

Abstract
Two models have been proposed to explain the requirement for CD40 signaling in CD8 T cell responses. The first model suggests that CD4 T cells activate antigen-presenting cells (APCs) through CD40 signaling (APC licensing). In turn, licensed APCs are able to prime naive CD8 T cells. The second model suggests that CD154-expressing CD4 T cells activate CD40-bearing CD8 T cells directly. Although the requirement for CD40 in APC licensing can be bypassed by inflammatory responses to pathogens that activate APCs directly, the second model predicts that CD8 responses to all antigens will be dependent on CD40 signaling. Here we determined which model applies to CD8 responses to influenza. We demonstrate that optimal CD8 T cell responses to influenza are dependent on CD40 signaling, however both primary and secondary responses to influenza require CD40 expression on non–T cells. Furthermore, CD40−/− CD8 T cells proliferate and differentiate to the same extent as CD40+/+ CD8 T cells in response to influenza, as long as they have equal access to CD40+/+ APCs. Thus, CD4 T cells do not activate influenza-specific CD8 cells directly through CD40 signaling. Instead, these data support the classical model, in which CD4 T cells provide help to CD8 T cells indirectly by activating APCs through CD40.
Keywords: CD40, influenza, CD8 T cell, memory
Introduction
The interaction of CD40 on B cells with its ligand, CD154, on CD4 T cells is an essential step in generating effective humoral immune responses to T-dependent antigens (1). CD40 signaling also regulates the response of CD8 T cells to a variety of antigens (2–5). For example, systemic administration of CD40 agonists in vivo substitutes for CD4 T cell help (3–5), augments CD8 responses to weak antigens (6), enables cross-priming of CD8 cells to soluble antigens (5), and converts tolerogenic antigens into immunogenic antigens (7, 8). The adjuvant-like effects of systemic CD40 signaling are thought to be mediated through the activation of CD40-bearing APCs (3), which up-regulate costimulatory ligands and produce inflammatory cytokines in response to CD40 triggering (9, 10). Although the activation of APCs by CD40 provides a plausible mechanism for the effects of systemic CD40 signaling on CD8 T cell responses, this explanation was recently challenged by data showing that CD4 T cells provide help directly to CD40-bearing CD8 T cells (2). These data suggest that the development of memory CD8 T cells is similar to the development of memory B cells, in which CD40 signaling initiates a cell-autonomous program of differentiation leading to the formation of functional memory cells (11).
Interestingly, CD8 T cell responses to bacterial or viral antigens do not absolutely require CD40 signaling (12–16), possibly due to the inflammatory nature of the pathogens, which directly activate APCs through Toll-like receptors. However, despite the ability of inflammatory antigens to activate APCs in a CD40-independent manner, CD8 responses to viral or bacterial antigens are impaired in the absence of CD40 signaling (12–16). Consistent with this, recent data shows that the development of CD8 memory cells to Listeria or vaccinia antigens is dependent on CD4 T cell help even though these pathogens induce strong inflammatory responses (17–19). Although the mechanism of CD4 help was not elucidated, it is possible that CD154–CD40 interactions between CD4 and CD8 T cells may play a role. If this is true, then CD40−/− CD8 T cells should be unable to differentiate into effective memory cells, regardless of the nature of the antigen and regardless of whether APCs express CD40.
In this report we used BM chimeric mice, in which CD40 expression is absent from all hematopoietic cells or is specifically absent from the T cell compartment, to demonstrate that CD40 expression on non–T cells, but not on T cells, is required for normal CD8 responses to influenza. Furthermore, when compared side by side, CD40+/+ and CD40−/− CD8 T cells exhibited equivalent proliferative and functional abilities in both primary and secondary responses to influenza. This is in stark contrast to CD40−/− B cells, which were intrinsically defective and did not proliferate or differentiate in response to antigen. Thus, unlike B cells, which have a direct requirement for CD40 signaling, CD8 T cells require CD40 signaling on non–T cells for both primary and secondary responses to influenza. These results are consistent with a role for CD40 signaling in APC licensing rather than a role for direct CD40 signaling to CD8 T cells.
Materials and Methods
Mice, BM Chimeras, and Infections.
CD154−/− mice, TCRβ−/− δ−/− mice, and Ly5.1 mice were purchased from The Jackson Laboratory. C57BL/6 (WT) and CD40−/− mice were bred at the Trudeau Institute. Recipient TCRβ−/−δ−/− mice or Ly5.1 mice were irradiated (950 rads) and reconstituted with BM as described in Results and Discussion. Mice were allowed to reconstitute for at least 6 wk before infection. Primary infections were performed intranasally with 100 egg infectious units of influenza A/PR8/34 (PR8) in 100 μl. Secondary infections were performed intranasally with A/HK-x31 (X31) at 5,000 egg infectious units in 100 μl. All experimental procedures involving animals were approved by the Trudeau Institute's Institutional and Animal Care Use Committee and performed according to the guidelines set by the National Research Council.
Antibodies, Intracellular Cytokine Staining, and Flow Cytometry.
Tissues were disrupted by passage through wire mesh and live leukocytes were obtained by density gradient centrifugation using Lympholyte-Poly (Cedarlane). Cells were incubated in 3% FCS in PBS containing 10 μg/ml 2.4G2 to block Fc receptor binding, followed by the addition of fluorochrome-conjugated antibodies or MHC class I tetramers. The H-2Db tetramer containing influenza nucleoprotein (NP)366–374 peptide was generated by the Trudeau Institute's Molecular Biology Core Facility. For intracellular cytokine staining, cells were cultured at 5 × 106/ml in 200 μl complete RPMI containing 10 μg/ml brefeldin A and 10 U/ml IL-2 and stimulated with NP366–374 peptide at 1 μg/ml for 5 h. Simulated cells were then washed, blocked with 2.4G2, and probed with anti-CD8 before fixation in 4% paraformaldehyde in PBS. Fixed cells were washed with 0.1% Triton X-100, 3% FCS in PBS, and probed with FITC-labeled anti-IFNγ in the same buffer. Antibodies were purchased from BD Biosciences. FITC-conjugated peanut agglutinin was purchased from Sigma-Aldrich. Flow cytometry was performed on a dual laser FACSCalibur™ available through the Flow Cytometry Core Facility at the Trudeau Institute.
Results and Discussion
The CD8 T Cell Response to Influenza Is CD40 Dependent.
To determine whether CD40 is required for CD8 responses to influenza, WT and CD40−/− mice were infected with PR8 and the accumulation of influenza-specific T cells in the lungs was assayed by flow cytometry. As shown in Fig. 1 A, the frequency of NP-specific CD8 T cells was lower in the lungs of CD40−/− mice relative to WT mice at day 9 and dramatically lower at day 12 (Fig. 1 A). The frequency of CD8 T cells that produced IFNγ in response to NP peptide was also reduced in CD40−/− mice (Fig. 1 B). As shown in Fig. 1 C, the number of NP-specific tetramer-binding T cells as well as the number of NP-specific, IFNγ-producing CD8 cells was about twofold lower in CD40−/− mice than in WT mice on day 9. This difference was more dramatic on day 12, when the numbers of NP-specific tetramer-binding and IFNγ-producing CD8 T cells in the lungs of CD40−/− mice had dropped and were about eightfold lower than those in the lungs of WT mice. Similar differences were observed in spleen and draining LN. Thus, the primary CD8 T cell response to influenza is progressively impaired in CD40−/− mice compared with WT mice.
Figure 1.

CD40 is required for optimal CD8 T cell responses to influenza. (A) CD40−/− and WT mice were infected with PR8 and cells from the lungs were analyzed on days 9 and 12. The percentage of NP-specific CD8 cells is indicated. (B) Cells from the lungs of infected animals were stimulated with NP366–374 peptide and the production of IFNγ was measured by intracellular staining. The percentage of IFNγ+ CD8 cells is indicated. (C) The total numbers of NP-specific, tetramer-binding CD8 cells (top) and NP-specific, IFNγ-producing CD8 cells (bottom) are shown for both WT mice (black bars) and CD40−/− mice (gray bars). This experiment contained five mice per group per time point and was performed three times.
Generation of BM Chimeric Mice.
To determine whether CD40 or CD154 are required on T cells for normal CD8 responses, we generated BM chimeric mice, in which T cells selectively lacked CD40 or CD154. In the first group (Fig. 2 A), TCRβ−/−δ−/− mice were reconstituted with a mix of TCRβ−/−δ−/− BM and C57BL/6 BM. As shown in the dot plots of Fig. 2 A, the majority of B cells and DCs in these chimeras, known as WT chimeras, express CD40. In the second group (Fig. 2 B), TCRβ−/−δ−/− mice were reconstituted with CD40−/− BM. As shown in the dot plots of Fig. 2 B, the majority of B cells and DCs in these mice, known as CD40-KO chimeras, lack CD40. In the third group (Fig. 2 C), TCRβ−/−δ−/− mice were reconstituted with a mix of TCRβ−/−δ−/− BM and CD40−/− BM. Because the TCRβ−/−δ−/− BM is genetically unable to produce T cells, all the T cells in the recipient mice must be derived from the CD40−/− donor BM. In contrast, the majority of non–T cells in these mice are derived from the TCRβ−/−δ−/− donor BM and are CD40+/+. As shown in the dot plots of Fig. 2 C, 90% of the B cells and 76% of the DCs express CD40 in these mice, which are known as CD40-T cell KO (CD40-TKO chimeras). In the final group (Fig. 2 D), TCRβ−/−δ−/− mice were reconstituted with a mix of TCRβ−/−δ−/− BM and CD154−/− BM. Because the TCRβ−/−δ−/− BM cannot produce T cells, all the T cells in the chimeric mice are derived from the CD154−/− BM, whereas the majority of non–T cells are derived from the CD154+/+ TCRβ−/−δ−/− BM. As expected, the B cells and DCs in these mice, known as CD154-TKO chimeras, express CD40.
Figure 2.

Generation of chimeric mice. Irradiated TCRβ−/−δ−/− recipient mice were reconstituted with (A) TCRβ−/−δ−/− BM and C57BL/6 BM, (B) CD40−/− BM, (C) TCRβ−/−δ−/− BM and CD40−/− BM, or (D) TCRβ−/−δ−/− BM and CD154−/− BM. The proportion of B cells, DCs, and T cells of each genotype that are expected to repopulate the chimeras is indicated. Cells from the LNs (top) and lungs (bottom) of chimeric mice were analyzed on day 10 after infection to determine the actual frequency of CD40-expressing B cells and DCs. The percentage of B cells or DCs expressing CD40 is indicated.
Given that CD40 signaling is important for humoral immunity (1), we first tested whether the loss of CD40 or CD154 on T cells affected the B cell response. Therefore, we infected chimeric mice with influenza and assayed the frequency of responding B cells on day 10 after infection. As shown in Fig. 3 A, left, a high frequency of germinal center (GC) B cells was observed in the draining LNs of WT and CD40-TKO chimeras on day 10 after infection, whereas very few GC B cells were observed in the LNs of CD40-KO or CD154-TKO chimeric mice. The frequencies of CD138+ antibody-secreting plasma cells and IgG2 isotype-switched cells were also much higher in the WT and CD40-TKO chimeras than in the CD40-KO or CD154-TKO chimeras (Fig. 3 A). Thus, the lack of CD40 on all cells (CD40-KO mice) or the lack of CD154 exclusively on T cells (CD154-TKO mice) leads to impaired B cell responses to influenza. However, the loss of CD40 on T cells did not adversely affect the B cell response.
Figure 3.

Optimal CD8 T cell responses to influenza require CD40 expression on hematopoietic cells, but not on T cells. (A) Chimeric mice were infected with PR8 and the frequency of PNAhi FAShi GC B cells (left) as well as the frequency of CD138hi plasma cells and IgG2+ B cells (right) in the draining LNs of chimeric mice was analyzed on day 10. All plots show cells gated on CD19 expression. The numbers refer to the percentage of B cells that have the indicated phenotype. (B) NP-specific CD8 T cells were enumerated on days 10 and 49. Memory mice were infected on day 50 with X31, and the responding memory CD8 T cells were analyzed on day 56. All plots show cells gated on CD8 expression. The numbers refer to the percentage of NP-specific CD8 cells. (C) The total numbers of NP-specific CD8 T cells in the lungs of chimeric mice were calculated for each time point. This experiment contained five mice per group per time point and was performed two times. The samples in A are from the same mice as the day 10 samples in B.
Optimal CD8 Responses Require CD40 Expression on Hematopoietic Cells, but Not on T Cells.
We also assayed the frequency of NP-specific T cells in the influenza-infected chimeric mice on day 10 (the peak of the primary response), at day 49 (resting memory), and at day 56, which is 6 d after a challenge infection with the heterotypic influenza virus, X31. As shown in Fig. 3 B, numerous NP-specific CD8 T cells were observed in WT and CD40-TKO chimeric mice on day 10, whereas fewer of these cells were observed in CD40-KO and CD154-TKO chimeric mice at this time. A similar trend was observed at days 49 and 56, which is 6 d after challenge. As shown in Fig. 3 C, the number of NP-specific T cells in CD40-KO and CD154-TKO mice was greatly reduced compared with those in WT mice. In contrast, the number of NP-specific T cells in CD40-TKO mice was only modestly reduced relative to those in WT mice. Thus, it appears that the loss of CD40 on all hematopoietic cell types (CD40-KO mice) and the loss of CD154 on T cells leads to reduced CD8 responses (Fig. 3 B), however the loss of CD40 expression on T cells only (CD40-TKO) had only a minor effect on primary or secondary CD8 responses to influenza.
The slightly reduced CD8 response in the CD40-TKO mice relative to that in WT mice (Fig. 3 B) could reflect an intrinsic defect in CD40−/− T cells, however it could also be related to the lack of CD40 expression on 20% of the APCs in CD40-TKO mice (Fig. 2 C). To address this, we next generated BM chimeras in which the response of CD40+/+ lymphocytes could be compared with CD40−/− lymphocytes side by side in the same hosts and each population would have equal access to CD40-bearing APCs and CD154-expressing CD4 T cells. Thus, we reconstituted WT mice with a 50:50 mix of BM from Ly5.1+ WT donors and Ly5.2+ CD40−/− donors. As a control, we reconstituted WT mice with a 50:50 mix of BM from Ly5.1+ WT donors and Ly5.2+ WT donors. The chimeric mice were infected with influenza and the response of CD40+/+ and CD40−/− B and T cells was measured. As expected, the total CD19+ B cell population in the LNs of chimeric mice is composed of B cells from both CD40+/+ and CD40−/− donors (Fig. 4 A). In addition, both the WT/WT chimeras and the CD40−/−/WT chimeras generated similar frequencies of GC B cells after primary influenza infection (Fig. 4 B). However, although the GC B cell population from the CD40−/−/WT chimeras consisted exclusively of cells from the WT donor (Fig. 4 C, left), the GC B cell population from the WT/WT chimera consisted of cells from both Ly5.1 and Ly5.2 genotypes (Fig. 4 C, right). Similarly, although the CD138+ antibody-secreting plasma cells generated in the CD40−/−/WT chimeras were almost exclusively derived from WT donor cells, the CD138+ antibody-secreting plasma cells in the WT/WT chimera were derived from cells of both donors (Fig. 4 D). Thus, the generation of effector B cells is strongly dependent on CD40 expression on B cells.
Figure 4.
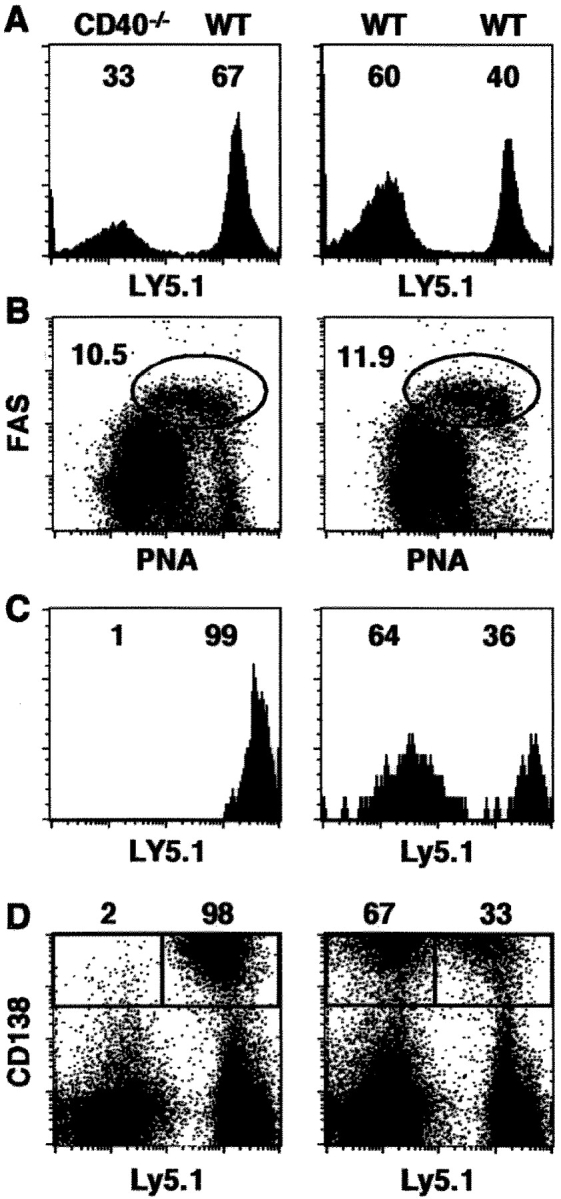
B cells require CD40 expression to respond to influenza. Chimeric mice were infected with PR8 and the B cell response in the LNs was analyzed on day 10. (A) The contribution of Ly5.1 and Ly5.2 donor cells to the total CD19+ B cell population is shown. (B) GC B cells were identified by coexpression of peanut agglutinin and FAS. The percentage of B cells with a GC phenotype is indicated next to the circled areas. (C) The contribution of Ly5.1 and Ly5.2 donor cells to the GC B cell population is shown. (D) The contribution of Ly5.1 and Ly5.2 donor cells to the CD138+ plasma cell population is shown. This experiment contained five mice per group and was performed two times.
Next, we examined the response of influenza-specific CD8 T cells in the chimeric mice after influenza infection. As shown in Fig. 5 A, the total CD8 T cell population in the spleens and lungs of these chimeras was split nearly equally between the cells from the Ly5.1+ and Ly5.2+ donors at day 10 of the primary response, day 49 of the resting memory response, and at day 56, which is 6 d after challenge. In addition, the NP-specific CD8 T cells were also derived nearly equivalently from both the WT and CD40−/− donor cell types at each time (Fig. 5 B). Thus, there was no preferential expansion of WT cells over CD40−/− cells at any time as would be expected if CD40 signaling directly to CD8 T cells was important in this response. Furthermore, as shown in Fig. 5 C, the ability of NP-specific memory CD8 T cells to produce IFNγ upon restimulation was independent of whether the cells were derived from the CD40+/+ or CD40−/− donors. Therefore, the function of NP-specific CD40+/+ and CD40−/− memory CD8 T cells is equivalent when they are primed in the same environment, indicating that the generation of influenza-specific effector and memory CD8 T cells is entirely independent of CD40 expression on CD8 T cells.
Figure 5.
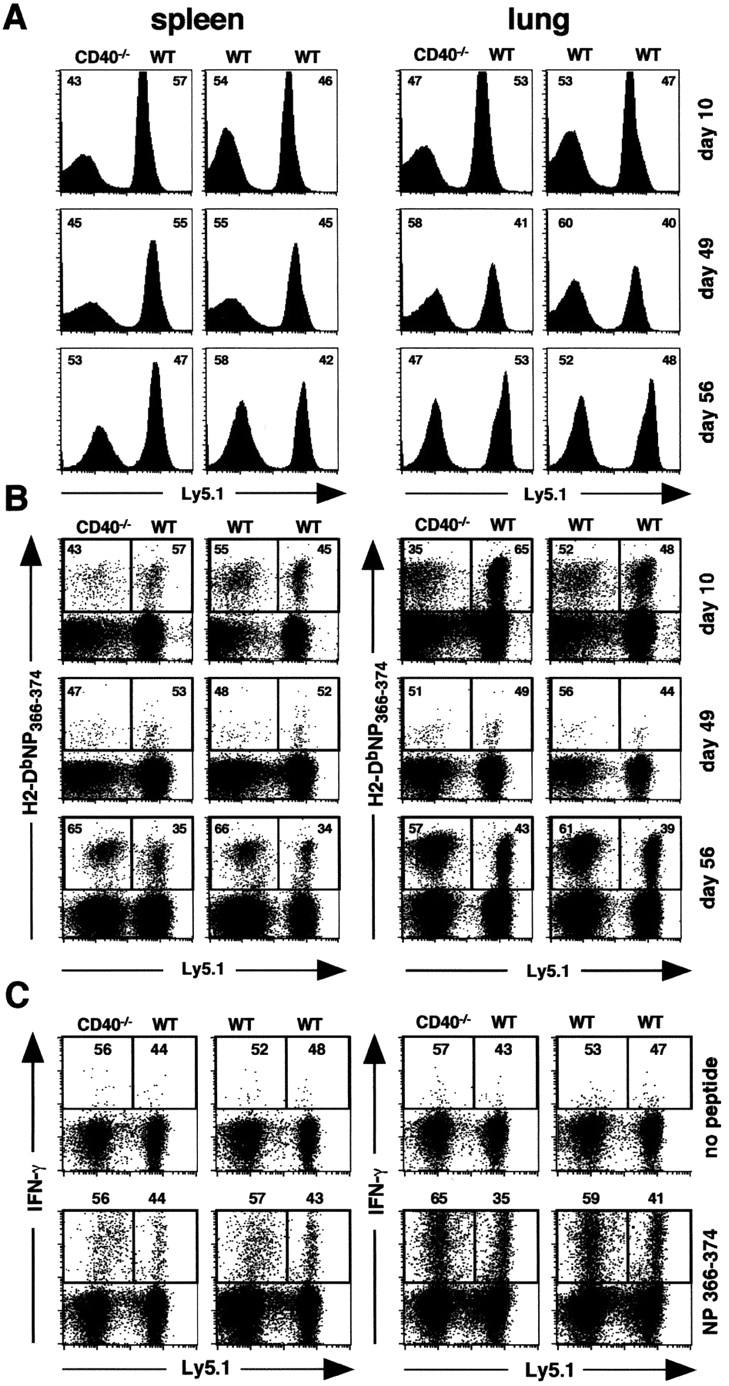
CD40−/− CD8 T cells develop into memory cells with normal proliferative and functional capacity. Chimeric mice were infected with PR8 on day 0 and analyzed on days 10 and 49. Memory mice were infected with X31 on day 50 and analyzed on day 56. (A) T cells were gated on CD8 and the contribution of Ly5.1 and Ly5.2 cells to the CD8 population was determined by Ly5.1 expression. The numbers in the upper corners of each plot refer to the percentage of total CD8 cells that are either LY5.1+ or Ly5.2+. (B) The contribution of Ly5.1 and Ly5.2 cells to the NP tetramer-binding CD8 population was determined by Ly5.1 expression. The numbers in the upper corners of each plot refer to the percentage of NP-specific CD8 cells that are either LY5.1+ or Ly5.2+. (C) The contribution of Ly5.1 and Ly5.2 cells to the NP-specific, IFNγ-producing CD8 population on day 56 was determined by Ly5.1 expression. The numbers in the upper corners of each plot refer to the percentage of NP-specific CD8 cells that are derived from Ly5.2 donor (left) and the Ly5.1 donor (right). This experiment contained five mice per group per time point and was performed two times. The day 10 samples in this experiment are from the same mice as the samples in Fig. 4.
These data show that although CD8 responses to influenza are facilitated by CD40 signaling, CD40 expression is not required on CD8 T cells. In fact, CD40+/+ and CD40−/− CD8 T cells make equivalent primary and secondary CD8 T cell responses when cells of both genotypes have equal access to CD40+/+ APCs. Consistent with conventional models of T cell–APC interactions (20), we find that CD8 responses to influenza require CD40 expression on non–T cells and CD154 expression on T cells. Thus, our results are consistent with a role for CD40 signaling in APC activation, but not with a role for CD40 signaling directly to CD8 T cells.
One obvious difference between our studies and the studies that showed a requirement for CD40 expression on T cells is the nature of the antigen to which the CD8 T cells are responding. The studies of Bourgeois et al. (2) examined CD8 responses to the male HY antigen, which is not intrinsically inflammatory. In contrast, influenza induces an acute inflammatory response and has the ability to directly induce the maturation of DCs into potent APCs (3, 21, 22). Thus, the requirement for CD40 signaling directly to CD8 T cells might be bypassed by the production of inflammatory cytokines that act directly on CD8 cells. Despite this possibility, our data show that primary and secondary CD8 responses to influenza are impaired in the absence of CD40. This is consistent with a variety of reports demonstrating that CD8 responses to lymphocytic choriomeningitis virus (12, 13), recombinant vaccinia virus (14), recombinant adeno-associated virus (15), or antigens coadministered with Listeria (16) are also impaired in the absence of CD40 signaling, even though each of these pathogens triggers a strong inflammatory response. Thus, the inflammatory nature of a particular antigen does not entirely compensate for the absence of CD40 signaling. In fact, CD40 signaling appears to provide a uniquely potent adjuvant-like effect that cannot be mimicked by inflammatory mediators, such as LPS (23). Although it is possible that this adjuvant-like effect could be due to CD40 signaling directly to CD8 T cells in some systems, our data show that in response to influenza, the effects of CD40 signaling on CD8 responses are mediated indirectly through CD40 signals to non–T cells.
In summary, we find that although CD154–CD40 interactions are important for CD8 T cell responses to influenza, CD40 signaling directly to CD8 T cells is not required for the generation of primary effector CD8 T cells or for the development and function of memory CD8 T cells. In contrast, CD40 signaling on non–T cells is necessary for optimal CD8 T cell responses. These data are consistent with the model of APC licensing (3–5), in which CD4 help for CD8 responses is provided indirectly, rather than the model proposed by Bourgeois et al. (2), in which CD4 T cells signal to CD8 T cells directly through CD40–CD154 interactions.
Acknowledgments
The authors wish to thank Dr. Frances E. Lund and Dr. David L. Woodland for helpful discussions.
This work was supported in part by National Institutes of Health grant AI-43589 to T.D. Randall and by the Trudeau Institute.
References
- 1.Kawabe, T., T. Naka, K. Yoshida, T. Tanaka, H. Fujiwara, S. Suematsu, N. Yoshida, T. Kishimoto, and H. Kikutani. 1994. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1:167–178. [DOI] [PubMed] [Google Scholar]
- 2.Bourgeois, C., B. Rocha, and C. Tanchot. 2002. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 297:2060–2063. [DOI] [PubMed] [Google Scholar]
- 3.Ridge, J.P., F. Di Rosa, and P. Matzinger. 1998. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer. Nature. 393:474–477. [DOI] [PubMed] [Google Scholar]
- 4.Schoenberger, S.P., R.E.M. Toes, E.I.H. van der Voort, R. Offringa, and C.J.M. Melief. 1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 393:480–483. [DOI] [PubMed] [Google Scholar]
- 5.Bennett, S.R.M., F.R. Carbone, F. Karamalis, R.A. Flavell, J.F.A.P. Miller, and W.R. Heath. 1998. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 393:478–480. [DOI] [PubMed] [Google Scholar]
- 6.Rolph, M.S., and S.H. Kaufmann. 2001. CD40 signaling converts a minimally immunogenic antigen into a potent vaccine against the intracellular pathogen Listeria monocytogenes. J. Immunol. 166:5115–5121. [DOI] [PubMed] [Google Scholar]
- 7.Sotomayor, E.M., I. Borrello, E. Tubb, F.M. Rattis, H. Bien, Z. Lu, S. Fein, S. Schoenberger, and H.I. Levitsky. 1999. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 5:780–787. [DOI] [PubMed] [Google Scholar]
- 8.Diehl, L., A.T. den Boer, S.P. Schoenberger, E.I. van der Voort, T.N. Schumacher, C.J. Melief, R. Offringa, and R.E. Toes. 1999. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat. Med. 5:774–779. [DOI] [PubMed] [Google Scholar]
- 9.Caux, C., C. Massacrier, B. Vanbervliet, B. Dubois, C. Van Kooten, I. Durand, and J. Banchereau. 1994. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 180:1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koch, F., U. Stanzl, P. Jennewein, K. Janke, C. Heufler, E. Kampgen, N. Romani, and G. Schuler. 1996. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J. Exp. Med. 184:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanchot, C., and B. Rocha. 2003. CD8 and B cell memory: same strategy, same signals. Nat. Immunol. 4:431–432. [DOI] [PubMed] [Google Scholar]
- 12.Borrow, P., A. Tishon, S. Lee, J. Xu, I.S. Grewal, M.B. Oldstone, and R.A. Flavell. 1996. CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. J. Exp. Med. 183:2129–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borrow, P., D.F. Tough, D. Eto, A. Tishon, I.S. Grewal, J. Sprent, R.A. Flavell, and M.B. Oldstone. 1998. CD40 ligand-mediated interactions are involved in the generation of memory CD8(+) cytotoxic T lymphocytes (CTL) but are not required for the maintenance of CTL memory following virus infection. J. Virol. 72:7440–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu, Z., L. Yuan, X. Zhou, E. Sotomayor, H.I. Levitsky, and D.M. Pardoll. 2000. CD40-independent pathways of T cell help for priming of CD8+ cytotoxic T lymphocytes. J. Exp. Med. 191:541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang, Y., N. Chirmule, G. Gao, and J. Wilson. 2000. CD40 ligand-dependent activation of cytotoxic T lymphocytes by adeno-associated virus vectors in vivo: role of immature dendritic cells. J. Virol. 74:8003–8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamilton, S.E., A.R. Tvinnereim, and J.T. Harty. 2001. Listeri monocytogenes infection overcomes the requirement for CD40 ligand in exogenous antigen presentation to CD8 T cells. J. Immunol. 167:5603–5609. [DOI] [PubMed] [Google Scholar]
- 17.Shedlock, D.J., and H. Shen. 2003. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 300:337–339. [DOI] [PubMed] [Google Scholar]
- 18.Sun, J.C., and M.J. Bevan. 2003. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 300:339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janssen, E.M., E.E. Lemmens, T. Wolfe, U. Christen, M.G. von Herrath, and S.P. Schoenberger. 2003. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 421:852–856. [DOI] [PubMed] [Google Scholar]
- 20.Mackey, M.F., R.J. Barth, Jr., and R.J. Noelle. 1998. The role of CD40/CD154 interactions in the priming, differentiation, and effector function of helper and cytotoxic T cells. J. Leukoc. Biol. 63:418–428. [DOI] [PubMed] [Google Scholar]
- 21.Cella, M., F. Facchetti, A. Lanzavecchia, and M. Colonna. 2000. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat. Immunol. 1:305–310. [DOI] [PubMed] [Google Scholar]
- 22.Brimnes, M.K., L. Bonifaz, R.M. Steinman, and T.M. Moran. 2003. Influenza virus–induced dendritic cell maturation is associated with the induction of strong T cell immunity to a coadministered, normally nonimmunogenic protein. J. Exp. Med. 198:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelleher, M., and P.C. Beverley. 2001. Lipopolysaccharide modulation of dendritic cells is insufficient to mature dendritic cells to generate CTLs from naive polyclonal CD8+ T cells in vitro, whereas CD40 ligation is essential. J. Immunol. 167:6247–6255. [DOI] [PubMed] [Google Scholar]