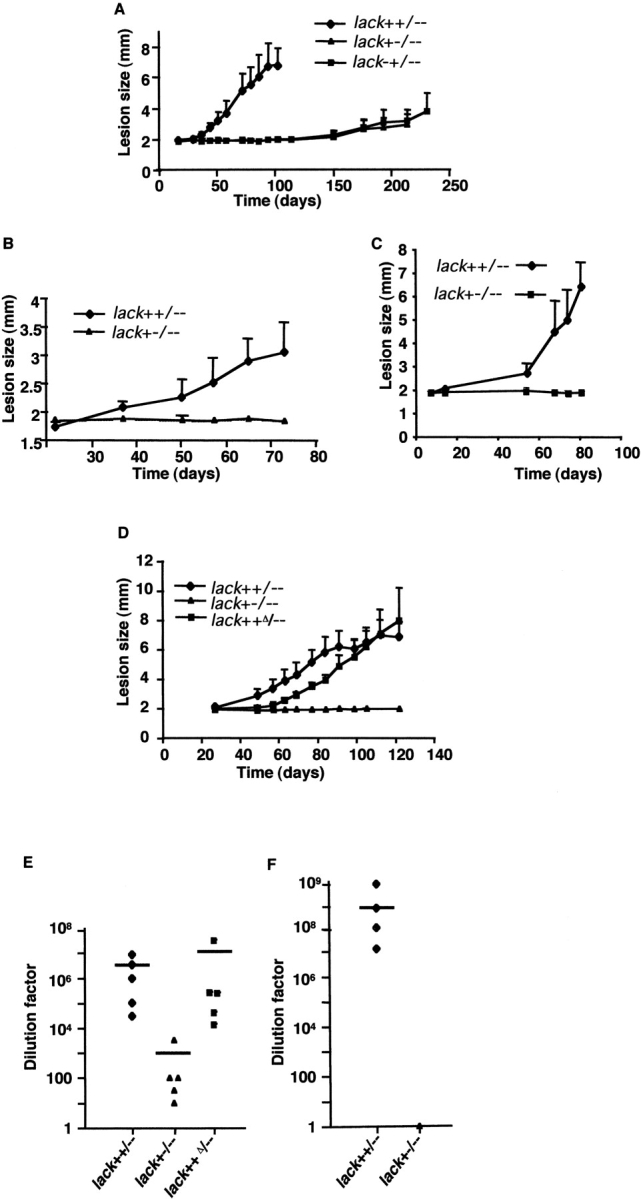
Lesion development and parasite burden after inoculation of lack-targeted L. major. Wild-type BALB/c mice (A and D), TCR Cα−/− BALB/c mice (B), or RAG-2–deficient BALB/c mice (C) were infected in the hind footpad with 2 × 106 lack-targeted L. major using the indicated strains (four to five mice per data point). Lesion development in the left footpad was monitored using calipers. Parasites recovered from the draining popliteal lymph nodes of wild-type BALB/c mice infected for 125 d (E) or RAG-2–deficient BALB/c mice infected for 80 d (F) with the indicated lack-targeted parasites. Bars indicate averaged parasite dilution factors (three to five mice per group). Dilution factors for individual mice are also shown: ♦, L. major lack++/−−; ▴, L. major lack+−/−−; ▪, L. major lack++Δ/−. Similar data (not depicted) were obtained for L. major lack−+/−−.
Because initial activation of the LACK-reactive CD4 T cell response has been associated with susceptibility of BALB/c mice to L. major (2, 3), we next analyzed the early T cell response after infection with the various promastigotes to assess whether LACK deficiency was attenuating virulence by failure to activate LACK-specific CD4 T cells. Our previous analysis determined that the period 96 h after infection corresponded with maximal LACK-reactive T cell expansion in the draining lymph nodes (6). As assessed using LACK peptide/I-Ad MHC class II tetramers, however, no differences were apparent in the expansion of LACK-reactive T cells in the early response to these various lack mutants (Fig. 5) .
Figure 5.

Analysis of the LACK-specific CD4 T cell response to lack-targeted L. major. 2 × 106 wild-type or lack-targeted parasites were inoculated into the hind footpads of BALB/c mice. After 96 h, lymphocytes from the draining popliteal lymph nodes were assayed for expansion of LACK-specific CD4 T cells using MHC class II I-Ad/LACK peptide tetramers as previously described (reference 6). Bars, mean frequencies (two to four mice) of LACK-specific cells in popliteal lymph nodes for the designated parasite strains; •, individual mice.
Next, we used infection of T cell–deficient (TCR-Cα–deficient) BALB/c mice to investigate whether the attenuated phenotype of the single copy mutants was independent of the adaptive T cell immune response. Although lesion development was slowed, even using lack++/−− parasites, consistent with previous observations of experimental leishmaniasis using T cell–deficient animals (38, 39), single copy lack parasites continued to demonstrate a highly attenuated disease phenotype (Fig. 4 B). Comparable data were obtained after inoculation of RAG-2–deficient BALB/c mice that are deficient in both T and B cells (Fig. 4 C). Analysis of parasite burdens in the infected RAG-2–deficient mice showed substantial diminution in the recovery of lack+−/−− parasites as compared with lack++/−− parasites (Fig. 4 F). Thus, as assessed both by lesion size and parasite recovery, lack single copy mutants display a marked deficiency in the capacity to cause progressive infection in vivo, even in highly immunodeficient animals on a genetically susceptible background.
Growth in Macrophages and at Elevated Temperature In Vitro.
The finding that single copy lack parasites displayed impaired virulence suggested that LACK might be required for efficient growth in macrophages, the major target cell for parasitization. To address this issue, bone marrow–derived macrophages were prepared and incubated with the various mutant parasites for 16 h. After washing, the numbers of infected macrophages were similar in comparing the various mutant strains, suggesting that the lack mutations do not affect initial entry into macrophages (Fig. 6 A). After 96 h, however, the percentage of infected macrophages, particularly of macrophages containing large numbers of organisms, was reduced using single copy lack parasites, and this was partially overcome using the reconstituted lack++Δ/– parasites (Fig. 6 B). Microscopically counting the numbers of amastigotes present in macrophages after 96 h of in vitro culture confirmed that single copy mutant parasites essentially did not replicate over a period when lack++/−− organisms had increased almost fourfold (Fig. 6 C).
Figure 6.
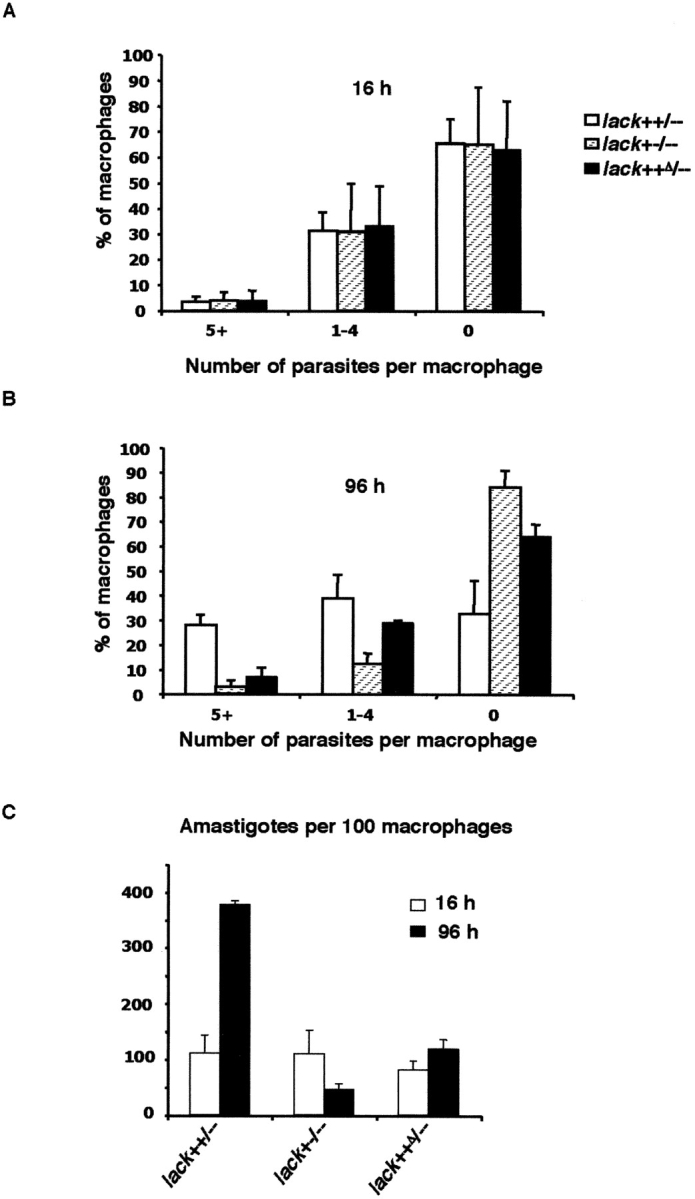
Infection of mouse macrophages with lack-targeted L. major in vitro. BALB/c bone marrow–derived macrophages were incubated with indicated lack-targeted L. major promastigotes using three parasites per macrophage. After 16 h at 35°C, the monolayers were washed to remove extracellular parasites and stained immediately (A), or washed and then incubated for an additional 80 h at 35°C and stained (B). Monolayers were scored for frequency (percent) of macrophages infected with either five or more amastigotes (5+), one to four amastigotes (1–4), or uninfected (0). In C, mean numbers of intracellular amastigotes per 100 macrophages are indicated at 16 and 96 h after infection using the indicated organisms. For each infection, ∼200 macrophages were analyzed. Data are averages from two experiments.
The attenuated development of lesions in vivo and the impaired capacity to infect macrophages in vitro might reflect a general inability to adapt to growth at the elevated temperatures of the vertebrate host. As noted previously, growth of each of these variant parasites was similar at 27°C in vitro, reaching comparable densities by day 6 (Fig. 7 A). When raised to 37°C in vitro, both lack++/−− and lack++Δ/– parasites suffered loss of viability, although organisms reached a maximum density of ∼3 × 106 parasites/ml. Single copy lack+−/−− parasites suffered a greater initial loss of viability after raising the culture temperature, and reached maximal densities approximately twofold lower than double copy organisms (Fig. 7 B). As such, some of the loss of virulence in single copy lack L. major may reflect impaired capacity to grow at elevated temperature.
Figure 7.

Proliferation of lack-targeted promastigotes at 27 and 37°C. lack-targeted promastigotes of the indicated genotypes were cultured at either 27 or 37°C and monitored for proliferation as indicated. Data represent averages from two experiments.
Discussion
Although the importance of WD repeat proteins has been demonstrated in a range of physiologic settings, the functions of these proteins in the context of infectious diseases remain incompletely characterized. The LACK protein of L. major is the only protozoan WD repeat protein for which studies suggest a pathogenic role (2, 3, 5). These studies have implicated an epitope in LACK as the immunodominant focus for the aberrant Th2-mediated susceptibility of BALB/c mice to L. major (2, 3, 5–6). Immune responses to the LACK antigen have been documented in human leishmaniasis (40) and experimental studies have confirmed an immunoprotective role for LACK when used as a vaccine against L. major (8–10). Despite this interest, genetic and biochemical information regarding the role of LACK in the parasite life cycle remains unknown, and thus provoked this study to create LACK-deficient L. major.
RACK-like proteins are highly conserved in eukaryotes, including the kinetoplastids, L. major and Trypanosoma brucei (24). The tandem arrangement of the two lack genes characterized in this study parallels that found for the two L. infantum lack genes, Li36-1 and Li36-2 (23). Similarly, the free-living trypanosomatid, C. fasciculata, contains two copies of a rack homologue, cack (24). The reiterated occurrence of these tandemly paired WD repeat genes might be functionally important in trypanosomatid protozoa. Sequencing of the two L. major lack genes, designated lack1 and lack2, revealed predicted proteins that were identical.
Tandem duplications of homologous genes are common in Leishmania and may represent a mechanism that ensures coordinated production of sufficient protein levels required through the parasite life cycle (41, 42). Indeed, our L. major mutants that lacked one intact, two copy lack allele, were severely attenuated in the mammalian stage of their life cycle. Despite the ability to target and select individual lack genes on each allele, lack-null parasites were never isolated, suggesting, although not formally proving, that LACK is required for parasite viability. A single haploid gene copy, either lack1 or lack2, was sufficient to generate parasites that grew comparably to wild-type L. major promastigotes in vitro, including the capacity to modify surface carbohydrates during stationary phase growth. Despite the requirements for these modifications, termed metacyclogenesis, in establishing intracellular infection, single copy lack1 or lack2 parasites remained essentially avirulent, even when introduced into highly immunodeficient mice, a finding that correlated with attenuated growth and replication as intracellular amastigotes in macrophages in vitro.
The comparable phenotype of single copy lack1 and lack2 parasites, together with the effective rescue of single copy lack1 parasites by replacement of the lack2 coding region in cis with an XhoI-tagged lack coding region, suggests that threshold levels of LACK are required in amastigotes and such thresholds require coordinate regulation across both genes on an allele. The regions flanking the identical lack1 and lack2 coding sequences diverge, suggesting divergent mechanisms for regulating the respective proteins. The inability of single copy lack parasites to thrive in the intracellular habitat may reflect limitations on LACK protein thresholds required to establish parasitism or disruption of regulatory regions responsive to the phagolysosomal environment. In either case, the identical nature of the coding sequences suggests that the defect lies in the levels or regulation of LACK protein, rather than in any qualitative differences in the lack1 or lack2 gene products. The lack1 and lack2 transcripts, as well as LACK protein, were expressed at comparable levels in L. major promastigotes and amastigotes (1 and unpublished data), and LACK protein levels, although not absent, were consistently reduced in the single copy mutants (Fig. 2 A). In contrast to studies with L. infantum and the related kinetoplastid, C. fasciculata, where this WD protein was found to associate to distinct subcellular domains (23, 24), fluorescent imaging of L. major promastigotes expressing a GFP-LACK fusion protein (Fig. 2 B) demonstrated LACK throughout the parasite cytosol. Such generalized cytosolic localization leaves open the possibility that LACK might be involved in a range of physiologic pathways in L. major.
Studies examining the potential role of LACK as a candidate vaccine antigen for leishmaniasis have documented important functions for not only CD4 T cells, but also for CD8 T cells for sustained immunity (10, 43). Despite the wealth of data demonstrating that LACK is a dominant target of the immune response during infection, the single copy lack parasites were highly attenuated in the complete absence of an adaptive immune response. The phenotype of L. major lack+−/−− is similar, at least in part, to the phenotype of lpg2-deficient L. major (44). These mutant L. major fail to persist in sandflies and macrophages, and cause minimal disease when inoculated into mice, but can be recovered in low numbers months after infection, even from asymptomatic mice. Such findings suggest that parasite persistence and pathogenesis are separable events and may reflect residence of parasites in other host cell types, such as fibroblasts (45), in addition to macrophages. Such attenuated organisms may not only provide insights into the mechanisms of parasite persistence, but may have potential for the development of live Leishmania vaccines.
The loss of virulence upon partial loss of LACK protein suggests that drugs capable of impeding the activity of LACK might be highly efficacious in treating leishmaniasis, even if effects on LACK function are incomplete. Further studies of the biochemical pathways affected by LACK will be necessary to delineate fully the role of this protein in Leishmania and other kinetoplastids, but, as confirmed by the genetic studies here, justify the interest in this family of WD repeat proteins as potential targets for therapeutic intervention.
Acknowledgments
The authors thank J.K. Schwartz and S.M. Beverley for the Leishmania codon usage-biased GFP gene; T. Jakobsen for help with densitometry; the University of California San Francisco Biomolecular Resource Center for help with DNA sequencing; N. Flores-Wilson for animal care; E. McEachron for help with DNA cloning; C. MacArthur for flow cytometry expertise; and laboratory members for critically reading the manuscript.
This work is supported by National Institute of Allergy and Infectious Diseases 26918 from the National Institutes of Health and the Howard Hughes Medical Institute. R.M. Locksley is an Ellison Medical Foundation Senior Scholar in Global Infectious Disease.
Abbreviations used in this paper: GFP, green fluorescent protein; PKC, protein kinase C; PNA, peanut agglutinin.
References
- 1.Mougneau, E., F. Altare, A.E. Wakil, S. Zheng, T. Coppola, Z.E. Wang, R. Waldmann, R.M. Locksley, and N. Glaichenhaus. 1995. Expression cloning of a protective Leishmania antigen. Science. 268:563–566. [DOI] [PubMed] [Google Scholar]
- 2.Launois, P., I. Maillard, S. Pingel, K.G. Swihart, I. Xenarios, H. Acha-Orbea, H. Diggelmann, R.M. Locksley, H.R. MacDonald, and J.A. Louis. 1997. IL-4 rapidly produced by Vβ4 Vα8 CD4+ T cells instructs Th2 development and susceptibility to Leishmania major in BALB/c mice. Immunity. 6:541–549. [DOI] [PubMed] [Google Scholar]
- 3.Launois, P., K.G. Swihart, G. Milon, and J.A. Louis. 1997. Early production of IL-4 in susceptible mice infected with Leishmania major rapidly induces IL-12 unresponsiveness. J. Immunol. 158:3317–3324. [PubMed] [Google Scholar]
- 4.Pingel, S., P. Launois, D.J. Fowell, C.W. Turck, S. Southwood, A. Sette, N. Glaichenhaus, J.A. Louis, and R.M. Locksley. 1999. Altered ligands reveal limited plasticity in the T cell response to a pathogenic epitope. J. Exp. Med. 189:1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Julia, V., M. Rassoulzadegan, and N. Glaichenhaus. 1996. Resistance to Leishmania induced by tolerance to a single antigen. Science. 274:421–423. [DOI] [PubMed] [Google Scholar]
- 6.Stetson, D.B., M. Mohrs, V. Mallet-Designe, L. Teyton, and R.M. Locksley. 2002. Rapid expansion and IL-4 expression by Leishmania-specific naïve helper T cells in vivo. Immunity. 17:191–200. [DOI] [PubMed] [Google Scholar]
- 7.Etges, R., J. Bouvier, and C. Bordier. 1986. The major surface protein of Leishmania promastigotes is a protease. J. Biol. Chem. 261:9098–9101. [PubMed] [Google Scholar]
- 8.Tapia, E., E. Perez-Jimenez, L. Lopez-Fuertes, R. Gonzalo, M.M. Gherardi, and M. Esteban. 2003. The combination of DNA vectors expressing IL-12 + IL-18 elicits high protective immune response against cutaneous leishmaniasis after priming with DNA-p36/LACK and the cytokines, followed by a booster with a vaccinia virus recombinant expressing p36/LACK. Microbes Infect. 5:73–84. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalo, R.M., J.R. Rodriguez, D. Rodriguez, G. Gonzalez-Aseguinolaza, V. Larraga, and M. Esteban. 2001. Protective immune response against cutaneous leishmaniasis by prime/booster immunization regimens with vaccinia virus recombinants expressing Leishmania infantum p36/LACK and IL-12 in combination with purified p36. Microbes Infect. 3:701–711. [DOI] [PubMed] [Google Scholar]
- 10.Gurunathan, S., D.L. Sacks, D.R. Brown, S.L. Reiner, H. Charest, N. Glaichenhaus, and R.A. Seder. 1997. Vaccination with DNA encoding the immunodominant LACK parasite antigen confers protective immunity to mice infected with Leishmania major. J. Exp. Med. 186:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neer, E.J., C.J. Schmidt, R. Nambudripad, and T.F. Smith. 1994. The ancient regulatory-protein family of WD-repeat proteins. Nature. 371:297–300. [DOI] [PubMed] [Google Scholar]
- 12.Lambright, D.G., J. Sondek, A. Bohm, N.P. Skiba, H.E. Hamm, and P.B. Sigler. 1996. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 379:311–319. [DOI] [PubMed] [Google Scholar]
- 13.Smith, T.F., C. Gaitatzes, K. Saxena, and E.J. Neer. 1999. The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci. 24:181–185. [DOI] [PubMed] [Google Scholar]
- 14.Ron, D., C.H. Chen, J. Caldwell, L. Jamieson, E. Orr, and D. Mochly-Rosen. 1994. Cloning of an intracellular receptor for protein kinase C: a homolog of the β subunit of G proteins. Proc. Natl. Acad. Sci. USA. 91:839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mochly-Rosen, D., and A.S. Gordon. 1998. Anchoring proteins for protein kinase C: a means for isozyme selectivity. FASEB J. 12:35–42. [PubMed] [Google Scholar]
- 16.Mochly-Rosen, D., B.L. Smith, C.H. Chen, M.H. Disatnik, and D. Ron. 1995. Interaction of protein kinase C with RACK1, a receptor for activated C kinase: a role in beta protein kinase C mediated signal transduction. Biochem. Soc. Trans. 23:596–600. [DOI] [PubMed] [Google Scholar]
- 17.Dell, E.J., J. Connor, S. Chen, E.G. Stebbins, N.P. Skiba, D. Mochly-Rosen, and H.E. Hamm. 2002. The βγ subunit of heterotrimeric G proteins interacts with RACK1 and two other WD repeat proteins. J. Biol. Chem. 277:49888–49895. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez, M.M., D. Ron, K. Touhara, C.H. Chen, and D. Mochly-Rosen. 1999. RACK1, a protein kinase C anchoring protein, coordinates the binding of activated protein kinase C and select pleckstrin homology domains in vitro. Biochemistry. 38:13787–13794. [DOI] [PubMed] [Google Scholar]
- 19.Chang, B.Y., K.B. Conroy, E.M. Machleder, and C.A. Cartwright. 1998. RACK1 a receptor for activated C kinase and a homolog of the beta subunit of G proteins, inhibits activity of src tyrosine kinases and growth of NIH 3T3 cells. Mol. Cell. Biol. 18:3245–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang, B.Y., M. Chiang, and C.A. Cartwright. 2001. The interaction of Src and RACK1 is enhanced by activation of protein kinase C and tyrosine phosphorylation of RACK1. J. Biol. Chem. 276:20346–20356. [DOI] [PubMed] [Google Scholar]
- 21.Liliental, J., and D.D. Chang. 1998. Rack1, a receptor for activated protein kinase C, interacts with integrin beta subunit. J. Biol. Chem. 273:2379–2383. [DOI] [PubMed] [Google Scholar]
- 22.Welburn, S.C., and N.B. Murphy. 1998. Prohibitin and RACK homologues are up-regulated in trypanosomes induced to undergo apoptosis and in naturally occurring terminally differentiated forms. Cell Death Differ. 5:615–622. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez-Aseguinolaza, G., S. Taladriz, A. Marquet, and V. Larraga. 1999. Molecular cloning, cell localization and binding affinity to DNA replication proteins of the p36/LACK protective antigen from Leishmania infantum. Eur. J. Biochem. 259:909–916. [DOI] [PubMed] [Google Scholar]
- 24.Taladriz, S., G. Gonzalez-Aseguinolaza, A. Marquet, and V. Larraga. 1999. Cloning, molecular analysis and differential cell localization of the p36 RACK analogue antigen from the parasite protozoon Crithidia fasciculata. FEBS Lett. 443:375–380. [DOI] [PubMed] [Google Scholar]
- 25.Korchak, H.M., and L.E. Kilpatrick. 2001. Roles for βII-protein kinase C and RACK1 in positive and negative signaling for superoxide anion generation in differentiated HL60 cells. J. Biol. Chem. 276:8910–8917. [DOI] [PubMed] [Google Scholar]
- 26.Tardif, M., M. Savard, L. Flamand, and J. Gosselin. 2002. Impaired protein kinase C activation/translocation in Epstein-Barr virus-infected monocytes. J. Biol. Chem. 277:24148–24154. [DOI] [PubMed] [Google Scholar]
- 27.Pingel, S., Z.E. Wang, and R.M. Locksley. 1998. Distribution of protein kinase C isoforms after infection of macrophages with Leishmania major. Infect. Immun. 66:1795–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joshi, P.B., D.L. Sacks, G. Modi, and W.R. McMaster. 1998. Targeted gene deletion of Leishmania major genes encoding developmental stage-specific leishmanolysin (GP63). Mol. Microbiol. 27:519–530. [DOI] [PubMed] [Google Scholar]
- 29.Joshi, P.B., B.L. Kelly, S. Kamhawi, D.L. Sacks, and W.R. McMaster. 2002. Targeted gene deletion in Leishmania major identifies leishmanolysin (GP63) as a virulence factor. Mol. Biochem. Parasitol. 120:33–40. [DOI] [PubMed] [Google Scholar]
- 30.Ha, D.S., J.K. Schwartz, S.J. Turco, and S.M. Beverley. 1996. Use of the green fluorescent protein as a marker in transfected Leishmania. Mol. Biochem. Parasitol. 77:57–64. [DOI] [PubMed] [Google Scholar]
- 31.Medina-Acosta, E., and G.A.M. Cross. 1993. Rapid isolation of DNA from trypanosomatid protozoa using a simple ‘mini-prep’ procedure. Mol. Biochem. Parasitol. 59:327–329. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 545 pp.
- 33.Fowell, D.J., K. Shinkai, X.C. Liao, A.M. Beebe, R.L. Coffman, D.R. Littman, and R.M. Locksley. 1999. Impaired NFATc translocation and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity. 11:399–409. [DOI] [PubMed] [Google Scholar]
- 34.Philpott, K.L., J.L. Viney, G. Kay, S. Rastan, E.M. Gardiner, S. Chae, A.C. Hayday, and M.J. Owen. 1992. Lymphoid development in mice congenitally lacking T cell receptor αβ-expressing cells. Science. 256:1448–1452. [DOI] [PubMed] [Google Scholar]
- 35.Descoteaux, A., G. Matlashewski, and S.J. Turco. 1992. Inhibition of macrophage protein kinase C-mediated protein phosphorylation by Leishmania donovani lipophosphoglycan. J. Immunol. 149:3008–3015. [PubMed] [Google Scholar]
- 36.Miller, S.I., and D.F. Wirth. 1988. Trans-splicing in Leishmania enriettii and identification of ribonucleoprotein complexes containing the spliced leader and U2 equivalent RNAs. Mol. Cell. Biol. 8:2597–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacks, D.L., T.N. Brodin, and S.L. Turco. 1990. Developmental modification of the lipophosphoglycan from Leishmania major promastigotes during metacyclogenesis. Mol. Biochem. Parasitol. 42:225–234. [DOI] [PubMed] [Google Scholar]
- 38.Soong, L., C.H. Chang, J. Sun, B.J. Longley, Jr., N.H. Ruddle, R.A. Flavell, and D. McMahon-Pratt. 1997. Role of CD4+ T cells in pathogenesis associated with Leishmania amazonensis infection. J. Immunol. 158:5374–5383. [PubMed] [Google Scholar]
- 39.Belkaid, Y., S. Mendez, R. Lira, N. Kadambi, G. Milon, and D. Sacks. 2000. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 165:969–977. [DOI] [PubMed] [Google Scholar]
- 40.Bourreau, E., G. Prevot, J. Gardon, R. Pradinaud, H. Hasagewa, G. Milon, and P. Launois. 2002. LACK-specific CD4(+) T cells that induce gamma interferon production in patients with localized cutaneous leishmaniasis during an early stage of infection. Infect. Immun. 70:3122–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelly, B.L., S.D. Dyall, J.P. Warner, J. Tang, and D.F. Smith. 1995. Chromosomal organization of a repeated gene cluster expressed in mammalian stages of Leishmania. Gene. 163:145–149. [DOI] [PubMed] [Google Scholar]
- 42.Voth, B.R., B.L. Kelly, P.B. Joshi, A.C. Ivens, and W.R. McMaster. 1998. Differentially expressed Leishmania major gp63 genes encode cell surface leishmanolysin with distinct signals for glycosylphosphatidylinositol attachment. Mol. Biochem. Parasitol. 93:31–41. [DOI] [PubMed] [Google Scholar]
- 43.Gurunathan, S., L. Stobie, C. Prussin, D.L. Sacks, N. Glaichenhaus, A. Iwasaki, D.J. Fowell, R.M. Locksley, J.T. Chang, C.Y. Wu, et al. 2000. Requirements for the maintenance of Th1 immunity in vivo following DNA vaccination: a potential immunoregulatory role for CD8+ T cells. J. Immunol. 165:915–924. [DOI] [PubMed] [Google Scholar]
- 44.Späth, G.F., L. Lye, H. Segawa, D.L. Sacks, S.J. Turco, and S.M. Beverley. 2003. Persistence without pathology in phosphoglycan-deficient Leishmania major. Science. 301:1241–1243. [DOI] [PubMed] [Google Scholar]
- 45.Bogdan, C., N. Donhauser, R. Döring, M. Röllinghoff, A. Diefenbach, and M.G. Rittig. 2000. Fibroblasts as host cells in latent leishmaniosis. J. Exp. Med. 191:2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]