

Abstract
Helicobacter pylori toxin, VacA, damages the gastric epithelium by erosion and loosening of tight junctions. Here we report that VacA also interferes with T cell activation by two different mechanisms. Formation of anion-specific channels by VacA prevents calcium influx from the extracellular milieu. The transcription factor NF-AT thus fails to translocate to the nucleus and activate key cytokine genes. A second, channel-independent mechanism involves activation of intracellular signaling through the mitogen-activated protein kinases MKK3/6 and p38 and the Rac-specific nucleotide exchange factor, Vav. As a consequence of aberrant Rac activation, disordered actin polymerization is stimulated. The resulting defects in T cell activation may help H. pylori to prevent an effective immune response leading to chronic colonization of its gastric niche.
Keywords: MAP kinase signaling cascades, immunosuppression, host–pathogen interactions, calcium signaling
Introduction
Chronic infection by the gastric pathogen Helicobacter pylori causes peptic ulcer disease and is associated with gastric cancer (1). H. pylori produces a protein exotoxin, VacA, which causes vacuolar degeneration of epithelial cells in vitro and is believed to play an important role in the erosion of the gastric epithelium leading to ulcer formation (2, 3). VacA binds to target cells and then is slowly internalized to the cytoplasm where it is found associated with internal membrane-bound compartments (4, 5). Vacuole biogenesis requires the activity of the Rab7 GTPase and the V-type ATPase. VacA treatment also causes reduction in transepithelial resistance in epithelial monolayers in vitro (6). In addition, VacA interferes with cells of the immune system by inhibiting antigen processing in APCs, resulting in altered epitope presentation and recognition by effector cells (7), suggesting that VacA may contribute to the capacity of H. pylori to evade immune surveillance and chronically colonize the gastric mucosa.
More recently, VacA has been shown to form anion-selective channels both in artificial membranes (8) and in patch-clamped epithelial cells (9), which can be blocked by chloride channel inhibitors. Both vacuolating activity and transepithelial resistance reduction are suppressed by chloride channel inhibitors, and it has been suggested that anion channel formation could account for all of the biological activity of VacA (9). Several lines of evidence indicate that the initial interaction of VacA with its target cells is through a high affinity receptor (5, 10), which facilitates its interaction with the cell membrane to form the channels which are subsequently internalized. Three cell surface proteins have been implicated as specific receptors for VacA. In some cell types, the receptor-like tyrosine phosphatase RPTPβ has been shown to be required for cell vacuolation by VacA(11) and gastric damage (12). More recently, a second tyrosine phosphatase, RPTPα, has also been shown to act as a VacA receptor (13). Furthermore, some evidence implicates the epidermal growth factor receptor in VacA-induced pathology (14). On the other hand, both high affinity, saturable binding and low affinity, nonsaturable binding of VacA to some cell types has been described (10). Since VacA can interact directly with artificial membranes in the absence of specific receptors (15), it is likely that VacA interaction with target cells is complex and may involve both specific receptors and direct interaction with lipid components of the membrane.
VacA is released from the bacteria as high molecular weight homooligomers of a ∼90-kD polypeptide observable as flower-like structures by electron microscopy (16). In the hydrophobic environment of artificial membranes, VacA forms hexameric structures, which are likely to represent the ion channels (17). After release from the bacteria, each 90-kD oligomer undergoes slow proteolytic cleavage at a specific site in an exposed loop to produce two fragments of ∼35 and 58 kD (18). DNA transfection experiments have demonstrated that a NH2-terminal fragment of the protein containing the 35-kD fragment plus ∼110 amino acids of the 58-kD fragment (p58) are necessary and sufficient for vacuolation (19). On the other hand, p58 produced in H. pylori binds epithelial cells with similar kinetics to the holotoxin but lacks vacuolating activity and, in fact, does not enter the cell (20). Fig. 1 A shows the structure of the vacA gene and protein.
Figure 1.
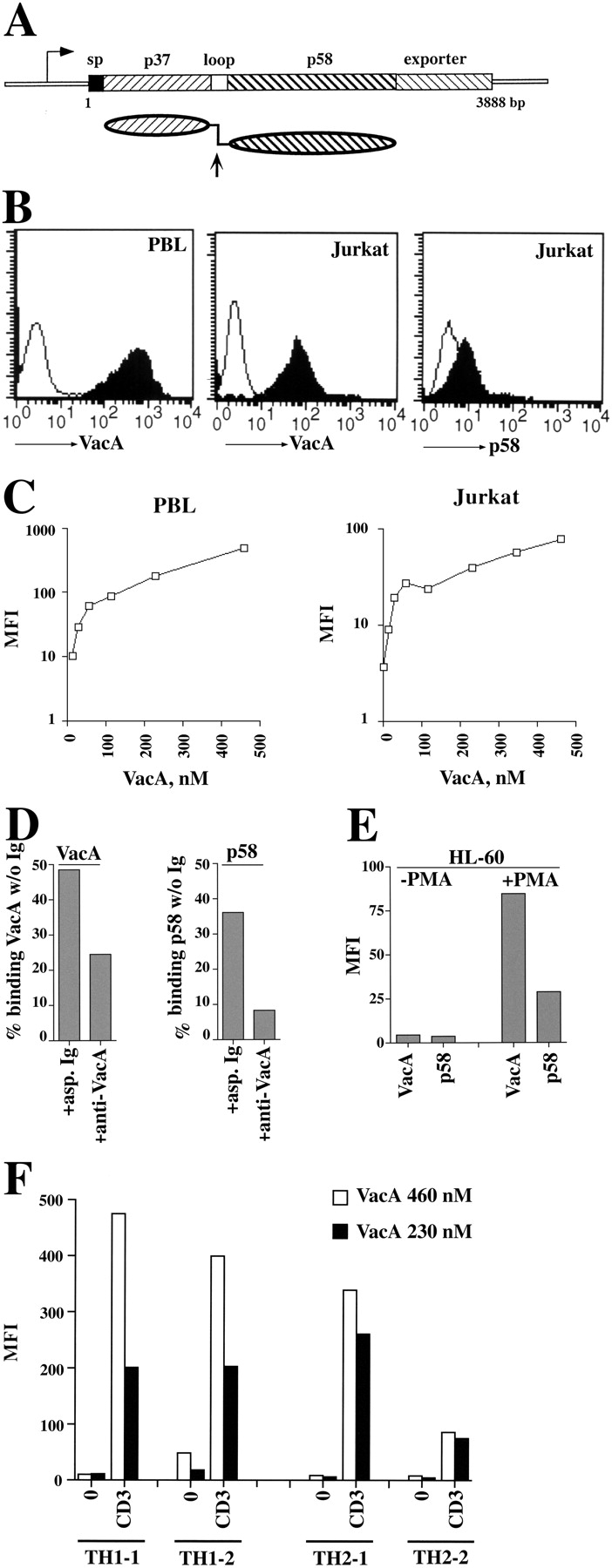
VacA binding on Jurkat T cells and human PBL and Th cells. (A) Schematic representation of the vacA gene and VacA protein. The 3,888-bp vacA gene from H. pylori strain CCUG17874 is represented showing regions encoding the signal peptide (sp), the 37-kD (p37) and 58-kD (p58) natural proteolytic fragments separated by the short hydrophilic region containing the cleavage site (loop), and the outer membrane autotransporter (exporter). Below is represented the mature secreted protein showing the two domains connected by a flexible loop. The arrow indicates the site of proteolytic cleavage between p37 and p58. (B) Flow cytometric analysis of VacA and p58 binding to purified human PBL (left) or Jurkat T cells (middle, right). VacA and p58 concentrations were 40 μg/ml (460 nM) and 80 μg/ml (1,365 nM), respectively. (C) Titration of VacA binding to PBL (left) or Jurkat cells (right) by flow cytometry. The data are expressed as mean fluorescence intensity (MFI). (D) Residual VacA or p58 binding to Jurkat cells after immunodepletion with either neutralizing anti-VacA or irrelevant (asp) Ig (starting VacA/p58 concentration 20 μg/ml, Ig 100 μg/ml). The data, obtained by flow cytometry, are expressed as the percentage of binding of the same concentration of untreated VacA/p58. (E) Flow cytometric analysis of VacA/p58 (40 μg/ml) binding to HL-60 cells, either untreated or incubated for 48 h with carrier or 20 ng/ml PMA. (F) Flow cytometric analysis of VacA binding to Th1 and Th2 cells, either unstimulated (0) or after 16-h stimulation with plastic-bound anti-CD3 mAb (CD3). Two representative Th1/Th2 clones are shown.
Although mice immunized with a variety of H. pylori antigens, including VacA, are protected from challenge, the robust immune response induced in man during natural H. pylori infection fails to prevent chronic colonization (21). The objective of this study was to assess whether, in addition to its activity on APCs, VacA might interfere with the immune system by interacting with T lymphocytes. The results demonstrate that VacA interferes with T cell activation by two independent mechanisms, only one of which is blocked by chloride channel inhibitors. The second activity, which appears independent of internalization as it is also induced by p58, may involve interaction with a specific receptor.
Materials and Methods
Cells, Reagents, Plasmids, and Antibodies.
Cells included the T lymphoma Jurkat line, a stably transfected Jurkat line expressing luciferase under the control of a trimer of the distal NF-AT binding site on the IL-2 promoter (22), the human leukemic line HL-60, and human PBL. HL-60 cells were used as such or following differentiation into macrophage-like cells by treatment for 48 h with 20 ng/ml PMA (23). PBMCs were purified from whole blood by density gradient centrifugation and subsequently depleted of monocytes by adherence. Monocytes were recovered for further analysis. Neutrophils were enriched by centrifugation on Dextran. Polarized human CD4+ Th clones were generated as described (24). Clones producing IFN-γ and TNF-β were categorized as Th1, and clones producing IL-4 and IL-5 were categorized as Th2. They were maintained in culture by periodical restimulation (every 12 d) with PHA (2.5 μg/ml) and rIL-2 (25 U/ml; Eurocetus) in the presence of irradiated PBMCs as feeder cells. IL-2 was replaced every 3 d to allow growth. All VacA binding experiments were performed on Th clones 1 d before restimulation.
VacA was purified from the culture supernatant of H. pylori strain CCUG17874 by ion exchange chromatography and size exclusion as described (25). VacA was activated by a short treatment at pH 2.0 before use (26). The 58-kD COOH-terminal fragment of VacA which extends from amino acid 330 to the end of the mature protein was purified from the culture supernatant of a H. pylori strain from which the DNA sequences encoding most of the NH2-terminal portion had been removed by allelic exchange (20). In some experiments, VacA (10–40 μg/ml) was preincubated for 30 min on ice with 100 μg/ml neutralizing anti-VacA or irrelevant rabbit Ig before addition to the cells. Alternatively, VacA/p58 (20 μg/ml) was immunodepleted by preincubation for 30 min on ice with 100 μg/ml neutralizing anti-VacA or irrelevant Ig and protein A–Sepharose (Amersham Biosciences). After removal of immune complexes by centrifugation, the supernatant was tested for residual activity. A23187 was from Roche Diagnostics SpA, ionomycin and 5-nitro-2-(3-phenylpropylamino)-benzoic acid (NPPB) were from Sigma-Aldrich, and Fluo-3 was from Molecular Probes Europe BV.
The anti-VacA mAb was made from a hybridoma (C1G9) produced from a mouse immunized with native highly purified VacA (27). The antibody, which recognizes a conformational epitope in the native toxin and the 58-kD COOH-terminal fragment, was protein G purified from the hybridoma supernatant. Neutralizing rabbit anti-VacA Ig were described previously (25). Phosphospecific antibodies recognizing the active forms of p38, Erk1/2, and MKK 3/6 were from Cell Signaling Technology, anti-p38 and anti-Erk antibodies were from Santa Cruz Biotechnology, antiphosphotyrosine mAb, anti–cyclooxygenase (COX)-2 polyclonal antibodies, and anti-Vav poly- and monoclonal antibodies were from Upstate Biotechnology. Fluorochrome-labeled anti-CD69 mAb and annexin V were from Becton Dickinson. IgG from OKT3 (anti-CD3; American Type Culture Collection) hybridoma supernatants were protein G purified. Unlabeled secondary antibodies were from Cappel, peroxidase-labeled antibodies were from Amersham Biosciences, and FITC-labeled anti–mouse antibodies were from DAKO SpA. Sepharose-conjugated GST-Pak and anti-Rac mAb were from Upstate Biotechnology, and PE-labeled phalloidin was from Sigma-Aldrich.
Transfections, Confocal Microscopy, and Flow Cytometry.
Jurkat cells were transiently transfected with the plasmid pEGFP/NFAT-1D (28) using a modification of the DEAE/dextran procedure as described (29). Each experiment was performed at least three times. Cells were treated for 30 min or 6 h at 37°C with 500 ng/ml A23187. Alternatively, anti-CD3 mAb was cross-linked for the same times at 37°C with secondary antibody. When VacA or p58 was used, cells were incubated for 1 h on ice with 5–40 μg/ml VacA or 40 μg/ml p58 and then further incubated on ice for 30 min with excess anti-VacA mAb in the presence or absence of anti-CD3 mAb. After washing, cells were added with either secondary antibodies and, when required, A23187, and further incubated at 37°C as described above. To analyze cortical actin reorganization, cells were either treated with anti-CD3 mAb or VacA/anti-VacA mAb as described above, and bound antibodies were cross-linked with secondary antibodies for 15 min at 37°C. Cells were then fixed and permeabilized in Orthopermeafix™ (Orthodiagnostic Systems Inc.) for 45 min at RT. After washing, cells were incubated in the dark for 40 min with 0.77 μM FITC-labeled phalloidin. Confocal microscopy was performed on a Leica Microsystems confocal microscope.
VacA/p58 binding was analyzed by indirect immunofluorescence. Cells were incubated on ice for 1 h with different concentrations of VacA/p58. After washing, cells were incubated with anti-VacA mAb followed, after a further wash, by FITC-labeled anti–mouse antibodies, and processed for flow cytometry. Negative controls included either VacA/p58 or anti-VacA mAb, each in combination with fluorochrome-labeled secondary antibodies. CD69 surface expression was quantitated using FITC-labeled anti-CD69 mAb. Samples were processed using a FACScan flow cytometer (Becton Dickinson). Calcium flux was measured as described using fluo-3 (30). Fluorescence was recorded continuously for 512 s. To analyze the effect of VacA/p58 on calcium influx induced by A23187, fluo-3–loaded cells, resuspended in HBSS added with Mg2+ and Ca2+, were incubated for 20 min at RT with 20 μg/ml VacA or 40 μg/ml p58. After washing, anti-VacA mAb was added and incubated for 10 min at RT before addition of A23187. When NPPB was used, fluo-3–loaded cells were preincubated for 2 min with inhibitor or carrier before subsequent additions. Apoptosis was analyzed by flow cytometry by labeling cells with FITC-labeled annexin V and 50 μg/ml propidium iodide as described (31) and gating on annexin V+PI− cells.
Activations, Immunoprecipitations, Immunoblots, and Luciferase Assays.
For immunoprecipitation and immunoblot experiments, activations by TCR–CD3 ligation in solution were performed as described by binding of anti-CD3 mAb on ice and subsequent cross-linking for 1–5 min with secondary antibodies at 37°C (29). When VacA or p58 were used, cells were incubated for 1 h on ice with different concentrations of these proteins, washed, and incubated on ice for a further 30 min with anti-VacA mAb in the presence or absence of anti-CD3 mAb followed, after a wash, by cross-linking with secondary antibodies for at 37°C. Alternatively, anti-CD3 mAb and excess secondary antibodies were added simultaneously and cross-linked for 5 min at 37°C. When VacA or p58 were used, cells were incubated for 30 min at 37°C with VacA and excess anti-VacA mAb, followed by a further 5 min at 37°C with excess secondary antibodies. Nonactivated samples were processed as above with either secondary antibodies or anti-VacA mAb and secondary antibodies. Cells (2 × 106 cells/sample) were lysed in 1% (vol/vol) Triton X-100 in 20 mM Tris-HCl, pH 8, 150 mM NaCl (in the presence of 0.2 mg/ml Na orthovanadate, 1 μg/ml pepstatin, leupeptin, and aprotinin and 10 mM PMSF) and resolved by SDS-PAGE. Alternatively, postnuclear supernatants from 2.5–5 × 107 cells/sample were immunoprecipitated using the appropriate polyclonal antibodies and protein A–Sepharose. Rac activity was measured by in vitro binding assays of postnuclear supernatants from 107 cells/sample using a GST–Pak1 fusion, which specifically pulls down GTP-bound, active Rac followed by immunoblot with an anti-Rac mAb. Immunoblots were performed using peroxidase-labeled secondary antibodies and a chemiluminescence detection kit (Pierce Chemical Co.).
NF-AT/luciferase reporter Jurkat cells were activated by CD3 cross-linking on secondary antibody-coated plates as described (29) using anti-CD3 Ab alone or in combination with VacA or p58. In the latter case, cells were incubated for 1 h on ice with VacA or p58, then added with anti-VacA mAb, incubated a further 30 min on ice, and plated on secondary antibody-coated plates. When required, NPPB or carrier was added 10 min before activation. Cells were collected 6–8 h after activation and processed for luciferase assays as described (22). All samples were in duplicate, and each experiment was repeated three to five times. Induction of CD69 expression by CD3 ligation in the presence or absence of VacA/p58 was performed as above, but cells were processed for flow cytometry 16–24 h after activation. Analysis of COX-2 expression was performed on neutrophils and macrophages treated with VacA for 24 h as described for T cells. LPS activation of macrophages was performed by incubation for 10 min at 37°C with 10 μg/ml LPS.
Immunohistochemistry.
The use of gastric tissue for this study was approved by the Institutional and Ethical Review Boards and written informed consent was obtained from each patient before gastric sampling. Gastric specimens were formalin fixed from five patients infected by H. pylori type I strains and five healthy controls. Peroxidase staining was conducted with the ABC elite kit from Vector Laboratories. Mouse anti–phospho-p38 mAb (28B10) was from Cell Signaling Technology.
Results
VacA Binds to T Cells.
Fig. 1 shows VacA binding to T cells. Indirect immunofluorescence labeling and flow cytometric analysis of VacA-treated cells using a mAb against a conformational epitope in the p58 domain (27) revealed extensive binding both to Jurkat T cells and to freshly isolated PBL (Fig. 1 B) including both CD4+ and CD8+ subsets (not depicted). A dose–response curve showed a rapid increase in VacA binding at low concentrations followed by a slower increase which did not reach saturation (Fig. 1 C). These kinetics most likely reflect the previously described interactions with a high affinity receptor and low affinity, nonsaturable interactions (10). As shown for other cell types (20), p58 also bound to Jurkat cells albeit at lower levels than intact VacA (Fig. 1 B). That the binding activity revealed is in fact due to VacA or p58 has been confirmed by immunodepletion experiments using a VacA-specific neutralizing antiserum raised against intact native VacA (25) (Fig. 1 D). Furthermore, VacA and p58 binding to HL-60 cells increased substantially after treatment with phorbol ester consistent with the increase in sensitivity to VacA described by Padilla et al. (23), further supporting the specificity of the assay (Fig. 1 E). VacA binding to human Th1 and Th2 clones, allowed to come to rest in the absence of IL-2, was also detectable. Interestingly, TCR–CD3 ligation on both Th1 and Th2 clones resulted in increase of VacA binding by up to 50-fold (Fig. 1 F).
VacA Inhibits T Cell Activation.
Cell surface expression of the T cell activation marker CD69 is highly up-regulated by TCR–CD3 ligation. Fig. 2 shows that treatment with VacA completely inhibited the CD3-induced expression of CD69 on the T cell surface. Incubation with p58 resulted in reduction of CD69 expression (Fig. 2 A, right). Furthermore, VacA treatment inhibited CD69 expression on both CD4+ and CD8+ subsets of human PBL (Fig. 2 B). Hence VacA appears to effectively block TCR–CD3-dependent T cell activation. The inhibitory activity of VacA was not due to effects on cell viability nor to triggering of apoptosis as assayed by annexin V and propidium iodide staining (not depicted).
Figure 2.

VacA inhibits the induction of CD69 surface expression in response to TCR–CD3 engagement. (A) Flow cytometric analysis of CD69 expression on Jurkat cells activated for 16 h by CD3 cross-linking in the presence or absence of either 20 μg/ml VacA or 40 μg/ml p58. (B) CD69 expression on human PBL activated for 24 h by CD3 cross-linking in the presence or the absence of 20 μg/ml VacA. Cells were counterstained with fluorochrome-labeled anti-CD4 or anti-CD8 mAb, and CD69 expression was measured on gated CD4+ (left) or CD8+ (right) T cells.
VacA Inhibits NF-AT Activation.
NF-AT is a key transcription factor which becomes active within the first hour after TCR triggering and is essential for T cell activation and their subsequent capacity to express IL-2 and other cytokines (32). We assessed the capacity of VacA to inhibit this early stage of T cell activation. As a readout for NF-AT activation, we used a Jurkat cell line stably transfected with a luciferase gene under the control of a synthetic NF-AT–dependent promoter (22). VacA inhibited activation of the reporter by CD3 cross-linking in a dose–responsive fashion (Fig. 3 A). Cells were quite sensitive to VacA, which caused ∼80% reduction of luciferase activity at a concentration of ∼50 nM, similar to that required to achieve full vacuolation in epithelial cell lines. Inhibition of NF-AT activation was independent of VacA cross-linking with antibody (Fig. 3 C), was reduced significantly by preincubation with neutralizing anti-VacA antiserum (<30% residual inhibition at 20 μg/ml), and heat-inactivation of VacA abrogated the inhibitory effect (Fig. 3 C). No vacuoles were visible in the VacA-treated cells, although vacuolation may not have been detected because of the small size of the cytoplasm (not depicted). Pretreatment of the cells with the chloride channel inhibitor NPPB completely abrogated the effect of VacA such that CD3-dependent luciferase activity was not significantly different from that induced in the absence of VacA (Fig. 3 E). These data indicate that the effect of VacA is specific and dependent on its capacity to form ion channels in the cell membranes.
Figure 3.
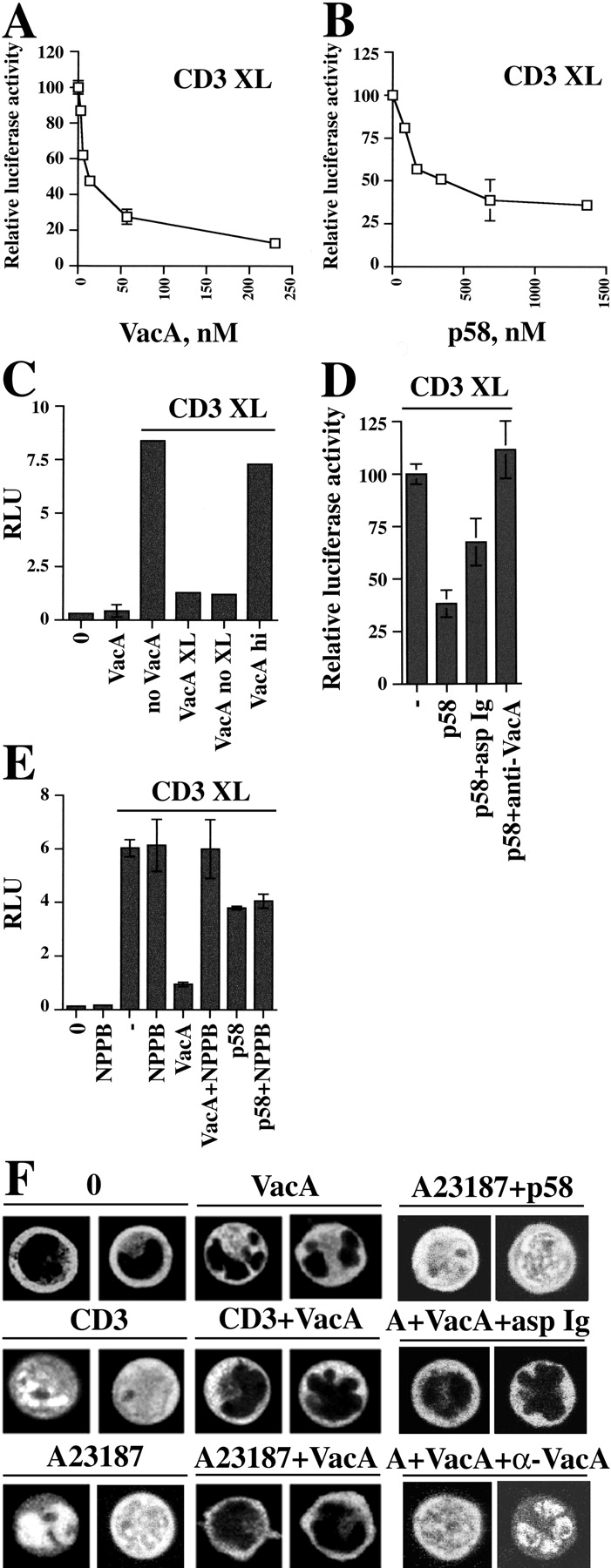
Inhibition of CD3-dependent NF-AT activation and nuclear translocation by VacA. Relative luciferase activity in NFAT/luciferase reporter Jurkat cells activated by CD3 cross-linking (CD3 XL) in the presence of different concentrations of VacA (A) or p58 (B). Luciferase activity induced by CD3 cross-linking in the absence of VacA was taken as 100%. (C) Relative luciferase units (RLU) in reporter Jurkat cells activated by CD3 cross-linking in the presence or absence of 20 μg/ml VacA. VacA was either cross-linked using anti-VacA mAb and plate-bound secondary antibodies (VacA XL) or bound to Jurkat cells and plated on secondary antibodies without anti-VacA mAb (VacA noXL). VacA hi, heat-inactivated VacA (boiled 5 min), cross-linked. (D) Relative luciferase activity (percentage of activity triggered by CD3 cross-linking) in reporter Jurkat cells activated by CD3 cross-linking in the presence or absence of 20 μg/ml p58. p58 was used either as such or after preincubation with 100 μg/ml neutralizing anti-VacA (p58+α-VacA) or irrelevant Ig (p58+asp Ig) and cross-linked using anti-VacA mAb and plate-bound secondary antibodies. (E) Relative luciferase units (RLU) in reporter Jurkat cells activated by CD3 cross-linking in the presence or absence of 20 μg/ml VacA or 80 μg/ml p58 and NPPB (100 μM). VacA was cross-linked using anti-VacA mAb and plate-bound secondary antibodies. NPPB was added 10 min before anti-CD3 mAb or VacA. (F) Confocal microscopy of Jurkat cells transiently transfected with a NFAT–GFP expression construct, either unstimulated (0) or after treatment with 500 ng/ml A23187 or CD3 cross-linking for 30 min, either in the absence or the presence of 40 μg/ml VacA/p58. In the neutralization assay, VacA (10 μg/ml) was used either as such or after preincubation with 100 μg/ml neutralizing anti-VacA (A+VacA+α-VacA) or irrelevant Ig (A+VacA+asp Ig). A23187 induced nuclear translocation in ∼100% of cells. In the presence of VacA, no cells showed nuclear staining. Pretreatment of VacA with neutralizing anti-VacA antiserum resulted in >80% translocation versus <15% translocation in the presence of nonspecific Ig (number of fluorescent cells scored ≥50).
To confirm this hypothesis p58, which has no vacuolating activity and is not internalized in epithelial cells (20), was tested. Treatment of Jurkat cells with p58 at concentrations similar to those used for VacA had negligible effect on CD3-induced NF-AT activity. At higher concentrations, p58 treatment resulted in some inhibition, reaching 50–60% inhibition at 10-fold higher concentration (Fig. 3 B). The inhibition was specific to p58, since it was completely abrogated by preincubation with neutralizing anti-VacA antiserum (Fig. 3 D). However, inhibition of chloride channels with NPPB failed to reverse the p58 effect (Fig. 3 E), suggesting that VacA may have two different effects on T cell activation, of which only one is dependent on ion channel formation and internalization.
NF-AT Fails to Translocate to the Nucleus in the Presence of VacA.
Two independent intracellular signals triggered by the TCR–CD3 complex are required for NF-AT activation (32). On the one hand, an increase in [Ca2+] activates the phosphatase calcineurin which dephosphorylates the cytoplasmic form of NF-AT, resulting in its accumulation in the nucleus. On the other hand, triggering of tyrosine kinases (PTKs) leads to activation of the Ras/mitogen-activated protein kinase (MAPK) signaling pathway and phosphorylation of both the AP-1 transcription factor and nuclear NF-AT, which cooperate to activate transcription. The effect of VacA on NF-AT nuclear translocation was assessed using Jurkat cells transiently transfected with a construct encoding a NF-AT–GFP fusion protein. The results are presented in Fig. 3 F. In untreated cells, fluorescence was detected in the cytoplasm but was completely excluded from the nucleus. CD3 cross-linking resulted in translocation of fluorescence to the nucleus. VacA completely blocked the nuclear translocation of NF-AT induced by anti-CD3 cross-linking. Inhibition of translocation persisted even after 6-h treatment (unpublished data). Remarkably, VacA blocked NF-AT translocation induced by A23187, a powerful calcium ionophore which results in massive increase of [Ca2+] independently of TCR signaling. Preincubation of VacA with neutralizing antiserum abrogated the VacA-dependent block of translocation. Of note, p58 did not affect CD3-dependent NF-AT translocation. Thus, while the dramatic effects of VacA on NF-AT translocation explain the complete abrogation by VacA of NF-AT–dependent luciferase expression, a different mechanism appears to underlie the inhibitory activity of p58 on NF-AT activation.
VacA Blocks Increases in Intracellular Calcium.
Jurkat cells were loaded with the calcium-sensitive fluorescent dye fluo-3 then treated with A23187 and VacA. In the absence of VacA, A23187 caused a massive increase in intracellular [Ca2+], which was strongly inhibited by VacA (Fig. 4 A). VacA also inhibited calcium flux caused by a second carboxylic ionophore, ionomycin (not depicted). Hence, the inhibition of agonist-induced NF-AT translocation to the nucleus by VacA can be attributed to its capacity to block the increase in [Ca2+]i. In agreement with its lack of effect on NF-AT translocation, p58 did not affect calcium mobilization induced by A23187 (Fig. 4 B). In the presence of NPPB, VacA had no effect on the A23187-induced increase in [Ca2+] (Fig. 4 C), suggesting that the opening of anion channels can influence calcium influx and that these channels contribute to the regulation of intracellular [Ca2+].
Figure 4.

Anion channel-dependent inhibition of calcium mobilization by VacA. (A) Flow cytometric analysis of intracellular calcium flux in fluo-3–loaded Jurkat cells treated with 500 ng/ml A23187 in the absence or the presence of 20 μg/ml VacA. MFI is shown in the top corner of each panel. (B) Flow cytometric analysis of intracellular calcium flux in fluo-3–loaded Jurkat cells treated with 500 ng/ml A23187 in the absence or the presence of 20 μg/ml VacA or 40 μg/ml p58. The data are expressed as fold increase of fluorescence after addition of A23187. (C) Flow cytometric analysis showing the MFI of fluo-3–loaded Jurkat cells after treatment with A23187 (A, 500 ng/ml) and 20 μg/ml VacA in the presence or absence of NPPB (50 μM).
Although the block of TCR-induced [Ca2+] by VacA is sufficient to explain its inhibition of T cell activation, the fact that p58 also inhibits induction of CD69 expression and NF-AT activation independently of anion channel formation and calcium influx indicates that VacA may have two independent activities on these cells. Since p58 binds to cells but is not internalized (20), its activity may be due to interaction with cell surface receptors and activation of intracellular signaling pathways.
VacA Triggers Signaling to p38 Stress Kinase.
TCR signaling through the TCR–CD3 complex results in rapid PTK activation which leads to phosphorylation of several cytoplasmic proteins. This cascade leads to activation of the Ras/MAPK pathway and subsequently to phosphorylation and activation of transcription factors such as AP-1 which interacts with nuclear NF-AT to form a functional heterodimer. In addition, these signals lead to actin cytoskeleton rearrangements necessary for the functional interaction between T cell and APC (33). Interestingly, treatment of Jurkat cells with VacA resulted in substantial increase of tyrosine phosphoproteins independent of TCR triggering (Fig. 5 A), suggesting that VacA affects the activity of PTKs. However, the pattern of phosphotyrosine-containing bands was different to that achieved by activation through the TCR. In fact, a discrete subset of the proteins phosphorylated after TCR triggering were phosphorylated in response to VacA.
Figure 5.

Activation of protein tyrosine phosphorylation and p38 activation by VacA. (A) Antiphosphotyrosine immunoblot of postnuclear supernatants from Jurkat cells either nonactivated (0) or activated by cross-linking of CD3 or 40 μg/ml VacA. (B–F) Immunoblot analysis of postnuclear supernatants from Jurkat cells, nonactivated (0) or activated by either TCR–CD3 (CD3) or VacA or p58 cross-linking in the presence or absence of 100 μM NPPB. hi, heat-inactivated VacA; no XL, VacA and secondary antibodies without anti-VacA mAb. The concentration of VacA used in B (right) and F was 40 and 5 μg/ml, respectively. In E, VacA or p58 were tested either as such or after immunodepletion with neutralizing anti-VacA or irrelevant (asp) Ig (starting VacA/p58 concentration 20 μg/ml, Ig 100 μg/ml). Similar differences could be observed when cells where treated with VacA or p58 in the presence of neutralizing or control Ig (not depicted). Each filter was probed by immunoblot with anti–phospho-Erk (B) or anti–phospho-p38 (C–F) antibodies. After stripping, all filters were reprobed with control antibodies as indicated.
Two major classes of MAPK, the “classical” MAPK represented by Erk 1/2 and the related stress-activated kinase p38 become phosphorylated in response to TCR triggering, and both are essential for T cell activation (34). We have assessed the effect of VacA treatment on these kinases using antibodies which recognize specifically the phosphorylated, active forms of the proteins. Treatment of Jurkat cells with VacA had no effect on either the basal or CD3-induced phosphorylation of Erk1/2 (Fig. 5 B). However VacA did result in substantial increase in tyrosine phosphorylation of the stress-activated kinase p38 even in the absence of CD3 cross-linking. Fig. 5 C shows anti–phospho-p38 blots of protein extracts from Jurkat cells treated with different VacA concentrations. p38 was activated to the level achieved by CD3 cross-linking by incubation with <1 μg/ml VacA, which is considerably less than that required for NF-AT inhibition in Jurkat cells or vacuolation in HeLa cells. No effect on p38 phosphorylation was observed when cells were treated with heat-inactivated VacA (Fig. 5 C). Of note, p58 also caused activation of p38 in Jurkat cells with a dose response similar to that of VacA (Fig. 5 D). Preincubation with anti-VacA neutralizing antibodies or immunodepletion of VacA/p58 reduced the effect elicited by both VacA and p58 (Fig. 5 E and not depicted). The capacity of p58 to activate p38 indicates that p38 activation is independent of the chloride channel activity of VacA. In support of this, NPPB did not affect p38 activation by VacA (Fig. 5 F).
Major activators of p38 in T cells are the serine-threonine kinases MKK3/6 which in turn are linked to transmembrane signaling molecules through Vav, an exchange factor specific for Rho family GTPases (35, 36). VacA treatment of Jurkat cells resulted in phosphorylation of these molecules. Fig. 6 shows anti–phospho-MKK3/6 blots of cell extracts (A) and antiphosphotyrosine blots of Vav-specific immunoprecipitates (B) from Jurkat cells treated with VacA. VacA induced substantial phosphorylation of MKK3/6 and a prolonged Vav phosphorylation for up to 15 min. Vav activation by VacA was not inhibited by NPPB (Fig. 6 C), indicating that this effect is also independent of its chloride channel activity. These data strongly support the notion that VacA interacts with a cell surface receptor which is linked to the Vav-MKK3/6-p38 signaling pathway.
Figure 6.
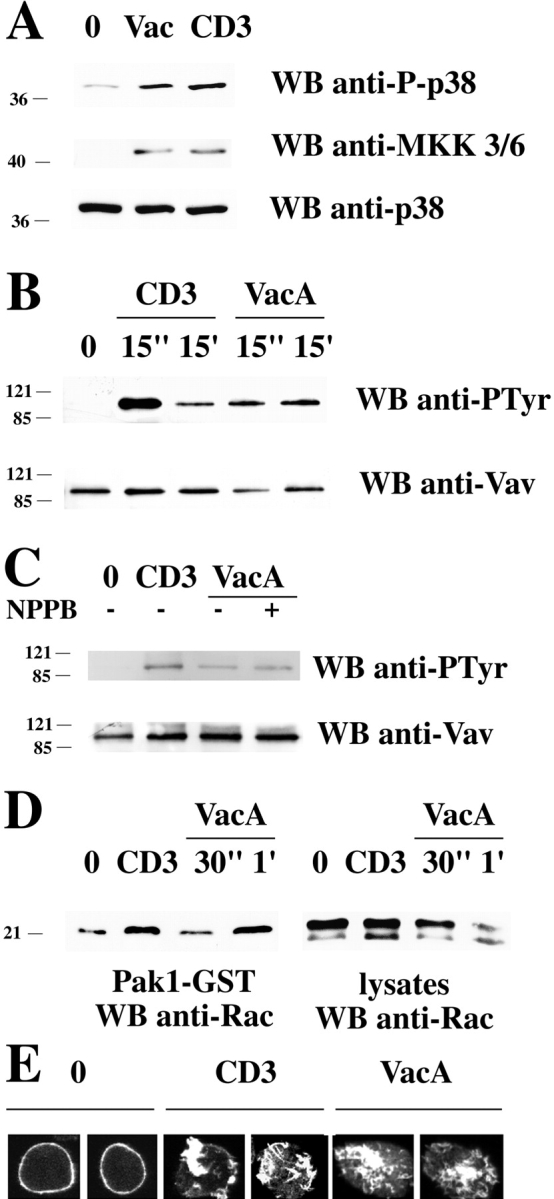
Activation of MKK3/6 and the Vav/Rac pathway of actin cytoskeleton reorganization by VacA. (A) Immunoblot analysis of postnuclear supernatants from Jurkat cells, nonactivated (0) or activated by either TCR–CD3 (CD3) or VacA (40 μg/ml) cross-linking, with anti–phospho-p38 and anti–phospho-MKK3/6 antibodies. (B) Immunoblot analysis using antiphosphotyrosine mAb of Vav-specific immunoprecipitates from Jurkat cells activated by TCR–CD3 or VacA cross-linking as in A for 15 s or 15 min (C) Immunoblot analysis using antiphosphotyrosine mAb of Vav-specific immunoprecipitates from Jurkat cells activated by TCR–CD3 or VacA cross-linking as in A for 15 s in the presence or absence of 100 μM NPPB (1-h pretreatment before addition of VacA). (D) Immunoblot analysis with anti-Rac mAb of in vitro binding assays of postnuclear supernatants from Jurkat cells using agarose-conjugated Pak1-GST. Equal amounts of postnuclear supernatants from the same samples were separated on the same gel (right). Cells were either nonactivated (0) or activated by cross-linking of CD3 (30 s) or 40 μg/ml VacA (30 s and 1 min). All filters were stripped and reprobed with control antibodies as indicated. (E) Confocal microscopy of FITC-phalloidin–labeled Jurkat cells. Cells were either nonactivated (0) or activated by cross-linking of CD3 or 20 μg/ml VacA for 15 min.
VacA Induces Rac1-dependent Cytoskeleton Rearrangement.
Vav is coupled to reorganization of the actin cytoskeleton through its exchange activity on the small GTPase Rac (35). Rac1 has also been implicated in VacA-induced vacuolation in epithelial cells (37). Fig. 6 D shows the results of assays of Rac activity in VacA-treated T cells. VacA treatment substantially increased Rac activity within 1 min of treatment. The effect of this activation could also be observed directly by its effect on cortical actin distribution in VacA-treated cells. Fig. 6 E shows fluorescence micrographs of VacA or anti-CD3–treated cells stained with phalloidin which specifically binds F-actin. Both VacA and CD3 cross-linking caused massive actin reorganization. This effect of VacA was not inhibited by NPPB (not depicted).
p38 Is Activated in H. pylori–colonized Gastric Mucosa.
To assess the in vivo relevance of p38 activation, gastric sections from H. pylori–infected patients were stained for phosphorylated p38. Gastric biopsies were obtained from five H. pylori–infected subjects and five healthy H. pylori–negative volunteers. Phospho-p38 was detected in glandular epithelial and lymphomononuclear-like cells in sections from all five H. pylori–infected patients but not from any of the healthy H. pylori–noninfected subjects (Fig. 7 A).
Figure 7.
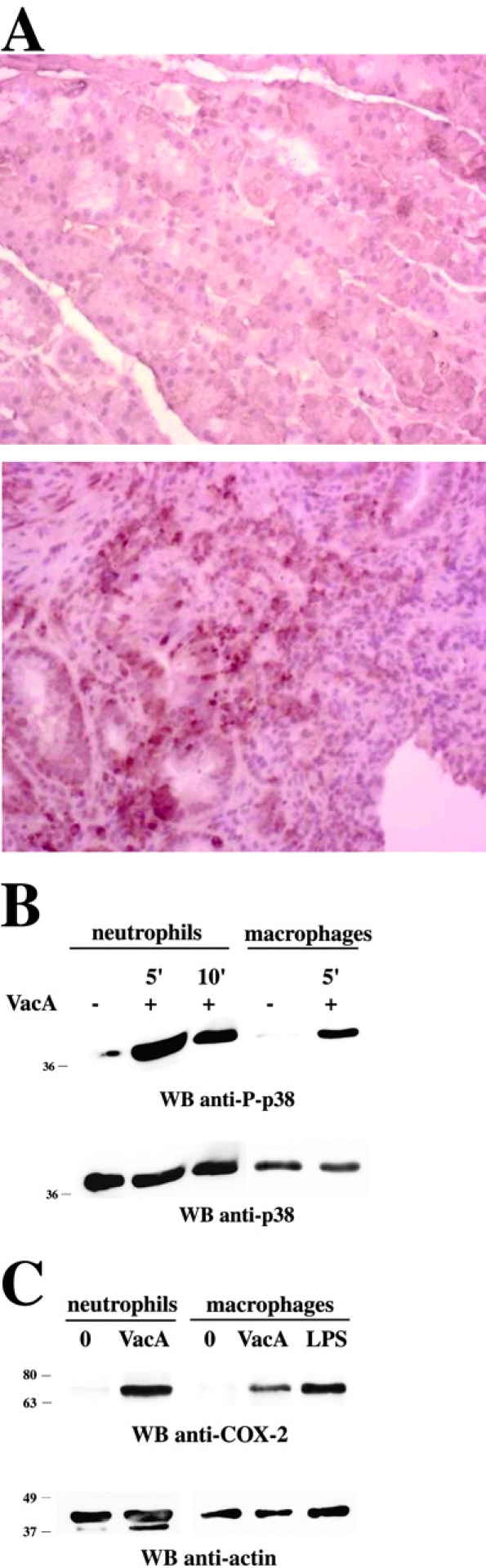
p38 activity is detectable in gastric biopsies of H. pylori–positive patients and in VacA-treated neutrophils and macrophages. (A) Immunohistochemistry of activated p38 in gastric mucosa of H. pylori–infected patient. Phosphorylated p38 antigens are identified by brown peroxidase staining. No staining was observed in the absence of primary antibody (not depicted). (Top) Healthy H. pylori–negative control (no positive anti–phospho-p38 staining). (Bottom) H. pylori–infected patient (positive anti–phospho-p38 staining). Original magnification 40×. (B) Immunoblot analysis with anti–phospho-p38 antibodies of postnuclear supernatants from purified human neutrophils or macrophages treated with 20 μg/ml VacA for 5 or 10 min as indicated. (C) Immunoblot analysis with anti–COX-2 antibodies of postnuclear supernatants from purified human neutrophils or macrophages treated with 20 μg/ml VacA for 24 h. Macrophages activated with 10 μg/ml LPS were used as a positive control. After stripping, all filters were reprobed with control antibodies as indicated.
VacA Activates p38 in Neutrophils and Macrophages.
Since extensive staining of infiltrating inflammatory cells was observed, we questioned whether VacA was capable of activating p38 in other immune cells. Neutrophils and macrophages were enriched from human blood and treated with VacA, and p38 activation was assayed by immunoblot analysis of cell lysates with anti–phospho-p38 antibodies. In both cell types, VacA treatment resulted in substantial activation of p38 (Fig. 7 B), indicating that activation of this kinase is not limited to T cells but also occurs in other cell types known to infiltrate the gastric epithelium during H. pylori colonization. Heat-inactivated VacA had no effect (not depicted).
A consequence of p38 activation in macrophages and neutrophils is increased expression of COX-2, which contributes to inflammation by production of proinflammatory prostanoids (38). VacA treatment induced increased expression of COX-2 in these cells (Fig. 7 C). Together, these data suggest that VacA may also play a role in H. pylori–induced gastric inflammation.
Discussion
Two considerations emerge from the data reported here. On the one hand, we describe signaling pathways which are modulated by VacA in T cells and which may be relevant to the interaction of VacA with other cell types in vivo as clearly exemplified by the induction of p38 activity and COX-2 expression in neutrophils and macrophages, as well as the presence of phospho-p38 in gastric biopsies from H. pylori–positive patients. On the other hand, the profound effect on T cell activation caused by VacA may play a role in vivo in preventing an appropriate protective immune response against infection by H. pylori and thus contribute to the chronicity of colonization.
The ability of VacA to block modulation of intracellular calcium is likely to have profound effects on many cell types in vivo. In T cells, calcium influx from extracellular sources is essential for cell activation, proliferation, and activation of effector functions (39). The major channels employed by T cells to elicit the prolonged increase in [Ca2+] in response to TCR triggering are the store-operated calcium (CRAC) channels. The driving force for calcium influx through CRAC channels and by carboxylic ionophores is membrane potential. The most likely explanation of the inhibitory activity of VacA on calcium influx is that it causes membrane depolarization by opening unregulated anion channels (8). This is supported by the fact that the chloride channel blocker NPPB restored calcium flux in the presence of VacA.
We have demonstrated a second activity of VacA on T cells which is independent of its channel-forming capacity and involves activation of a subset of the PTKs normally activated by TCR engagement. This leads to activation of p38 stress kinase but not of Erk1/2. NPPB had no effect on this activity, and the channel-defective p58 mutant was fully active in these assays. It should be noted that maximal p38 kinase activation was achieved at VacA concentrations well below those required to inhibit NF-AT through channel formation and hence could reflect an important response to limited VacA.
PTK triggering is usually triggered by cell surface receptors although forced coalescence of lipid rafts by cross-linking of cholera toxin bound to GM1 ganglioside in rafts can mimic TCR signaling (40, 41). Both these mechanisms could play a role in VacA activation of PTK activity. VacA has been shown recently to interact with lipid rafts in cells of a gastric epithelial line; however, VacA-induced raft coalescence was not demonstrated (42). On the other hand, VacA interacts with the receptor tyrosine phosphatases, RTPTα and RTPTβ (11, 13), in gastric cell lines, and these receptors are known to be present on the surface of T cells. Surprisingly, however, gastric cells isolated from RTPTβ−/− mice were equally susceptible to VacA vacuolation as cells from WT siblings, although gastric epithelial damage by VacA was significantly reduced in the knockout mice (43). Tyrosine phosphatases are involved in both activation and feedback control of the PTK cascade in T cells (44). Thus, the partial activation of kinases in T cells by VacA could be due to activation of RPTPα/β or other VacA receptor molecules.
The capacity of VacA to induce intracellular signaling and to activate p38 in inflammatory cells such as neutrophils and macrophages may play an important role in H. pylori pathogenesis. It is of note that the concentration of VacA required to activate p38 in T cells was much lower than that required for the channel-dependent activity or that required for epithelial cell vacuolation. Thus, the channel-dependent activity may be more relevant close to the VacA-producing bacteria, whereas the signaling activity may be more relevant at sites further removed where concentrations are lower. VacA-triggered signaling through RPTPβ was also demonstrated in gastric epithelial cells. In these experiments, VacA caused detachment of epithelial cells from the basement membrane in WT mice but not RPTPβ−/− mice (43). This phenomenon may be related to the Rac-dependent actin cytoskeleton rearrangement described here.
Regardless of the mechanism of kinase activation by VacA, the rearrangement of actin cytoskeleton induced through Vav/Rac activation outside of the normal context of T cell activation is likely to interfere with the ordered rearrangement required for the formation of the immunological synapse between T cells and APCs (45). Actin reorganization is essential also for the coalescence of lipid rafts containing the TCR–CD3 complex and important signaling molecules and the resulting triggering of the kinase cascade (41, 46). Subversion of signaling to the actin cytoskeleton by modification of Rho family GTPases, resulting in their inhibition or activation, is in fact a common denominator among bacterial toxins, including Clostridium difficile toxin B, C. sordellii lethal toxin, and cytotoxic necrotizing factor-1 of uropathogenic Escherichia coli strains (47).
Infection with H. pylori induces a vigorous systemic immune response which is, however, insufficient to prevent colonization of its gastric niche. Disease is associated with a Th1 response in the gastric mucosa, and this is believed to be partially responsible for the gastric inflammation characteristic of H. pylori–induced disease (24). Both oral and systemic immunization with purified antigens and an appropriate adjuvant can nevertheless confer protection against challenge in animal models and can even force eradication of chronically colonized mice in a therapeutic setting (48). This would suggest that H. pylori can either avoid evoking the appropriate immune response on infection or has mechanisms by which to control the response at least locally. VacA has been demonstrated to interfere with antigen presentation in B cells by inhibiting correct proteolytic processing of internalized antigens and presentation of the resulting peptides by MHC (7). It has been suggested that this activity may play a role in immune evasion. Suppression or modification of T cell responses by VacA-dependent modulation of signaling during T cell activation or differentiation might represent an additional effective strategy of immune evasion by H. pylori.
Acknowledgments
The authors wish to thank F. Tombola for helpful suggestions and Daniella Rinaudo for help with neutrophil purification.
This work was supported by grants from the Ministero dell' Università e della Ricerca Scientifica e Tecnologica (PRIN), the European Commission (ICA4-1999-10010), the Associazione Italiana per la Ricerca sul Cancro, and Telethon (E.1161).
Note added in proof: While the revised version of this manuscript was being prepared, Gebert et al. (2003. Science. 301:1099–1102) reported a similar inhibitory activity of VacA on T cell activation, which could also be correlated to inhibition of nuclear translocation of NF-AT.
M. Boncristiano and S. Rossi Paccani contributed equally to this work.
D. Ilver's present address is Institute of Medical Biochemistry, Göteborg University, 40530 Göteborg, Sweden.
Abbreviations used in this paper: COX, cyclooxygenase; MAPK, mitogen-activated protein kinase; NPPB, 5-nitro-2-(3-phenylpropylamino)-benzoic acid.
References
- 1.Blaser, M.J. 1998. Helicobacter pylori and gastric diseases. BMJ. 316:1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Telford, J.L., A. Covacci, P. Ghiara, C. Montecucco, and R. Rappuoli. 1994. Unravelling the pathogenic role of Helicobacter pylori in peptic ulcer: potential new therapies and vaccines. Trends Biotechnol. 12:420–426. [DOI] [PubMed] [Google Scholar]
- 3.Cover, T.L. 1996. The vacuolating cytotoxin of Helicobacter pylori. Mol. Microbiol. 20:241–246. [DOI] [PubMed] [Google Scholar]
- 4.Garner, J.A., and T.L. Cover. 1996. Binding and internalization of the Helicobacter pylori vacuolating cytotoxin by epithelial cells. Infect. Immun. 64:4197–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massari, P., R. Manetti, D. Burroni, S. Nuti, N. Norais, R. Rappuoli, and J.L. Telford. 1998. Binding of the Helicobacter pylori vacuolating cytotoxin to target cells. Infect. Immun. 66:3981–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papini, E., B. Satin, N. Norais, M. de Bernard, J.L. Telford, R. Rappuoli, and C. Montecucco. 1998. Selective increase of the permeability of polarized epithelial cell monolayers by Helicobacter pylori vacuolating toxin. J. Clin. Invest. 102:813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molinari, M., M. Salio, C. Galli, N. Norais, R. Rappuoli, A. Lanzavecchia, and C. Montecucco. 1998. Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin VacA. J. Exp. Med. 187:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tombola, F., C. Carlesso, I. Szabo, M. de Bernard, J.M. Reyrat, J.L. Telford, R. Rappuoli, C. Montecucco, E. Papini, and M. Zoratti. 1999. Helicobacter pylori vacuolating toxin forms anion-selective channels in planar lipid bilayers: possible implications for the mechanism of cellular vacuolation. Biophys. J. 76:1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szabo, I., S. Brutsche, F. Tombola, M. Moschioni, B. Satin, J.L. Telford, R. Rappuoli, C. Montecucco, E. Papini, and M. Zoratti. 1999. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 18:5517–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, W.C., H.J. Wang, and C.H. Kuo. 2001. Two distinctive cell binding patterns by vacuolating toxin fused with glutathione S-transferase: one high-affinity m1-specific binding and the other lower-affinity binding for variant m forms. Biochemistry. 40:11887–11896. [DOI] [PubMed] [Google Scholar]
- 11.Yahiro, K., T. Niidome, M. Kimura, T. Hatakeyama, H. Aoyagi, H. Kurazono, K. Imagawa, A. Wada, J. Moss, and T. Hirayama. 1999. Activation of Helicobacter pylori VacA toxin by alkaline or acid conditions increases its binding to a 250-kDa receptor protein-tyrosine phosphatase beta. J. Biol. Chem. 274:36693–36699. [DOI] [PubMed] [Google Scholar]
- 12.Fujikawa, A., D. Shirasaka, S. Yamamoto, H. Ota, K. Yahiro, M. Fukada, T. Shintani, A. Wada, N. Aoyama, T. Hirayama, et al. 2003. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat. Genet. 33:375–381. [DOI] [PubMed] [Google Scholar]
- 13.Yahiro, K., A. Wada, M. Nakayama, T. Kimura, K. Ogushi, T. Niidome, H. Aoyagi, K. Yoshino, K. Yonezawa, J. Moss, and T. Hirayama. 2003. Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor. J. Biol. Chem. 278:19183–19189. [DOI] [PubMed] [Google Scholar]
- 14.Seto, K., Y. Hayashi-Kuwabara, T. Yoneta, H. Suda, and H. Tamaki. 1998. Vacuolation induced by cytotoxin from Helicobacter pylori is mediated by the EGF receptor in HeLa cells. FEBS Lett. 431:347–350. [DOI] [PubMed] [Google Scholar]
- 15.Pagliaccia, C., X.M. Wang, F. Tardy, J.L. Telford, J.M. Ruysschaert, and V. Cabiaux. 2000. Structure and interaction of VacA of Helicobacter pylori with a lipid membrane. Eur. J. Biochem. 267:104–109. [DOI] [PubMed] [Google Scholar]
- 16.Reyrat, J.M., R. Rappuoli, and J.L. Telford. 2000. A structural overview of the Helicobacter cytotoxin. Int. J. Med. Microbiol. 290:375–379. [DOI] [PubMed] [Google Scholar]
- 17.Czajkowsky, D.M., H. Iwamoto, T.L. Cover, and Z. Shao. 1999. The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc. Natl. Acad. Sci. USA. 96:2001–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Telford, J.L., P. Ghiara, M. Dell'Orco, M. Comanducci, D. Burroni, M. Bugnoli, M.F. Tecce, S. Censini, A. Covacci, Z. Xiang, et al. 1994. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 179:1653–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Bernard, M., D. Burroni, E. Papini, R. Rappuoli, J. Telford, and C. Montecucco. 1998. Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infect. Immun. 66:6014–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reyrat, J.M., S. Lanzavecchia, P. Lupetti, M. de Bernard, C. Pagliaccia, V. Pelicic, M. Charrel, C. Ulivieri, N. Norais, X. Ji, et al. 1999. 3D imaging of the 58 kDa cell binding subunit of the Helicobacter pylori cytotoxin. J. Mol. Biol. 290:459–470. [DOI] [PubMed] [Google Scholar]
- 21.Telford, J.L., A. Covacci, R. Rappuoli, and P. Chiara. 1997. Immunobiology of Helicobacter pylori infection. Curr. Opin. Immunol. 9:498–503. [DOI] [PubMed] [Google Scholar]
- 22.Baldari, C.T., M.M. Di Somma, M.B. Majolini, C. Ulivieri, E. Milia, and J.L. Telford. 1998. NF-AT-luciferase reporter T cell lines as tools to screen immunosuppressive drugs. Biologicals. 26:1–5. [DOI] [PubMed] [Google Scholar]
- 23.Padilla, P.I., A. Wada, K. Yahiro, M. Kimura, T. Niidome, H. Aoyagi, A. Kumatori, M. Anami, T. Hayashi, J. Fujisawa, et al. 2000. Morphologic differentiation of HL-60 cells is associated with appearance of RPTPbeta and induction of Helicobacter pylori VacA sensitivity. J. Biol. Chem. 275:15200–15206. [DOI] [PubMed] [Google Scholar]
- 24.D'Elios, M.M., M. Manghetti, M. De Carli, F. Costa, C.T. Baldari, D. Burroni, J.L. Telford, S. Romagnani, and G. Del Prete. 1997. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J. Immunol. 158:962–967. [PubMed] [Google Scholar]
- 25.Manetti, R., P. Massari, D. Burroni, M. de Bernard, A. Marchini, R. Olivieri, E. Papini, C. Montecucco, R. Rappuoli, and J.L. Telford. 1995. Helicobacter pylori cytotoxin: importance of native conformation for induction of neutralizing antibodies. Infect. Immun. 63:4476–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Bernard, M., E. Papini, V. de Filippis, E. Gottardi, J. Telford, R. Manetti, A. Fontana, R. Rappuoli, and C. Montecucco. 1995. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J. Biol. Chem. 270:23937–23940. [DOI] [PubMed] [Google Scholar]
- 27.Reyrat, J.M., M. Charrel, C. Pagliaccia, D. Burroni, P. Lupetti, M. de Bernard, X. Ji, N. Norais, E. Papini, R. Dallai, et al. 1998. Characterisation of a monoclonal antibody and its use to purify the cytotoxin of Helicobacter pylori. FEMS Microbiol. Lett. 165:79–84. [DOI] [PubMed] [Google Scholar]
- 28.Plyte, S., M. Boncristiano, E. Fattori, F. Galvagni, S.R. Paccani, M.B. Majolini, S. Oliviero, G. Ciliberto, J.L. Telford, and C.T. Baldari. 2001. Identification and characterization of a novel nuclear factor of activated T-cells-1 isoform expressed in mouse brain. J. Biol. Chem. 276:14350–14358. [DOI] [PubMed] [Google Scholar]
- 29.Milia, E., M.M. Di Somma, F. Baldoni, R. Chiari, L. Lanfrancone, P.G. Pelicci, J.L. Telford, and C.T. Baldari. 1996. The aminoterminal phosphotyrosine binding domain of Shc associates with ZAP-70 and mediates TCR dependent gene activation. Oncogene. 13:767–775. [PubMed] [Google Scholar]
- 30.Ulivieri, C., S. Pacini, S. Bartalini, S. Valensin, J.L. Telford, and C.T. Baldari. 1999. Obligatory cross-talk with the tyrosine kinases assembled with the TCR/CD3 complex in CD4 signal transduction. Eur. J. Immunol. 29:2625–2635. [DOI] [PubMed] [Google Scholar]
- 31.Koopman, G., C.P. Reutelingsperger, G.A. Kuijten, R.M. Keehnen, S.T. Pals, and M.H. van Oers. 1994. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 84:1415–1420. [PubMed] [Google Scholar]
- 32.Rao, A., C. Luo, and P.G. Hogan. 1997. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15:707–747. [DOI] [PubMed] [Google Scholar]
- 33.Cantrell, D. 1996. T cell antigen receptor signal transduction pathways. Annu. Rev. Immunol. 14:259–274. [DOI] [PubMed] [Google Scholar]
- 34.Dong, C., R.J. Davis, and R.A. Flavell. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 20:55–72. [DOI] [PubMed] [Google Scholar]
- 35.Bustelo, X.R. 2000. Regulatory and signaling properties of the Vav family. Mol. Cell. Biol. 20:1461–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ono, K., and J. Han. 2000. The p38 signal transduction pathway: activation and function. Cell. Signal. 12:1–13. [DOI] [PubMed] [Google Scholar]
- 37.Hotchin, N.A., T.L. Cover, and N. Akhtar. 2000. Cell vacuolation induced by the VacA cytotoxin of Helicobacter pylori is regulated by the Rac1 GTPase. J. Biol. Chem. 275:14009–14012. [DOI] [PubMed] [Google Scholar]
- 38.Smith, W.L., D.L. DeWitt, and R.M. Garavito. 2000. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69:145–182. [DOI] [PubMed] [Google Scholar]
- 39.Lewis, R.S. 2001. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 19:497–521. [DOI] [PubMed] [Google Scholar]
- 40.Janes, P.W., S.C. Ley, and A.I. Magee. 1999. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 147:447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valensin, S., S.R. Paccani, C. Ulivieri, D. Mercati, S. Pacini, L. Patrussi, T. Hirst, P. Lupetti, and C.T. Baldari. 2002. F-actin dynamics control segregation of the TCR signaling cascade to clustered lipid rafts. Eur. J. Immunol. 32:435–446. [DOI] [PubMed] [Google Scholar]
- 42.Schraw, W., Y. Li, M.S. McClain, F.G. Van Der Goot, and T.L. Cover. 2002. Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. J. Biol. Chem. 277:34642–34650. [DOI] [PubMed] [Google Scholar]
- 43.Fujikawa, A., D. Shirasaka, S. Yamamoto, H. Ota, K. Yahiro, M. Fukada, T. Shintani, A. Wada, N. Aoyama, T. Hirayama, et al. 2003. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat. Genet. 33:375–381 (published erratum appears in Nat. Genet. 2003. 33:533. [DOI] [PubMed] [Google Scholar]
- 44.Plas, D.R., and M.L. Thomas. 1998. Negative regulation of antigen receptor signaling in lymphocytes. J. Mol. Med. 76:589–595. [DOI] [PubMed] [Google Scholar]
- 45.Acuto, O., and D. Cantrell. 2000. T cell activation and the cytoskeleton. Annu. Rev. Immunol. 18:165–184. [DOI] [PubMed] [Google Scholar]
- 46.Villalba, M., K. Bi, F. Rodriguez, Y. Tanaka, S. Schoenberger, and A. Altman. 2001. Vav1/Rac-dependent actin cytoskeleton reorganization is required for lipid raft clustering in T cells. J. Cell Biol. 155:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boquet, P. 1999. Bacterial toxins inhibiting or activating small GTP-binding proteins. Ann. NY Acad. Sci. 886:83–90. [DOI] [PubMed] [Google Scholar]
- 48.Del Giudice, G., A. Covacci, J.L. Telford, C. Montecucco, and R. Rappuoli. 2001. The design of vaccines against Helicobacter pylori and their development. Annu. Rev. Immunol. 19:523–563. [DOI] [PubMed] [Google Scholar]