

Abstract
T cell recruitment to elicit contact sensitivity (CS) requires a CS-initiating process mediated by B-1 cells that produce IgM, which activates complement to promote T cell passage into the tissues. We now show that Vα14i NKT cells induce B-1 cell activation likely by releasing IL-4 early postimmunization. The CS initiation process is absent in Jα18−/− and CD1d−/− NKT cell–deficient mice and is reconstituted by populations enriched for Vα14i NKT cells. Transfers are not effective if cells are derived from IL-4−/− mice. Staining with specific tetramers directly showed that hepatic Vα14i NKT cells increase by 30 min and nearly double by 2 h postimmunization. Transfer of immune B-1 cells also reconstitutes CS responses in NKT cell–deficient mice. The B-1 cells act downstream of the Vα14i NKT cells to restore CS initiation. In addition, IL-4 given systemically to Jα18−/− or CD1d−/− NKT cell–deficient mice reconstitutes elicitation of CS. Further, splenocytes from immune Jα18−/− mice produce less antigen (Ag)-specific IgM antibodies compared with sensitized WT mice. Together these findings indicate that very early after skin immunization Vα14i NKT cells are stimulated to produce IL-4, which activates B-1 cells to produce Ag-specific IgM, subsequently needed to recruit effector T cells for elicitation of CS responses.
Keywords: Vα14i NKT cells, B-1 cells, contact sensitivity, liver lymphocytes, IL-4
Introduction
Contact sensitivity (CS) is an example of delayed-type hypersensitivity (DTH) induced by haptens that conjugate to self-protein Ags and peptides in the skin. CS is mediated by local extravascular recruitment of Ag-specific circulating CS effector T cells that cause inflammatory tissue swelling, peaking 24 h after secondary skin Ag challenge.
To locally recruit sensitized T cells, an early 2-h initiating response is required (1). This initiating response is due to binding of Ag to specific IgM antibodies produced rapidly postimmunization by B-1 B cells (2). Local Ag–IgM complexes are generated leading to complement activation to initiate elicitation of CS by locally generating C5a to activate receptors (3) on mast cells (4, 5) and platelets (6, 7), resulting in release of vasoactive TNF-α (3, 8) and serotonin (4, 6, 9, 10). This stimulates endothelial expression of adhesion molecules (ICAM-1 and VCAM-1) (8) needed to recruit circulating CS effector T cells (11). These early events of elicitation are called “CS initiation,” since they are required to recruit CS effector T cells to mediate the 24-h component of CS.
NKT cells participate early in immune responses, expressing NK receptors and markers and a unique αβ-TCR (12). Mice dominantly express a conserved αβ-TCR with invariant Vα14Jα18 (formerly Jα281) paired mainly with Vβ 8.2, Vβ7, or Vβ2 chains (13). They are herein referred to as Vα14i NKT cells or iNKT cells, to distinguish from other NKT cells, and represent 30–40% of liver αβ-T cells but smaller numbers in lymphoid organs (12). iNKT cells are restricted to recognizing Ags bound in the MHC class I–like molecule CD1d expressed with β2 microglobulin on various APC (14). Ligand Ags are glycolipids such as α-galactosylceramide (α-GalCer) (15). The Vα14i NKT cells rapidly release cytokines, such as IL-4 or IFN-γ, very soon after TCR activation.
Here, we provide evidence that Vα14i NKT cells are important in the CS initiation process because they stimulate B-1 cells to produce IgM antibodies rapidly after immunization that eventually leads to local T cell recruitment to elicit CS. Hepatic NKT cells are preferentially activated to release IL-4, that together with immunizing Ag, coactivate the B-1 cells. It is noteworthy that Vα14i NKT cells, like B-1 cells, are subpopulations of innate cells. The rapid response in CS of B-1 cells and hepatic Vα14i NKT cells, both innate cells, illustrates their cooperative interaction bridging innate and adaptive immune responses.
Materials and Methods
Mice.
Specific pathogen-free male CBA/J, female BALB/c, female CD1d−/− (129/SvImJ), and WT controls were from The Jackson Laboratory. Breeders of BALB/c Jα18−/− mice (formerly Jα281−/−) were from Masaru Taniguchi (Chiba University, Chiba, Japan), and BALB/c Vα14 transgenics were from Albert Bendelac (University of Chicago, Chicago, IL). Groups of four to five mice at 6–12 wk of age were rested at least 1 wk under specific pathogen-free conditions. Experiments were performed according to guidelines of the Animal Care and Use Committee at Yale.
Reagents.
Picryl chloride (PCl, TNP-Cl) (provided by Nacalai Tesque, Inc.) was recrystallized twice and stored protected from light. Kirin Laboratories provided α-GalCer (16). A stock solution at 220 μg/ml was diluted to 0.5–4 μg/ml in 0.5% polysorbate-20 for injection in sterile pyrogen-free 0.9% NaCl (Abbott Labs).
Immunization and Elicitation of CS.
Mice were contact sensitized with 150 μl of 5% PCl in absolute ethanol and acetone (4:1) on the shaved chest, abdomen, and rear footpads. CS responses were elicited on day 4 by painting ears with 10 μl of 0.4% PCl in acetone and olive oil (1:1). Sensitization was performed on days −1 and 0 in 129S3/SvImJ mice. Ear thickness was measured with a micrometer (Mitutoyo) before challenge and then at 2 and 24 h by an observer unaware of experimental groups. Increases in ear thickness are expressed as the mean ± SE × 10−2 mm.
Isolation of Sensitized T Cells from Immune LN and Spleen.
Mice were killed on day 4, and axillary and inguinal LNs and spleens were processed to cell suspension used for transfers. To remove B cells, anti–mouse CD19 mAb (0.5 μg/106 cells) (BD Biosciences) was added at 4°C for 45 min, followed by washing and resuspension at 107 cells/ml, and treated with 1 ml of 1:5 diluted rabbit complement (PEL-FREEZ) for 45 min at 37°C, followed by extensive washing with cold PBS, and injected i.v. at 2–5 × 107 cells/mouse.
T cells were isolated by magnetic bead depletion of non–T cells using a Pan T Cell Isolation kit (MACS Miltenyi Biotec). Cells from LNs and spleen were passed through 30 μm nylon mesh to remove cell clumps, washed with degassed buffer (0.5% BSA, 2 mM EDTA, PBS pH 7.2), and stained with mixed biotinylated antibodies to Mac-1, B220, DX5 (NK marker), and Ter-119 (erythrocyte marker). Then, cells were combined with antibiotin microbeads at 4°C and passed through an LS column (108 cells) in a magnetic field of a MidiMACS separator. The columns were washed four times, and then effluent cells were collected, centrifuged, counted, and analyzed by flow cytometry (99% TCRβ+), resuspended in PBS, and injected i.v. at 2 × 107 per mouse.
Liver Cell Preparation to Obtain Enriched Mononuclear Cells (LMNCs).
After sacrifice, liver was PBS perfused via the portal vein until opaque, then strained (70 μm; Becton Dickinson), resuspended in 40% isotonic Percoll (Amersham Biosciences), and overlaid onto 60% isotonic Percoll. After centrifugation for 20 min at 900 g at 25°C, liver mononuclear cells (LMNCs) were isolated at the interface and the 40% Percoll and washed with RPMI 1640 (Life Technologies) plus 5% FBS (Gemini-Bio-Products). Viability was >90%, and ∼2 × 106 LMNCs were obtained per mouse.
Flow Cytometry and Binding of Tetramers to iNKT Cell TCR.
PE-labeled tetramer mouse CD1d–α-GalCer complexes that bind Vα14i NKT cell receptors and “unloaded” control CD1d without α-GalCer were prepared (13). LMNCs were resuspended in PBS staining buffer containing 2% BSA and 0.02% NaN3 and then incubated for 15 min at 4°C with blocking 2.4G2 anti-Fc mAb (BD Biosciences) and blocking neutravidin (Molecular Probes). After washing, LMNCs were stained with a mixture of FITC anti-TCRβ mAb (BD Biosciences) and PE-labeled CD1d–α-GalCer tetramers at 25°C for 20 min and then washed twice. Double tetramer and TCRβ-positive cells were identified using a FACS® Vantage SE (Becton Dickinson). A minimum of 5 × 104 events was acquired, and results were analyzed using Mac CellQuest (Becton Dickinson).
Sorting of Immune Lymphoid B-1 Cells.
Spleen and LN cells from 1-d PCl-sensitized donors were stained at 106–107/ml with anti-CD5–cychrome, and anti-CD19–FITC (BD Biosciences) at 0.025 μg per 106 cells for 30 min on ice and then washed with RPMI. Stained cells were sorted to obtain ± 98% enriched CD19+CD5+ B-1 cells, representing ∼1% of total cells, and 6.5 × 104 B-1 cells were injected i.v. per mouse.
Enzyme-linked Immunospot (ELISPOT) Assay for Anti-TNP IgM-producing Cells.
Spleen and LN cells were from 4-d 5% PCl immune mice and seeded in triplicate into 96-well filtration plates with Immobilon-P membranes (Millipore) at 2 × 106 cells/well, precoated with 50 μl of TNP3-BSA (100 μg/ml). Plates were incubated overnight at 37°C, cells were discarded, and wells were washed with PBS three times and three times with PBS containing 0.05% Tween-20, and incubated for 1 h at 25°C with 2 μg/ml of biotin-conjugated anti–mouse IgM mAb (BD Biosciences), followed by incubation with streptavidin–horseradish peroxidase (1:200; Vector Laboratories) for 1 h. Spots were developed by using 3-amino-9-ethylcarbazole as substrate, the reaction was stopped by washing, and wells were dried at 25°C in the dark. Membranes were removed, stuck on glass slides, and spots were enumerated under an inverted dissecting phase microscope and expressed per organ.
Statistics.
Statistics were performed using the paired two-tailed Student's t test. P < 0.05 was taken as the level of significance.
Results
Defective Elicitation of CS in NKT Cell–deficient Mice
We evaluated CS responses in CD1d−/− mice that are NKT cell deficient because CD1d is the Ag-presenting molecule required for Vα14i TCR+ cells to develop in the thymus (17). Contact-sensitized CD1d−/− mice had significantly inhibited 24-h CS responses (Fig. 1 A, right, Group C vs. A). Importantly, the 2-h initiating component of CS was profoundly inhibited in CD1d−/− compared with WT mice (Fig. 1 A, left, Group A vs. C) and reduced to baseline levels equivalent to nonimmunized and similarly PCl-challenged controls (Fig. 1 A, left, Group C vs. B and D). To determine if the major Vα14i NKT cell population was involved, CS responses were studied in Jα18−/− mice specifically deficient in this subset. Again, we found defective CS, with absent 2-h early responses (Fig. 1 B, left, Group C vs. A) and significantly impaired 24-h late responses (Fig. 1 B, right, Group C vs. A), similar to findings in CD1d−/− (Fig. 1 A).
Figure 1.


NKT-deficient mice have defective CS. (A) CD1d−/− mice (Group C) and WT CD1d+/+ controls (Group A) were skin sensitized with 5% PCl and on day 4 were ear challenged with 0.4% PCl. Nonimmune CD1d−/− and CD1d+/+ (Groups D and B) controls were similarly ear challenged. Ear swelling was measured at 2 h (left) and 24 h (right). The results represent pooling from three different experiments each with three mice per group. (B) Jα18−/− mice (Group C) and WT BALB/c controls (Group A) were sensitized and ear challenged. Nonimmune Jα18−/− (Group D) and BALB/c (Group B) controls were ear challenged similarly. 2- and 24-h ear swelling responses were measured. The results are from a representative experiment with five mice per group.
Liver Mononuclear Cells from WT Mice and Splenocytes from Vα14 Transgenic Mice Reconstitute Defective CS in Jα18−/− Mice.
To confirm the role of Vα14i NKT cells in CS responses more directly, we i.p. transferred nonimmune WT LMNCs into NKT cell–deficient Jα18−/− mice 1 d before immunization. Transfer of 1–2 × 106 LMNCs from nonimmune BALB/c mice resulted in reconstitution of 2- and 24-h CS ear responses in subsequently immunized Jα18−/− mice (Fig. 2 A, Group C vs. B), whereas transfer of LMNCs from nonimmune Jα18−/− mice without Vα14i NKT cells failed (Fig. 2 A, Group D vs. B and C).
Figure 2.


Reconstitution of CS in Jα18−/− mice by transfer of LMNCs. (A) BALB/c (Group A) and Jα18−/− mice (Groups B–F) were sensitized, and 4 d later were ear challenged and responses were measured at 2 and 24 h. 1 d before immunization, groups of Jα18−/−mice received 1–2 × 106 LMNCs i.p. from nonimmune BALB/c mice (Group C), Jα18−/− mice (Group D), IL-4−/− mice (Group E), or IFN-γ−/− mice (Group F). Responses in each group represent the result from subtraction of background ear swelling of a control nonimmune group challenged similarly. The results are the pooling of three different experiments each with four mice per group. 2-h response statistics: P < 0.01, Group A vs. B; P < 0.01, Group C vs. E; **P < 0.05, Group F vs. B. 24-h response statistics: P < 0.001, Group A vs. B; P < 0.01, Group C vs. E; NS, not significant, Group F vs. B. (B) BALB/c (Group A) and Jα18−/− mice (Groups B–E) were sensitized, ear challenged, and responses were measured at 2 and 24 h. 1 d before, groups of Jα18−/−mice received LMNCs i.p. from nonimmune WT BALB/c mice (Group C), or Vα14 transgenic mice (Group D), or splenocytes from Vα14 transgenic mice (Group E). Each transferred group received an equivalent number of Vα14i NKT cells according to the percentages of tetramer-positive T cells determined previously. The response in each group represents the result from subtraction of background ear swelling of a control nonimmune group challenged similarly. The results are from a representative experiment with four mice per group. 2-h response statistics: P < 0.02, Group A vs. B; **P < 0.01, Group B vs. D and B vs. E. 24-h response statistics: P < 0.005, Group A vs. B; ***P < 0.001, Group B vs. D; **P < 0.01, Group B vs. E.
Since Vα14i NKT cells can rapidly release IL-4 (12) and IL-4 can act on B-1 cells (18, 19), we hypothesized that Vα14i NKT cells acted in CS by release of IL-4. To test this hypothesis, we similarly transferred nonimmune LMNCs from IL-4−/− mice that failed to reconstitute defective CS responses in Jα18−/− recipient mice (Fig. 2 A, Group E) compared with Jα18−/− mice transferred with WT BALB/c LMNCs (Fig. 2 A, Group C). In contrast, LMNCs from IFN-γ−/− mice fully reconstituted the 2-h response (Fig. 2 A, left, Group F vs. B).
To test if Vα14i NKT cells from spleen also could reconstitute impaired CS in Jα18−/− mice, we similarly i.p. transferred nonimmune splenocytes or LMNCs from Vα14 transgenic mice that have greatly increased Vα14i NKT cells compared with BALB/c. (Fig. 2 B). Equivalent numbers of Vα14i NKT cells were transferred compared with WT LMNCs, according to percentages of tetramer positive T cells in the Vα14 transgenic LMNCs (65%) and splenocytes (51%). Results showed that either LMNCs or splenocytes from Vα14 transgenic mice reconstituted defective CS responses in Jα18−/−mice similar to nonimmune BALB/c LMNC NKT cells (Fig. 2 B, Group B vs. C–E).
Direct Stimulation of Liver Vα14i NKT Cells by Skin Contact Sensitization.
We tested for a more direct connection between cutaneous sensitization and Vα14i NKT cells by determining changes in the proportion of liver Vα14i NKT cells after skin immunization. PE-CD1d–α-GalCer tetramers were used to identify NKT cells in liver after PCl skin painting by flow cytometry (13). As positive control, we followed liver Vα14i NKT cell levels after i.v. injection of 4 μg α-GalCer (13) that caused initial increases followed by a rapid disappearance (Fig. 3 A).
Figure 3.
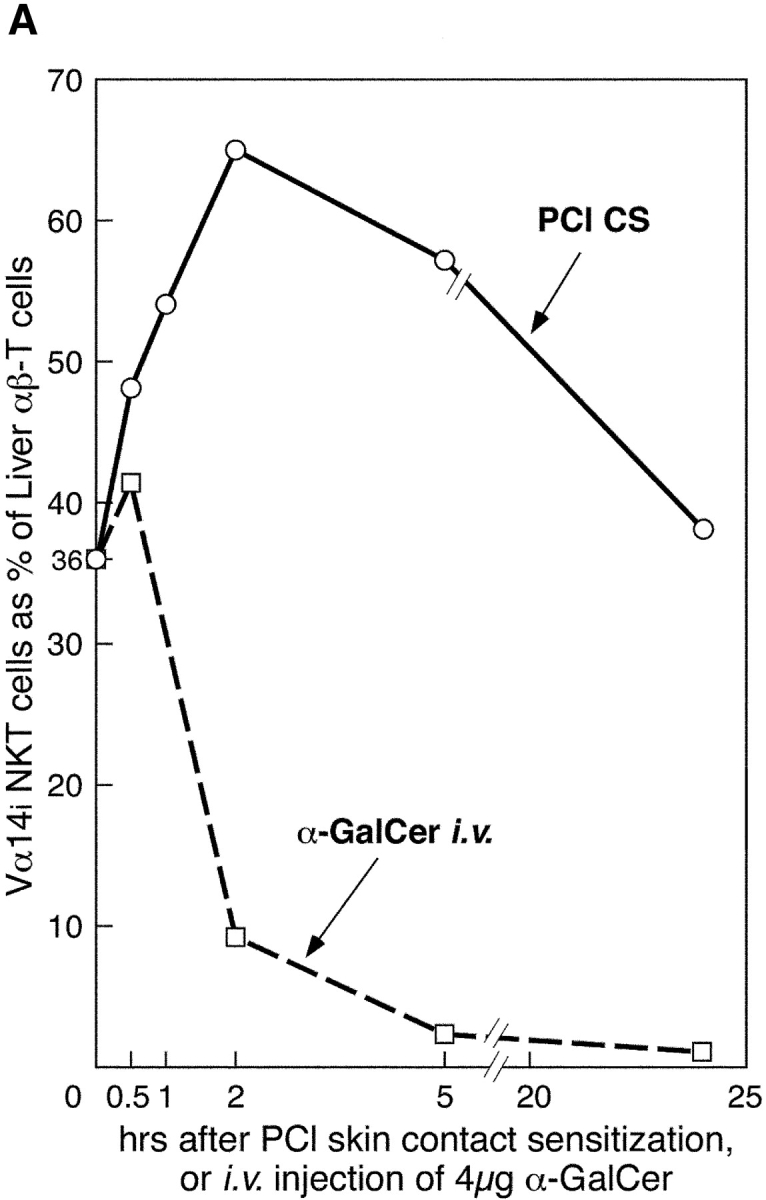

Liver NKT cells are activated early after PCl skin sensitization. (A) CBA/J mice were PCl skin sensitized or injected i.v. with 4 μg α-GalCer. Separate groups of mice were killed at 30 min, 1 h, 2 h, 5 h, or 24 h, and LMNCs were isolated and stained with FITC anti-TCRβ mAb and PE CD1d–α-GalCer tetramers and analyzed by flow cytometry to identify double positive Vα14i NKT cells. Percentages were determined based on the analysis of pooled LMNCs from four to five mice per group, except for 2- and 5-h points performed twice. (B) Dot plots show the percentage of tetramer-positive T cells from a representative experiment comparing nonimmune and 2-h skin immune CBA/J mice.
Remarkably, cutaneous PCl immunization also rapidly induced profound changes in liver Vα14i NKT cells but with very different kinetics (Fig. 3 A). By 30 min, Vα14i NKT cells rose from 36% to 48% (33% increase), and by 1 h rose further to 54% (50% increase), and peaked at near doubling to 65% of T cells by 2 h (81% increase) (Fig. 3 B), with a similar increase in total numbers of liver Vα14i NKT cells (not depicted). By 5 h, there was a decline to 58% and then to a normal level of 38% by 24 h.
Important negative controls showed that unloaded CD1d tetramers without α-GalCer binding in the groove did not nonspecifically bind to LMNCs from nonimmune or α-GalCer–treated or PCl contact-sensitized mice (Fig. 4 A). Thus, binding depended on fully loaded tetramers containing α-GalCer. Further, when the full tetramers were used both nonimmune and untreated Jα18−/− and CD1d−/− LMNCs, and those from sensitized Jα18−/− and CD1d−/− mice, had no binding to remaining LMNCs in these mice without NKT cells (Fig. 4 B, middle and right). Hence, CS did not induce LMNCs to nonspecifically bind the tetramers. In contrast, nonimmune BALB/c controls for Jα18−/− mice had 44% tetramer-positive cells in the liver, and nonimmune WT 129 controls for the CD1d−/− mice had 11% tetramer positive cells (Fig. 4 B, left). These control findings support the specificity of the rapid increase in tetramer binding to liver Vα14i NKT cells in PCl contact-sensitized WT mice (Fig. 3).
Figure 4.


CS does not induce nonspecific binding of tetramers to LMNCs. (A) Different groups of CBA/J mice were PCl skin sensitized or i.v. injected with 0.5 μg of α-GalCer, and 4 h later LMNCs were obtained and stained with CD1d-α-GalCer tetramers or PE-CD1d–unloaded tetramers to evaluate nonspecific binding. We analyzed pooled LMNCs from four mice per group. (B) Different groups of Jα18−/− or CD1d−/− mice and appropriate WT controls were PCl sensitized, and 2 h later LMNCs were obtained. The LMNCs were stained to analyze Vα14i NKT cells by flow cytometry. The numbers indicate the percentages of tetramer-positive liver TCRβ+ cells analyzing pooled LMNCs from four mice per group.
Comparison of NKT Cell Activation in Different Organs of PCl Contact-sensitized Mice.
We questioned if the rise in Vα14i NKT cells directly after skin immunization occurred preferentially in the liver. Thus, we used tetramers to analyze levels of Vα14i NKT cells in spleen and also in other sites of PCl contact-sensitized CBA/J mice at 2 h compared with nonimmune controls. Again, a near doubling of liver Vα14i NKT cells occurred (Fig. 5 A, top), whereas there were no increases in the spleen, peripheral LNs, or peritoneal cavity after PCl skin immunization (Fig. 5 A). To compare results at an earlier time point in another strain, we similarly measured the levels of Vα14i NKT cells in BALB/c mice 1 h after skin immunization. Similarly, only the hepatic Vα14i NKT cells increased as a percentage of αβ T cells, from 29% to 38% (Fig. 5 B, top), whereas the other sites did not show any significant changes (Fig. 5 B). Further, nonimmune Vα14 transgenic mice (BALB/c background) with higher basal levels of Vα14i NKT cells at these various sites also had only increases in the liver from 65% to 85% after skin immunization, and not at the other sites (Fig. 5 C).
Figure 5.



Vα14i NKT cells increase in liver after CS but not LNs, spleen, and peritoneal cavity. (A) CBA/J mice were PCl skin sensitized, and 2 h later mononuclear cells from liver, peritoneal cavity, spleen, and LNs were stained with FITC anti-TCRβ mAb and PE CD1d–α-GalCer tetramers and analyzed by flow cytometry for the percentage of tetramer-positive T cells compared with nonimmune mice determined by analysis of pooled mononuclear cells from three mice per group. BALB/c (B) and Vα14 transgenic mice (C) also were PCl immunized, and 1 h later mononuclear cells from the liver, peritoneal cavity, spleen, and LNs were stained and analyzed by flow cytometry.
Failure of CS Initiation Is the Cause of Defective CS Elicitation in NKT-deficient Mice.
The absent 2-h component of CS in both NKT cell–deficient strains suggests defective CS initiation because Vα14i NKT cells are needed to activate the CS-initiating B-1 cells. Thus, we compared the CS initiation process in 1-d PCl contact-sensitized recipient CD1d−/− vs. WT mice. At this early time, the B-1 cells are activated (2), but Ag-specific CS effector T cells are not yet immunized (1, 2, 20, 21). We transferred syngeneic 4-d PCl immune-isolated CS effector T cells that are known to require CS initiation to mediate late 24 h CS (2, 21). The isolated CS effector T cells were obtained by depleting B cells from 4-d PCl immune-mixed LN and spleen cells from WT mice using anti–CD-19 and complement treatment or by negative magnetic selection (99% TCRβ+).
As shown (2, 21, 22), the CS effector T cells could not transfer either 2- or 24-h components of CS to ear-challenged nonimmune control WT recipients (Fig. 6 A, Group A) without CS-initiating B-1 cells. However, when B-depleted T cells were instead transferred to 1-d PCl immune WT recipients, the 2- and 24-h CS were elicited (Fig. 2 A, Group B). In contrast, the same CS effector T cells failed to transfer 2- or 24-h CS responsiveness into 1-d immune CD1d−/− recipients (Fig. 6 A, Group C vs. B). The 1-d immune CD1d−/− recipients (Fig. 6 A, Group C) may not have generated sensitized CS-initiating B-1 cells, perhaps due to their NKT cell deficiency. Similar results were obtained with CS effector BALB/c T cells purified further to 99% TCRβ+ T cells by magnetic bead separation that could not transfer either 2- or 24-h components of CS to nonimmune BALB/c recipients (Fig. 6 B, Group A). However, when these purified T cells were instead transferred to 1-d PCl immune WT BALB/c recipients, the 2 and 24 h CS responses were elicited (Fig. 2 B, Group B). In important contrast, the same purified CS effector T cells failed to transfer 2- or 24-h CS responses into 1-d immune Jα18−/− recipients (Fig. 6 B, Group C vs. B). These data show that 1-d immune Jα18−/− or CD1d−/− recipients fail to generate CS initiation. We considered an alternate explanation that NKT cell–deficient mice have reduced B-1 cells but found no deficiency of peritoneal CD19+CD5+ B-1 cell numbers in these mice (not depicted).
Figure 6.


4-d immune WT T cells fail to transfer CS to 1-d immune NKT cell–deficient mice. (A) CD1d−/− mice (Group C) and CD1d+/+ WT controls (Group B) were skin sensitized and 1 d later received 4-d PCl immune-isolated T cells from WT CD1d+/+ mice and then were ear challenged 1 d later. The T cells were obtained by treating WT immune cells with anti-CD19 plus complement. Nonimmune CD1d+/+ mice (Group A) similarly received T cell transfers and were ear challenged. Nonimmune CD1d−/− (Group D) and CD1d+/+ mice (Group E) were challenged but received no transfers. (B) Jα18−/− mice (Group C) and BALB/c WT controls (Group B) were contact sensitized and 1 d later received 4-d PCl immune–isolated T cells from BALB/c mice and then were ear challenged 1-d later. The T cells (99% TCRβ+) were obtained by magnetic separation. Nonimmune BALB/c mice (Group A) similarly received T cell transfers and were ear challenged. Nonimmune Jα18−/− (Group D) and BALB/c mice (Group E) were challenged but received no transfers. In A and B, the ear responses were measured at 2 and 24 h from a representative experiment with four mice per group.
IL-4 Treatment Reconstitutes Early and Late Components of Defective CS in Sensitized CD1d−/− and Jα18−/− Mice.
We further evaluated whether defective CS initiation in NKT cell–deficient mice was due to a lack of IL-4 by using the same cell transfer protocol from Fig. 6. We attempted to reconstitute CS in CD1d−/− mice by injecting rIL-4 at the time of sensitization (Fig. 7 A). Positive controls again showed elicitation of 2- and 24-h CS (Fig. 7 A, Group A), and there was defective 2- and 24-h CS in 1-d PCl immune CD1d−/− mice (Fig. 7 A, Group B). Systemic i.v. injection of just 2 ng IL-4 during immunization did not cause significant reconstitution of 24-h CS (Fig. 7 A, Group C). 20 ng IL-4 led to reconstitution of 2- and 24-h CS responses (Fig. 7 A, Group D), whereas 200 ng of IL-4 reconstituted 2 h CS, but the 24-h response was reduced compared with the 20 ng–treated group (Fig. 7 A. Group E vs. D, right).
Figure 7.


IL-4 reconstitutes early and late CS responses in NKT cell–deficient mice. (A) WT 1-d PCl immune mice (Group A) were compared with 1-d immune CD1d−/− mice (Group B) as recipients of 4-d PCl immune T cells from CD1d+/+ mice. WT immune mice elicited 2 and 24 h CS (Group A), whereas the CD1d−/− mice did not (Group B). Groups C–E are identical to Group B. However, these 1-d immune CD1d−/− mice that received isolated immune T cells also were injected with different doses of rIL-4 during immunization. *P < 0.05; **P < 0.001. (B) WT BALB/c 1-d active PCl immune mice (Group B) were compared with 1-d immune Jα18−/− (Group C) as recipients of 4-d PCl immune magnetic-separated T cells from BALB/c mice. Group D is identical to Group C, but 20 ng i.v. of IL-4 was injected during immunization of the 1-d immune Jα18−/− recipient mice. In A and B, the results are mean ± SE from four mice per group.
We similarly attempted CS reconstitution by injecting IL-4 at the time of sensitization in Jα18−/− mice using the protocol in Fig. 6 B in which we transferred WT magnetic-isolated purified CS effector T cells (Fig. 7 B). As before, positive controls showed elicitation of 2- and 24-h CS (Fig. 7 B, Group B), but CS responses were defective in 1-d PCl immune Jα18−/− mice (Fig. 4 B, Group C). Again, systemic injection of 20 ng IL-4 i.v. during immunization led to complete reconstitution of 2- and 24-h CS in these 1-d PCl immune Jα18−/− mice (Group D). These results suggest that injection of IL-4 at immunization restored impaired CS initiation for eventual recruitment of effector T cells in immunized NKT cell–deficient CD1d−/− and Jα18−/− mice.
Sensitized CD1d−/− Mice Have Intact Immunized CS Effector T Cells: Reconstitution with Immune B-1 Cells.
The above transfer experiments did not examine if CS effector T cells were intact in NKT cell–deficient mice. Thus, immunized CD1d−/− mice were used as cell transfer recipients at day 4 when effector T cells become fully activated (1, 20, 21). Thus, in a new experiment, we tested if failure of CS initiation in NKT cell–deficient mice was due to an inability to activate B-1 cells. We transferred activated B-1 cells from 1-d PCl-immune WT donors, and the 4-d immune recipients were challenged 1 d later. Positive control 4-d actively immunized CD1d+/+ mice had normal elicitation of 2- and 24-h CS compared with nonimmune controls (Fig. 8 , Group A vs. F). Further, as shown previously (Fig. 1 A) 4-d immune CD1d−/− mice had absent 2-h CS and inhibited 24-h CS (Fig. 8, Group A vs. B vs. E). Remarkably, transfer of WT 1-d immune cells that contained sensitized B-1 cells into 4-d immune CD1d−/− recipients largely reconstituted 2- and 24-h CS (Fig. 8, Group C vs. B). Also, 98% purified B-1 cells from the 1-d PCl immune donors similarly reconstituted both early 2- and late 24-h components of CS (Fig. 8, Group D vs. B).
Figure 8.

Immune B-1 cells from CD1d+/+ mice reconstitute 2 and 24 h CS in PCl-sensitized CD1d−/− mice. WT CD1d+/+ controls (Group A) and CD1d−/− mice (Groups B–D) were contact sensitized. Then, 3 d later Group C received 1-d immune cells from CD1d+/+ mice, and Group D received sorted 1-d immune CD19+CD5+ B-1 cells from CD1d+/+ mice. On day 4, all groups were ear challenged. Nonimmune CD1d−/− mice (Group E) and CD1d+/+ mice (Group F) were also ear challenged. Group G is nonimmune WT mice that just received 1-d PCl immune cells. Ear swelling was measured at 2 and 24 h, and the results are from a representative experiment with four mice per group; *P < 0.05; **P < 0.01.
These results confirmed the competence of 4-d PCl-immune CD1d−/− mice to generate CS effector T cells (Fig. 8, Groups C and D) that in this case were recruited normally to mediate 24-h CS, when CS initiation was provided by transferred WT 1-d immune cells (Group C) or by purified B-1 cells (Group D). Control Group G shows that 1-d PCl immune cells from WT mice mediated 2-h, but not the 24-h ear swelling, because CS effector T cells had not yet developed (1, 20, 21). This documents that 24-h CS responses obtained in Fig. 8, Groups C and D, were not from transferred 1-d immune cells alone.
NKT Cell–deficient Mice Have Impaired Specific IgM Responses.
Results suggest that soon after CS immunization, Vα14i NKT cell–derived IL-4 activates B-1 cells to produce specific IgM. Thus, defective CS initiation in NKT cell–deficient mice may be explained by defective production of specific initiating IgM antibodies by the B-1 cells. To test this, we used ELISPOT to evaluate numbers of spleen cells producing anti-TNP IgM antibodies in Jα18−/− compared with WT BALB/c mice after PCl immunization. We found a smaller number of anti-TNP IgM–producing cells 4 d after PCl skin immunization in the Jα18−/− mice compared with WT BALB/c (Fig. 9) when 2- and 24-h ear responses were defective. Similar results were obtained with PCl immunized CD1d−/− vs. WT 129 S3/SvImJ mice (not depicted). These finding of impaired production of anti-TNP–specific IgM antibodies after CS in both strains of iNKT cell–deficient mice reinforced the idea that Vα14i NKT cells are needed to activate B-1 cells to produce CS-initiating IgM antibodies.
Figure 9.

NKT cell–deficient mice have an impaired specific IgM response. Jα18−/− and WT BALB/c mice were PCl skin immunized or sham immunized, and 4 d later their splenocytes were harvested and incubated in vitro to measure numbers of anti-TNP IgM-producing cells using an ELISPOT assay. The results are pooled consisting of seven mice per group from three experiments. The background IgM anti-BSA responses, obtained by counting number of spots in BSA-coated plates, were subtracted, as were responses in sham immune mice of each strain.
Together (Fig. 10) , the data indicate that very early after PCl skin sensitization there is rapid stimulation of hepatic Vα14i NKT cells required to activate CS-initiating B-1 cells to produce IgM antibodies, likely via production of IL-4.
Figure 10.

Summary schema: early activation of liver Vα14i NKT cells leads to initiation of systemic CS immunity. Cutaneous sensitization with PCl (TNP-Cl) is postulated to cause local release from the site of immunization of diverse ligands that act in three directions. First, rapidly after immunization uncharacterized glycolipid ligands stimulate Vα14i NKT cells likely in the liver to produce IL-4. Second, soluble hapten–self-protein and peptide complexes formed by conjugation of the hapten are rapidly released systemically. It is postulated that together the Vα14i NKT cell-derived IL-4 and Ag hapten–self complexes coactivate the B-1 B cells to produce hapten-specific IgM antibodies that circulate within 1 d after sensitization. Third, self-peptides complexed with surface MHC molecules of local APC, such as Langerhans cells, are conjugated with the immunizing hapten to form TNP-peptide–MHC complexes that induce maturation and activation of CS effector memory T cells in the LNs within 4 d after immunization. Subsequently, after secondary ear Ag challenge to elicit CS the local B-1–derived IgM antibodies form complexes with the challenge TNP Ag–self complexes. This activates local complement to generate C5a at the Ag challenge site, leading to release of vasoactive mediators from mast cells that alter the local endothelium, allowing recruitment of the Ag/MHC-restricted CS effector T cells to mediate the late classical 24-h CS response.
Discussion
We have shown for the first time that Vα14i NKT cells are required in CS, which is a prototype of acquired in vivo T cell–mediated immunity. These findings reinforce previous connections we established between the initiation of CS elicitation and other components of the innate immune system (2, 3, 6, 23–25). A new aspect is the very early and rapid stimulation of the Vα14i NKT cell subset within 1 h of immunization that induces initiating B-1 cell responses within 1 d and leads to subsequent recruitment of CS effector αβT cells to enable elicitation of classical 24 h CS. Moreover, the unique early and preferential expansion of hepatic Vα14i NKT cells after contact sensitization suggests that cutaneous CS immunization induced rapid and previously unrecognized systemic effects on iNKT cells, in addition to the known Ag uptake by APC to generate the classical CS effector T cells.
NKT Cell Involvement in CS Initiation.
It is known that only activation of Vα14i NKT cells via their TCR, by either exogenous nonspecific α-GalCer or by anti-CD3, rapidly results in production of high levels of serum IL-4 (26–28). Therefore, we postulate that Vα14i NKT cells are activated through their TCR to rapidly release IL-4 after contact skin immunization, which together with specific TNP-Ag released systemically from the contact skin sensitization site (29) coactivates B-1 cells within 1 d of immunization (2, 21, 30, 31). Finding impaired CS responses in two different strains of NKT cell–deficient mice provided strong evidence for a role of Vα14i NKT cells in CS.
Reconstitution of defective CS in Jα18−/− mice by transferring nonimmune WT or Vα14 transgenic hepatic or splenic MNC containing Vα14i NKT cells pointed to involvement of these cells in CS. Importantly, the role of IL-4 derived from Vα14i NKT cells was demonstrated specifically by failure of CS reconstitution using LMNCs from IL-4−/− mice. The role of Vα14i NKT cells was confirmed by the lack of reconstitution of CS with LMNCs from NKT cell–deficient Jα18−/− mice.
Although Vα14i NKT cells can also rapidly release high levels of IFN-γ (28, 32), this probably is not involved in early activation of CS-initiating B-1 cells. When we transferred LMNCs from IFN-γ−/− mice 1 d before immunization of Jα18−/− mice, there was a full reconstitution of the early 2-h response. However, it remains possible that Vα14i NKT cells release both IFN-γ and IL-4 in CS but that IL-4 predominantly plays an early facilitating role by activating B-1 cells.
Systemic treatment with 20 ng of IL-4 at the time of sensitization reconstituted CS responses in immunized CD1d−/− and Jα18−/− mice. Moreover, we further tested the postulate that Vα14i NKT cell–derived IL-4 is needed to activate CS-initiating B-1 cells. We showed that downstream-acting immune B-1 cells from contact-sensitized WT mice were able to reconstitute elicitation of CS in NKT cell–deficient CD1d−/− or Jα18−/− mice. This confirmed our hypothesis that an early wave of IL-4 release from Vα14i NKT cells rapidly after contact sensitization likely activates CS-initiating B-1 cells. Therefore, the requirement for Vα14i NKT cells was circumvented by providing the missing NKT cells, or IL-4, or immune B-1 cells presumably already activated by IL-4 in the immunized WT donors.
Others have shown that systemic injection of α-GalCer rapidly activates Vα14i NKT cell production of IL-4 to induce B cell activation (28). Our demonstration of NKT cell activation of B-1 cells to initiate CS expands the B cell helper role of NKT cells via IL-4 shown with Th2 cell–dependent B-2 cell IgE responses in murine asthma (33, 34), in an IL-18–dependent in vitro model (35), and in a human system (36).
Activation of Vα14i NKT Cells by Cutaneous CS.
Early increases in liver Vα14i NKT cells after PCl skin sensitization provided strong direct evidence for in vivo participation of Vα14i NKT cells early postimmunization in CS. In addition, the time course of hepatic Vα14i NKT cell changes after PCl sensitization was very different from i.v. injection of α-GalCer. Cutaneous sensitization caused a rapid onset and steady increase in Vα14i NKT cells followed by a slow return to basal levels by 24 h, perhaps due to weaker activation with no subsequent apoptosis. In contrast, α-GalCer induced a smaller rapid rise and then severe reduction that persisted for 24 h. This may be due to TCR-mediated activation–induced apoptosis (37, 38) or down modulation of Vα14 TCR.
Although Vα14i NKT cells were increased in the liver after CS, no similar increases in the spleen, LNs, or peritoneal cavity were detected by tetramer staining. This result showing a specific association of activation of liver NKT cells with skin sensitization was unexpected. Perhaps Vα14i NKT cells from other distant sites also are activated after contact immunization. Note that transferred splenocytes from Vα14 transgenic mice could also reconstitute CS in Jα18−/− mice. Thus, it cannot be excluded that effective activation of Vα14i NKT cells from these other sites for participation in CS is not always detected by tetramer staining. Conversely, the Vα14i NKT cells, or the APC in the liver, may have specific properties that could result in preferential activation after skin sensitization.
Postulated Mechanisms of Vα14i NKT Cell Activation in CS.
Studies of CS may lead to eventual identification of natural endogenous CD1d-binding ligands for the TCR of Vα14i NKT cells. During PCl skin contact sensitization, there may be release from the skin of endogenous glycolipids that may disperse systemically, particularly to the liver. These released endogenous ligands may bind CD1d on APC and activate Vα14i NKT cells to cause rapid production of IL-4. As a parallel, it is established in CS that skin-derived hapten protein and peptide Ag conjugates derived from contact immunization rapidly are dispersed throughout the body by the systemic circulation and influence the overall immune response (29).
The stimulus for Vα14i NKT cell activation in CS may be less intense compared with the artificial ligand α-GalCer. Indeed, in vivo administration of α-GalCer leads to hepatic damage within 1 d (38) and in vivo treatment with anti-CD3ɛ and IL-12–induced NKT cell apoptosis in the liver (39). Hence, physiological NKT cell activation, such as in CS, may be less powerful without apoptosis and depletion. Another difference between CS and α-GalCer effects on Vα14i NKT cells is the route of exposure, since α-GalCer was injected i.v. and PCl applied by contact skin painting.
Comparison of NKT Cell Involvement in CS to Other Systems.
There is a significant difference between our data showing involvement of Vα14i NKT cells in CS and their participation in several other systems (40–49). In most of these instances, Vα14i NKT cells are triggered by systemic injections of α-GalCer, which is an exogenous ligand of CD1d and the NKT cell Vα14Jα18 TCR. In contrast, we have shown that cutaneous immunization alone rapidly activates liver Vα14i NKT cells via presumed release of endogenous ligands different from but analogous to exogenous α-GalCer. Some infections may also generate endogenous activators that can deplete NKT cells (50). The unique participation of Vα14i NKT cells in CS suggests similar positive involvement of Vα14i NKT cells in other important responses may also be uncovered, as is true in allergic murine asthma models (33, 34).
Fig. 10 summarizes the processes we postulate after release of endogenous glycolipids that trigger Vα14i NKT cells to coactivate B-1 cells. It is noteworthy that CS is a paradigm for other T cell–dependent in vivo immune DTH-like mechanisms that mediate numerous diseases. Similar mechanisms to those we have described in CS may underlie protection in infections, tumors, autoimmune diseases, allergies, and asthma which also involve recruitment of T cells into the tissue site.
Acknowledgments
We are grateful to Marilyn Avallone for her secretarial skills and Claudia Keiner and Neelendu Dey for reviewing the manuscript.
This work was supported by the National Institutes of Health; DK-34989, AR-41942, AI-07174, and the American Academy of Allergy, Asthma and Immunology to P.W. Askenase; and CA-52511 to M. Kronenberg; and the Polish Committee of Scientific Research to M. Szczepanik. This work was also supported by a government grant (200410/00-8) from Brazil and Fujisawa/American Academy of Allergy Asthma and Immunology award.
Abbreviations used in this paper: α-GalCer, α-galactosylceramide; CS, contact sensitivity; DTH, delayed-type hypersensitivity; LMNC, liver mononuclear cell; PCl, picryl-chloride (TNP-Cl).
References
- 1.Van Loveren, H., and P.W. Askenase. 1984. Delayed-type hypersensitivity is mediated by a sequence of two different T cell activities. J. Immunol. 133:2397–2401. [PubMed] [Google Scholar]
- 2.Tsuji, R.F., M. Szczepanik, I. Kawikova, V. Paliwal, R.A. Campos, M. Akahira-Azuma, N. Baumgarth, L.A. Herzenberg, and P.W. Askenase. 2002. B-cell dependent T cell responses: IgM antibodies are required to elicit contact sensitivity. J. Exp. Med. 196:1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsuji, R.F., I. Kawikova, R. Ramabhadran, M. Akahira-Azuma, D. Taub, T.E. Hugli, C. Gerard, and P.W. Askenase. 2000. Early local generation of C5a initiates the elicitation of contact sensitivity by leading to early T cell recruitment. J. Immunol. 165:1588–1598. [DOI] [PubMed] [Google Scholar]
- 4.Askenase, P.W., S. Bursztajn, M.D. Gershon, and R.K. Gershon. 1980. T cell-dependent mast cell degranulation and release of serotonin in murine delayed-type hypersensitivity. J. Exp. Med. 152:1358–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kops, S.K., H. Van Loveren, R.W. Rosenstein, W. Ptak, and P.W. Askenase. 1984. Mast cell activation and vascular alterations in immediate hypersensitivity-like reactions induced by a T cell-derived antigen-binding factor. Lab. Invest. 50:421–434. [PubMed] [Google Scholar]
- 6.Geba, G.P., W. Ptak, G.M. Anderson, V. Paliwal, R.E. Ratzlaff, J. Levin, and P.W. Askenase. 1996. Delayed-type hypersensitivity in mast cell-deficient mice: dependence on platelets for expression of contact sensitivity. J. Immunol. 157:557–565. [PubMed] [Google Scholar]
- 7.Matsuda, H., H. Ushio, G.P. Geba, and P.W. Askenase. 1997. Human platelets can initiate T cell-dependent contact sensitivity through local serotonin release mediated by IgE antibodies. J. Immunol. 158:2891–2897. [PubMed] [Google Scholar]
- 8.McHale, J.F., O.A. Harari, D. Marshall, and D.O. Haskard. 1999. Vascular endothelial cell expression of ICAM-1 and VCAM-1 at the onset of eliciting contact hypersensitivity in mice: evidence for a dominant role of TNF-α. J. Immunol. 162:1648–1655. [PubMed] [Google Scholar]
- 9.Schwartz, A., P.W. Askenase, and R.K. Gershon. 1977. The effect of locally injected vasoactive amines on the elicitation of delayed-type hypersensitivity. J. Immunol. 118:159–165. [PubMed] [Google Scholar]
- 10.Gershon, R.K., P.W. Askenase, and M.D. Gershon. 1975. Requirement for vasoactive amines for production of delayed-type hypersensitvity skin reactions. J. Exp. Med. 142:732–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Askenase, P.W. 2001. Yes T cells, but three different T cells (alphabeta, gammadelta and NK T cells), and also B-1 cells mediate contact sensitivity. Clin. Exp. Immunol. 125:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Godfrey, D.I., K.J.L. Hammond, L.D. Poulton, M.J. Smyth, and A.G. Baxter. 2000. NKT cells: facts, functions and fallacies. Immunol. Today. 21:573–583. [DOI] [PubMed] [Google Scholar]
- 13.Matsuda, J.L., O.V. Naidenko, L. Gapin, T. Nakayama, M. Taniguchi, C.R. Wang, Y. Koezuka, and M. Kronenberg. 2000. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lantz, O., and A. Bendelac. 1994. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8− T cells in mice and humans. J. Exp. Med. 180:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawano, T., J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno, R. Nakagawa, H. Sato, E. Kondo, et al. 1997. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 278:1626–1629. [DOI] [PubMed] [Google Scholar]
- 16.Morita, M., K. Motoki, K. Akimoto, T. Natori, T. Sakai, E. Sawa, K. Yamaji, Y. Koezuka, E. Kobayashi, and H. Fukushima. 1995. Structure-activity relationship of alpha-galactosylceramides against B16-bearing mice. J. Med. Chem. 38:2176–2187. [DOI] [PubMed] [Google Scholar]
- 17.Mendiratta, S.K., W.D. Martin, S. Hong, A. Boesteanu, S. Joyce, and L. Van Kaer. 1997. CD1d1 mutant mice are deficient in natural T cells that promptly produce IL-4. Immunity. 6:469–477. [DOI] [PubMed] [Google Scholar]
- 18.Paul, W.E., and R.A. Seder. 1994. Lymphocyte responses and cytokines. Cell. 76:241–251. [DOI] [PubMed] [Google Scholar]
- 19.Erickson, L.D., T.M. Foy, and T.J. Waldschmidt. 2001. Murine B1 B cells require IL-5 for optimal T cell-dependent activation. J. Immunol. 166:1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Loveren, H., K. Kato, R. Meade, D.R. Green, M. Horowitz, W. Ptak, and P.W. Askenase. 1984. Characterization of two different Ly-1+ T cell populations that mediate delayed-type hypersensitivity. J. Immunol. 133:2402–2411. [PubMed] [Google Scholar]
- 21.Ptak, W., W.R. Herzog, and P.W. Askenase. 1991. Delayed-type hypersensitivity initiation by early-acting cells that are antigen mismatched or MHC incompatible with late-acting, delayed-type hypersensitivity effector T cells. J. Immunol. 146:469–475. [PubMed] [Google Scholar]
- 22.Ptak, W., G.P. Geba, and P.W. Askenase. 1991. Initiation of delayed-type hypersensitivity by low doses of monoclonal IgE antibody. Mediation by serotonin and inhibition by histamine. J. Immunol. 146:3929–3936. [PubMed] [Google Scholar]
- 23.Tsuji, R.F., G.P. Geba, Y. Wang, K. Kawamoto, L.A. Matis, and P.W. Askenase. 1997. Required early complement activation in contact sensitivity with generation of local C5-dependent chemotactic activity, and late T cell interferon γ: a possible initiating role of B cells. J. Exp. Med. 186:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Askenase, P.W., H. Van Loveren, S. Kraeuter-Kops, Y. Ron, R. Meade, T.C. Theoharides, J.J. Nordlund, H. Scovern, M.D. Gerhson, and W. Ptak. 1983. Defective elicitation of delayed-type hypersensitivity in W/Wv and SI/SId mast cell-deficient mice. J. Immunol. 131:2687–2694. [PubMed] [Google Scholar]
- 25.van Loveren, H., R. Meade, and P.W. Askenase. 1983. An early component of delayed-type hypersensitivity mediated by T cells and mast cells. J. Exp. Med. 157:1604–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshimoto, T., and W.E. Paul. 1994. CD4pos, NK1.1pos T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J. Exp. Med. 179:1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen, H., and W.E. Paul. 1997. Cultured NK1.1+ CD4+ T cells produce large amounts of IL-4 and IFN- gamma upon activation by anti-CD3 or CD1. J. Immunol. 159:2240–2249. [PubMed] [Google Scholar]
- 28.Kitamura, H., A. Ohta, M. Sekimoto, M. Sato, K. Iwakabe, M. Nakui, T. Yahata, H. Meng, T. Koda, S. Nishimura, et al. 2000. α-galactosylceramide induces early B-cell activation through IL-4 production by NKT cells. Cell. Immunol. 199:37–42. [DOI] [PubMed] [Google Scholar]
- 29.Pior, J., T. Vogl, C. Sorg, and E. MacHer. 1999. Free hapten molecules are dispersed by way of the bloodstream during contact sensitization to fluorescein isothiocyanate. J. Invest. Dermatol. 113:888–893. [DOI] [PubMed] [Google Scholar]
- 30.Ishii, N., K. Takahashi, H. Nakajima, S. Tanaka, and P.W. Askenase. 1994. DNFB contact sensitivity (CS) in BALB/c and C3H/He mice: requirement for early-occurring, early-acting, antigen-specific, CS-initiating cells with an unusual phenotype (Thy-1+, CD5+, CD3−, CD4−, CD8−, sIg−, B220+, MHC class II−, CD23+, IL-2R−, IL-3R+, Mel-14−, Pgp-1+, J11d+, MAC-1+, LFA-1+, and Fc γ RII+). J. Invest. Dermatol. 102:321–327. [DOI] [PubMed] [Google Scholar]
- 31.Ishii, N., Y. Sugita, H. Nakajima, S. Tanaka, and P.W. Askenase. 1995. Elicitation of nickel sulfate (NiSO4)-specific delayed-type hypersensitivity requires early-occurring and early-acting, NiSO4- specific DTH-initiating cells with an unusual mixed phenotype for an antigen-specific cell. Cell. Immunol. 161:244–255. [DOI] [PubMed] [Google Scholar]
- 32.Nishimura, T., H. Kitamura, K. Iwakabe, T. Yahata, A. Ohta, M. Sato, K. Takeda, K. Okumura, L. Van Kaer, T. Kawano, et al. 2000. The interface between innate and acquired immunity: glycolipid antigen presentation by CD1d-expressing dendritic cells to NKT cells induces the differentiation of antigen-specific cytotoxic T lymphocytes. Int. Immunol. 12:987–994. [DOI] [PubMed] [Google Scholar]
- 33.Akbari, O., P. Stock, E. Meyer, M. Kronenberg, S. Sidobre, T. Nakayama, M. Taniguchi, M.J. Grusby, R.H. DeKruyff, and D.T. Umetsu. 2003. Essential role of NK cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat. Med. 9:582–588. [DOI] [PubMed] [Google Scholar]
- 34.Lisbonne, M., S. Diem, A.C. Keller, J. Lefort, L.M. Araujo, P. Hachem, J. Fourneau, S. Sidobre, M. Kronenberg, M. Taniguchi, et al. 2003. Cutting edge: invariant Vα14 NKT cells are required for allergen-induced airway inflammation and hyperreactivity in an experimental asthma model. J. Immunol. 171:1637–1641 [DOI] [PubMed] [Google Scholar]
- 35.Yoshimoto, T., B. Min, T. Sugimoto, N. Hayashi, Y. Ishikawa, Y. Sasaki, H. Hata, K. Takeda, K. Okumura, L. Van Kaer, et al. 2003. Nonredundant Roles for CD1d-restricted natural killer T cells and conventional CD4+ T cells in the induction of immunoglobulin E antibodies in response to interleukin 18 treatment of mice. J. Exp. Med. 197:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galli, G., S. Nutti, S. Tavarini, L. Galli-Stampino, C. De Lalla, G. Casorati, P. Dellabona, and S. Abrignani. 2003. CD1d-restricted help To B cells by human invariant natural killer T lymphocytes. J. Exp. Med. 197:1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leite-de-Moraes, M.C., A. Herbelin, C. Gouarin, Y. Koezuka, E. Schneider, and M. Dy. 2000. Fas/Fas ligand interactions promote activation-induced cell death of NK T lymphocytes. J. Immunol. 165:4367–4371. [DOI] [PubMed] [Google Scholar]
- 38.Osman, Y., T. Kawamura, T. Naito, K. Takeda, L. Van Kaer, K. Okumura, and T. Abo. 2000. Activation of hepatic NKT cells and subsequent liver injury following administration of alpha-galactosylceramide. Eur. J. Immunol. 30:1919–1928. [DOI] [PubMed] [Google Scholar]
- 39.Eberl, G., and H.R. MacDonald. 1998. Rapid death and regeneration of NKT cells in anti-CD3epsilon- or IL-12-treated mice: a major role for bone marrow in NKT cell homeostasis. Immunity. 9:345–353. [DOI] [PubMed] [Google Scholar]
- 40.Singh, A.K., M.T. Wilson, S. Hong, D. Olivares-Villagomez, C. Du, A.K. Stanic, S. Joyce, S. Sriram, Y. Koezuka, and L. Van Kaer. 2001. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J. Exp. Med. 194:1801–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyamoto, K., S. Miyake, and T. Yamamura. 2001. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature. 413:531–534. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez-Aseguinolaza, G., C. de Oliveira, M. Tomaska, S. Hong, O. Bruna-Romero, T. Nakayama, M. Taniguchi, A. Bendelac, L. Van Kaer, Y. Koezuka, and M. Tsuji. 2000. alpha-galactosylceramide-activated Valpha 14 natural killer T cells mediate protection against murine malaria. Proc. Natl. Acad. Sci. USA. 97:8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Exley, M.A., N.J. Bigley, O. Cheng, S.M. Tahir, S.T. Smiley, Q.L. Carter, H.F. Stills, M.J. Grusby, Y. Koezuka, M. Taniguchi, and S.P. Balk. 2001. CD1d-reactive T-cell activation leads to amelioration of disease caused by diabetogenic encephalomyocarditis virus. J. Leukoc. Biol. 69:713–718. [PubMed] [Google Scholar]
- 44.Ishikawa, H., H. Hisaeda, M. Taniguchi, T. Nakayama, T. Sakai, Y. Maekawa, Y. Nakano, M. Zhang, T. Zhang, M. Nishitani, et al. 2000. CD4(+) V(alpha)14 NKT cells play a crucial role in an early stage of protective immunity against infection with Leishmania major. Int. Immunol. 12:1267–1274. [DOI] [PubMed] [Google Scholar]
- 45.Kumar, H., A. Belperron, S.W. Barthold, and L.K. Bockenstedt. 2000. Cutting edge: CD1d deficiency impairs murine host defense against the spirochete, Borrelia burgdorferi. J. Immunol. 165:4797–4801. [DOI] [PubMed] [Google Scholar]
- 46.Kakimi, K., L.G. Guidotti, Y. Koezuka, and F.V. Chisari. 2000. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med. 192:921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeda, K., Y. Hayakawa, L. Van Kaer, H. Matsuda, H. Yagita, and K. Okumura. 2000. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc. Natl. Acad. Sci. USA. 97:5498–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakagawa, R., I. Nagafune, Y. Tazunoki, H. Ehara, H. Tomura, R. Iijima, K. Motoki, M. Kamishohara, and S. Seki. 2001. Mechanisms of the antimetastatic effect in the liver and of the hepatocyte injury induced by alpha-galactosylceramide in mice. J. Immunol. 166:6578–6584. [DOI] [PubMed] [Google Scholar]
- 49.van der Vliet, H.J., B.M. von Blomberg, N. Nishi, M. Reijm, A.E. Voskuyl, A.A. van Bodegraven, C.H. Polman, T. Rustemeyer, P. Lips, A.J. van den Eertwegh, et al. 2001. Circulating V(alpha24+) Vbeta11+ NKT cell numbers are decreased in a wide variety of diseases that are characterized by autoreactive tissue damage. Clin. Immunol. 100:144–148. [DOI] [PubMed] [Google Scholar]
- 50.Biron, C.A., and L. Brossay. 2001. NK cells and NKT cells in innate defense against viral infections. Curr. Opin. Immunol. 13:458–464. [DOI] [PubMed] [Google Scholar]