

Abstract
Phenotype analysis of female mice lacking androgen receptor (AR) deficient (AR −/−) indicates that the development of mammary glands is retarded with reduced ductal branching in the prepubertal stages, and fewer Cap cells in the terminal end buds, as well as decreased lobuloalveolar development in adult females, and fewer milk-producing alveoli in the lactating glands. The defective development of AR −/− mammary glands involves the defects of insulin-like growth factor I–insulin-like growth factor I receptor and mitogen-activated protein kinase (MAPK) signals as well as estrogen receptor (ER) activity. Similar growth retardation and defects in growth factor–mediated Ras/Raf/MAPK cascade and ER signaling are also found in AR −/− MCF7 breast cancer cells. The restoration assays show that AR NH2-terminal/DNA-binding domain, but not the ligand-binding domain, is essential for normal MAPK function in MCF7 cells, and an AR mutant (R608K), found in male breast cancer, is associated with the excessive activation of MAPK. Together, our data provide the first in vivo evidence showing that AR-mediated MAPK and ER activation may play important roles for mammary gland development and MCF7 breast cancer cell proliferation.
Keywords: androgen receptor, knockout mice, mammary gland, breast cancer, MAPK
Introduction
The roles of androgen receptor (AR; reference 1), a transcriptional factor that belongs to the nuclear receptor superfamily (2), in female organs remains unclear. The lack of clarity could be due to the lack of in vivo models because male mice lacking functional AR are infertile; thus, they are unable to generate female AR knockout (AR −/−) offspring. Epidemiological analyses indicated some positive correlation between testosterone concentration and breast cancer incidence, although it is debatable that testosterone effects on breast cancer progression could also result from conversion to 17β-estradiol (E2) via aromatization in peripheral tissues (3, 4). However, other papers also suggested that androgens could negatively regulate the growth of mammary epithelial and breast cancer cells (5–7). AR is expressed in normal breast cells, and up to 85% of breast tumors are AR positive (8–10). Also, 25–82% of metastatic breast tumors, which are estrogen receptor (ER) and progesterone (P) receptor (PR) negative, still express a significant amount of AR (9, 11). However, the mechanisms by which AR influences breast cancer progression and mammary gland development remain unclear. To answer these long-term puzzles and to dissect the molecular mechanism of AR in breast cells, we applied different strategies to knock out the AR gene in female mice and breast cancer MCF7 cells. Our results demonstrate that AR plays an important role in the mammary gland development and breast cancer cell growth.
Materials and Methods
Generation of Female AR−/− Mice.
Construction of targeting vectors and generation of the chimera founder mice have been described previously (12). The strain of the mosaic founder was C57BL/6-129/SEVE. β-Actin is a housekeeping gene and is universally expressed in every tissue; therefore, the β-actin promoter-driven Cre (ACTB-Cre) will express and delete the floxed AR fragment in all the cells. The mating strategy is illustrated in brief in Fig. 1. The female AR −/− mice were genotyped by PCR, rather than by Southern blot analysis, as described in the figure legend and previously (12).
Figure 1.
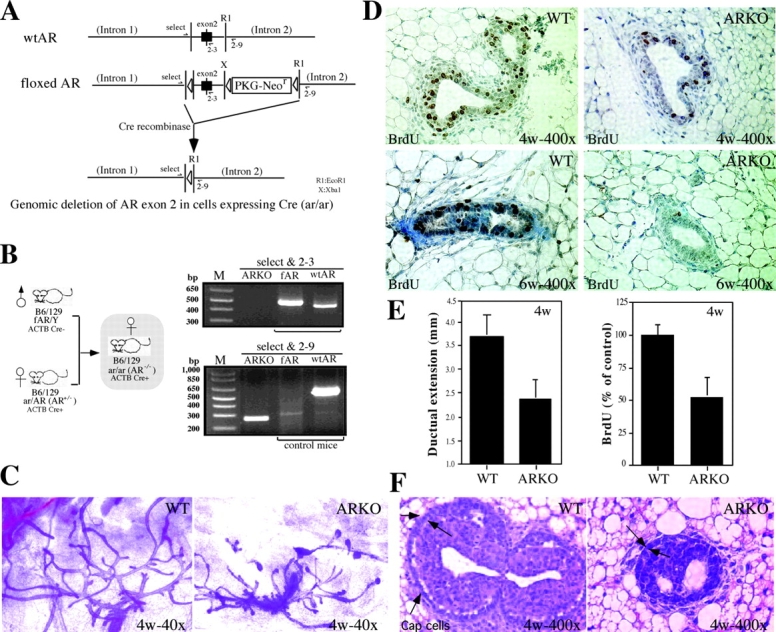
The generation and characterization of immature female AR −/− mice. (A) Gene targeting strategies. To generate female AR −/− mice, a Cre-lox strategy for conditional knockout was applied. The Cre-lox system uses the expression of P1 phage Cre recombinase to catalyze the excision of DNA located between flanking lox sites. (B) Breeding strategy of female AR −/− mice and genotyping of female AR −/− mice. Using the Cre-lox strategy, the targeted exon 2 of AR is not disrupted but floxed in the male mice. X and RI represent XbaI and EcoRI restriction enzyme sites, respectively. Thus, the AR functions normally in male mice, which can be bred with female AR+/− ACTB Cre+ mice and, thereby, generate homozygous female AR −/− mice. For examining the X chromosome with floxed AR, primers select and 2-3 are used. Select is located in the intron 1 with sequence 5′-GTTGATACCTTAACCTCTGC-3′. 2-3 is the 3′ end primer located in the exon 2 with the sequence 5′-CTTCAGCGGCTCTTTTGAAG-3′. This pair of primers will amplify a product with ∼510 bp for floxed AR and ∼460 bp for wt AR. For examining the AR knockout locus, primers select and 2-9 were used. 2-9 is located in intron 2 with the sequence 5′-CTTACATGTACTGTGAGAGG-3′. The PCR product size from this pair of primers would be ∼270 bp for AR knockout allele and ∼600 bp for wt AR allele. The expression of Cre and internal control IL-2 was also confirmed by PCR genotyping. (C) The defects in the ductal development of mammary gland in immature female AR −/− mice. Whole breast mounts from 4-wk-old female AR −/− mice show lessened extension of mammary ducts, as compared with age-matched AR +/+ mice. (D) The decreases in the percentage of BrdU-positive staining (brown) are observed in both 4- and 6-wk-old AR −/− mice. (E) Statistic analyses of the distance of ductal extension in female AR −/− and AR +/+ mice (left). Statistic analyses of BrdU staining signal in female AR −/− and AR +/+ mice (right, n = 5 for each group). (F) The number of Cap cells (arrows) in TEB of AR −/− mice is less than that in the AR +/+ mice.
Animal, Tissue Collection, and Real-time RT-PCR.
All animal experimentation was conducted in accordance with accepted standards of humane animal care. Tissues were fixed in buffered neutral formalin for 24 h. Fresh mammary gland tissues were frozen and stored in liquid nitrogen before RNA extraction. 3 μg of total RNA was reverse transcribed and subjected to real-time PCR using iCycle (Bio-Rad Laboratories), and the formulas used were described previously (13). Primer pair sequences were designed by Beacon Designer II software (Bio-Rad Laboratories). Primer pair sequences used for studying gene expression change are as follows: mouse PR, 5′-CAGATTCAGAAGCCAGCCAGAG-3′ and 5′-CCACAGGTAAGCACGCCATAG-3′; mouse hepatocyte growth factor (HGF), 5′-AGGTACGCTACGAAGTCTGTG-3′ and 5′-GGTGTGGTGTCTGCTGGTC-3′; mouse estrogen-responsive finger protein (Efp), 5′-ACCGCCTCTGTGTCTGATGG-3′ and 5′-GCCTACGCCGCAGAAGTTG-3′; mouse IGF-1, 5′-TGCTCTTCAGTTCGTGTG-3′ and 5′-ACATCTCCAGTCTCCTCAG-3′; mouse IGF-1R, 5′-GCCGACGAGTGGAGAAATC-3′ and 5′-CTTGGAGATGAGCAGGATGTG-3′; human Ki67, 5′-ATCGTCCCAGGTGGAAGAGTT-3′ and 5′-ATAGTAACCAGGCGTCTCGTGG-3′; human c-myc, 5′-CAGCTGCTTAGACGCTGGATTT-3′ and 5′-ACCGAGTCGTAGTCGAGGTCAT-3′; and human Bcl-2, 5′-CATGTGTGTGGAG-AGCGTCAA-3′ and 5′-GCCGGTTCAGGTACTCAGT-3′. β-Actin was used as an internal control.
Whole-mount Staining of Mammary Glands.
Whole mammary glands were spread on glass slides, fixed overnight in Carnoy's solution (Ethanol/CHCl3/acetic acid, 6:3:1), sequentially rehydrated by 100, 95, 70, and 50% ethanol and tap water, and stained in carmine red solution overnight until the whole gland became red. After staining, the tissue slides were dehydrated, cleared with xylene, and mounted.
Immunohistochemistry and Bromodeoxyuridine (BrdU) Staining.
Mammary glands were fixed overnight in buffered neutral formalin (VWR Scientific Products) at room temperature. The tissues were dehydrated by passing through 70, 85, 95, and 100% ethanol, cleared in xylene and 1:1 xylene/paraffin for 45 min, and embedded in paraffin. Tissue sections were cut at a 5–7-μm thickness and mounted onto Probe-On Plus charged slides (Fisher Scientific).
For immunohistochemistry, sections were heated at 55°C for at least 2 h, deparaffinized in xylene, rehydrated, and washed in Tris-buffered saline (TBS), pH 8.0. For antigen retrieval, slides were microwaved in 0.01 M sodium citrate, pH 6.0, immersed with 1% hydrogen peroxide in methanol for 30 min, and blocked with 20% normal goat serum in TBS for 60 min. After washing with PBS, sections were incubated for 90 min in different antibodies diluted 1:200 to 1:500 in TBS containing 1% BSA, followed by goat anti–rabbit biotinylated secondary antibody diluted 1:300 in TBS containing 1% BSA. Sections were incubated with avidin-biotin–peroxidase complex solution for 30 min, followed by development with diaminobenzidine peroxidase substrate kit (Vector Laboratories) for 5 min. Slides were counterstained with hematoxylin for 30 s, dehydrated, cleaned in xylene, and mounted. Primary antibody was replaced with normal rabbit IgG or 1% BSA in TBS for negative controls. Both of anti-phospho–mitogen-activated protein kinase (MAPK) and total MAPK antibodies were obtained from Cell Signaling Technology. BrdU labeling reagents and BrdU staining kit were purchased from Zymed Laboratories.
Steroid Hormone RIA.
For characterization of hormonal profiles, sera were collected by the intracardiac method from AR +/+ and AR −/− mice under ketamine and xylazine anesthesia (Sigma-Aldrich). Concentrations of E2 and P were determined using Coat-a-Count kits (Diagnostic Instruments).
Construction of the AR Targeting Vector and Generation of AR−/− MCF7 Cells.
The targeting vector for generating AR −/− MCF7 cells was constructed by replacing the SmaI–KpnI segments within the AR exon 1 with a promoterless neomycin cassette and inserting two flanking sequences, 5′ extending 1.1 kb into the human AR 5′ untranslated region (UTR) and 3′ extending 6.2 kb into the AR intron 1, on a pGEM-T easy vector (Promega). This promoterless neomycin cassette, containing a termination codon and a polyadenylation signal, was inserted in frame with AR ATG start codon. The flanking homologous sequences were generated by PCR using the genomic DNA from human LN Cap cells as a template. For generation of AR −/− MCF7 cells, parental MCF7 cells were transfected with the AatII-linearized AR targeting vector using SuperFect (QIAGEN) and selected with 400 μg/ml neomycin. The genotypes of surviving clones were screened by Southern blot analyses. The heterozygous clones (AR +/−) were picked up and subjected to the second gene targeting experiment using the same targeting vector. Clones were selected with a higher concentration of neomycin (1.25 mg/ml). The genotypes of surviving clones were screened again by Southern blot analyses.
Plasmids.
GAL4-Elk1, constitutively activated MEK1 (MEK-CA), Ras (Ras-CA), Raf (Raf-CA), dominant-negative Ras (Ras-DN) and Raf (Raf-DN), and MEK1 phosphatase CL-100 plasmids were provided by K.-L. Guan (University of Michigan Medical School, Ann Arbor, MI; reference 14). Constitutively activated Rac plasmid was a gift from J. Chernoff (Fox Chase Cancer Center, Philadelphia, PA; reference 15). The reporter genes (ARE)4-luciferase (Luc), ERE-Luc, and pG5-Luc used for monitoring AR, ER, and GAL4-Elk transactivation, respectively, were described previously (16). FLAG-tagged AR deletion mutant expression plasmids were constructed by inserting PCR-generated AR cDNA fragments into pCDNA3 vector (Invitrogen) containing FLAG tag. The natural promoter–driven (np)-AR plasmid was constructed by inserting 3.6 kb hAR promoter, the entire hAR 5′-UTR, full-length AR cDNA, and 310-bp 3′-UTRs followed by 280-bp bovine GH poly(A) signals into pBlueScript sk(−) vector (Stratagene). The AR small interfering RNA (siRNA) expression vector that expresses an siRNA-targeting AR in mammalian cells was constructed by digesting and inserting a double-strained polynucleotide 5′-GTCGGGCCCTATCCCAGTCCCACTTGCTCGAGCAAGTGGGACTGGGA- TAGGGCTTTTTGAATTCGC-3′ into the ApaI–EcoRI site of a DNA-based vector BS/U6 (17).
MTT Growth Assays.
104 cells seeded on 24-well plates were cultured with RPMI 1640 supplemented with 10% of charcoal dextran–stripped (CDS)–FBS for treatment with 0.1 nM E2 or 0.2% of heat-inactivated (HI)–FBS) for treatment with 100 ng/ml IGF-1 or 50 ng/ml epidermal growth factor (EGF). The cells were collected at indicated days for MTT assay according to the manufacturer's instructions (Sigma-Aldrich).
Soft-agar Colony Formation Assay.
2 × 104 cells suspended in 0.4% low melting agarose (FMC Corp.) were layered on top of 1 ml of 0.8% agarose in 6-well culture plates. Cells were incubated with 1 ml RPMI 1640 supplemented with 10% CDS-FBS for treatment with 0.1 nM E2 or 0.2% HI-FBS for treatment with 100 ng/ml heregulin-α (HRG-α). After 4 wk of incubation, the colonies were visualized by incubating with 1 mg/ml INT (Sigma-Aldrich) for 24 h and counted with VersaDoc Imaging System (Bio-Rad Laboratories).
Reporter Gene Assays.
Cells were plated in 96-well plates and plasmids at 0.5 μg per well were transfected into cells using SuperFect (QIAGEN). The medium was changed 2 h after transfection, and cells were cultured in media containing 10% CDS-FBS or 0.2% HI-FBS for 16 h, followed by treatment with 50 ng/ml EGF, 100 ng/ml IGF-1, 100 ng/ml HRG-α, 0.1 nM E2, or 1 nM DHT for another 16 h. 5 ng pRL-TK per well was used as internal control. Cells were harvested, and the Luc activity was analyzed using Dual-Luc Reporter Assay System (Promega).
Western Blots.
Cells were lysed with radioimmunoprecipitation assay buffer containing 0.5% NP-40 and Western blotted with anti-AR (NH-27), anti-ER, antiactin, anti–green fluorescent protein (GFP; Santa Cruz Biotechnology, Inc.), anti–total MAPK, and anti–phospho-MAPK (Cell Signaling Technology).
Statistical Analysis.
All data were analyzed by one-way analysis of variance using minitab statistical software (State College). Mean separation was accomplished using Fisher's pairwise comparison. Differences were considered significant at P < 0.05.
Results
Generation and Phenotype of Female AR−/− Mice.
Using a Cre-lox conditional knockout strategy by mating the floxed AR male mice with AR +/− ACTB Cre+ females, we were able to generate female AR −/− mice, genotyped as AR −/− ACTB Cre+ (Fig. 1, A and B) . Adult female mice with homologous deletion of AR appear healthy and develop normal external genitalia. Gross anatomical examination did not reveal obvious differences in the morphology of most organs between the AR +/+ and AR −/− littermates. The body weights are similar between the AR +/+ and AR −/− mice, but the thymus of female AR −/− mice is bigger than that of female AR +/+ or AR +/− mice. Several estrogen target organs, including mammary gland, ovary, oviduct, and uterus, were collected from 4-, 6-, and 12-wk-old mice. The weights of these organs are 15–23% less in female AR −/− mice as compared with their age-matched littermates.
Defects in Mammary Gland Development in Prepubertal and Pubertal Stages in Female AR−/− Mice.
First, we compared the morphology of mammary glands between immature (4- and 6-wk old) virgin AR +/+ and AR −/− mice. By the fourth to sixth week, the ductal system has ∼30–50% less extension in female AR −/− mice with reduced numbers and size of the terminal end buds (TEBs; Fig. 1, C and E). BrdU staining also revealed a 50% lower proliferation of AR −/− mammary glands, as compared with that of AR +/+ mice (Fig. 1, D and E). The number of Cap cells (18), which are responsible for the ductal extension from TEBs, were also reduced in female AR −/− mice (Fig. 1 F). Together, our results indicated that the mammary gland development is retarded in prepubertal and pubertal stages in female AR −/− mice.
Reduced Ductal Morphogenesis in the Mammary Glands of the Mature AR−/− Mice.
At maturity (8-, 16-, and 20-wk), we could see AR −/− mammary glands were filled with large bloated ducts terminating with bloated ends. It is also obvious that AR −/− mammary glands have fewer secondary and tertiary ductal branches as compared with age-matched AR +/− and AR +/+ mice (Fig. 2, A–C) . During the pregnancy stage, the retarded ductal branches in AR −/− mice are partially restored, yet compared with AR +/+ mice, the AR −/− mice mammary glands still have less milk-producing alveoli (Fig. 2 D). In agreement with these findings, we also observed shrunken ductal spaces in some AR −/− mice mammary glands in 16-wk-old or older mice (Fig. 2 E), and reluctant nursing behavior in some AR −/− mothers. The decreased milk-producing alveoli and shrunken ductal spaces may result in the lessened capacity for AR −/− mice to feed their offspring. In 20-wk-old mice, we found that mammary glands in some AR −/− mice underwent degeneration earlier than those of the AR +/+ mice (Fig. 2 C). Overall, Fig. 2 demonstrates that the lack of AR in female mice may retard the mammary gland development and affect the capacity of female AR −/− mice to feed their offspring.
Figure 2.
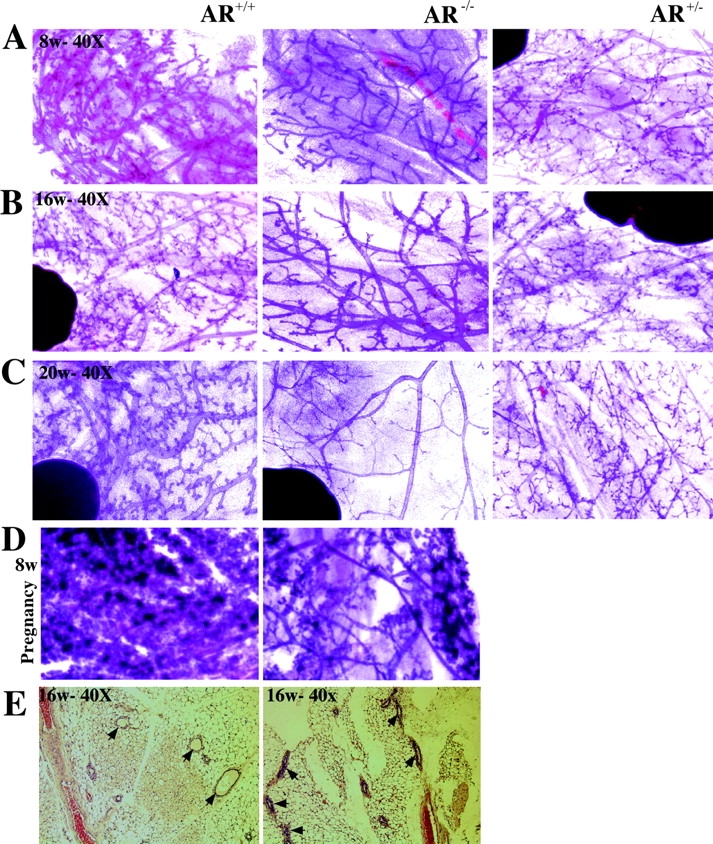
AR −/− mammary glands show the defects of the terminal branching and alveologenesis during maturity and pregnancy. Whole breast mounts from 8-, 16-, and 20-wk-old mature and 8-wk-old pregnant female mice were examined. (A–C) Less secondary and tertiary terminal branching in AR −/− mice, compared with AR +/+ and AR +/− mice. (C) Early degeneration occurs in AR −/− mammary glands in 20-wk-old mice. (D) The decreased milk-producing lobuloalveolar development in the 8-wk-old pregnant AR −/− mice. (E) Using hematoxylin and eosin staining, the results indicate that the shrunken ductal space occurs in some AR −/− mammary glands in 16–20-wk-old mice (n = 4 for each group).
Defects of MAPK Activity and Insulin-like Growth Factor I (IGF-I)–IGF-IR Pathway in AR−/− Mammary Glands.
Early works indicated that E2/ER, P/PR, and paracrine growth factors/MAPK signals may contribute to the growth and development of mammary glands (19–21). First, we examined the MAPK activity in 4-wk-old mice. The immunohistochemstaining data show that the overall phospho-MAPK expression is weaker in the mammary cells from AR −/− mice, as compared with AR +/+ mice (Fig. 3 A), although we could observe some ductal mammary cells with intensive phospho-MAPK staining in AR −/− mammary glands. The total MAPK protein expression is similar between AR −/− and AR +/+ mice, as shown in Fig. 3 B. We examined the upstream regulators of MAPK signals, IGF-I–IGF-IRs. We found that IGF-IR, but not IGF-I, mRNA expression is reduced by 46% in immature AR −/− mammary glands (Fig. 3 C), suggesting that the IGF-I–IGF-IR→MAPK signaling pathway may be defective in female AR −/− mice. We investigated the expression of the downstream target, cyclin D1, in AR −/− and AR +/+ mice using real-time quantitative PCR. The cyclin D1 mRNA expression was significantly reduced in female AR −/− mice (Fig. 3 C). Together, these data showing the reduction of IGF-IR, cyclin D1, and MAPK activity may suggest that the defects in the AR→IGF-I–IGF-IR→ MAPK→cyclin D1 signaling pathway might result in the retarded mammary gland development in female AR −/− mice. This is in agreement with early papers showing that IGF-I–IGF-IR is an important paracrine growth factor for mammary gland development (22, 23), and cyclin D1 is a downstream mediator of growth factor–induced mammary gland proliferation (24).
Figure 3.
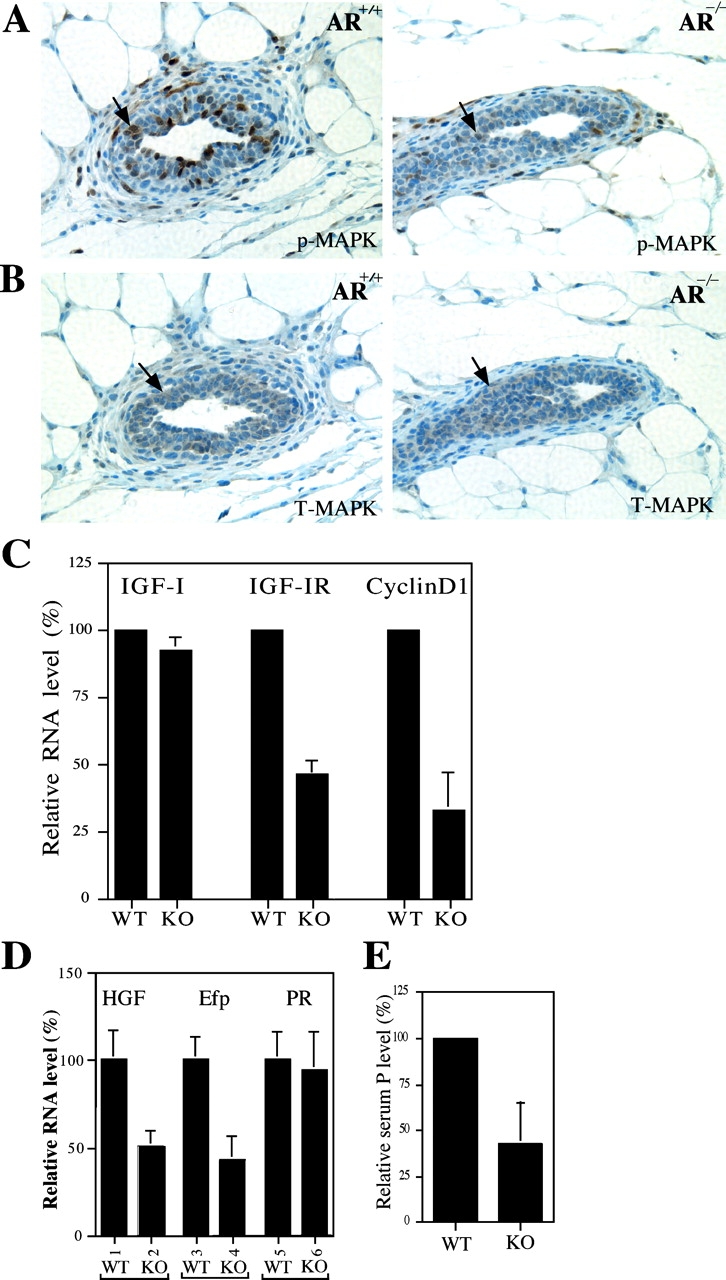
The reduced MAPK activity and mRNA expression of IGF-IR, HGF, and Efp in AR −/− mammary glands. (A) Reduced MAPK activities are observed in AR −/− mammary glands of 4-wk-old mice. Mammary glands were stained with anti–phospho-MAPK (p-MAPK) antibody. The decrease of positive staining (brown) indicates the reduced MAPK activity in AR −/− mammary glands. (B) The similar total MAPK (T-MAPK) staining results are observed in the mammary glands from 4-wk-old AR +/+ and AR −/− mice. Adjacent sections were used for p-MAPK and T-MAPK stainings. (C) The mRNA expression of IGF-IR, but not IGF-I, is reduced in AR −/− mice. Total RNA was extracted from 4-wk-old AR +/+ and AR −/− mice and quantitated by real-time RT-PCR. Cyclin D1, a proliferation indicator, is also reduced in mammary glands of female AR −/− mice. (D) The mRNA expressions of two ER target genes, HGF and Efp, are reduced in AR −/− mice. The expression of PR mRNA has no significant difference (bars 5 vs. 6). Total RNA was extracted from 4-wk-old AR +/+ and AR −/− mice injected with E2 (n = 5 for each group). (E) The serum levels of P were reduced in 12–16-wk-old adult female AR −/− mice.
Before systematic hormone function that occurs after puberty, growth factors, such as IGF-I, are the major contributing factors influencing the mammary gland development. IGF-I is a potent mitogen for mammary epithelial cells, and the ductal development can be stimulated by IGF-I. The mRNA for IGF-I and IGF-IR is expressed in mammary stroma and developing TEBs, and targeted deletion of IGF-IR inhibits normal TEB development before puberty (23). In the present paper, our results indicate that knock out of AR affects the mammary gland development before the puberty stage (Figs. 1 and 3), which suggests a possible disturbance in the growth factor pathway. Indeed, our results indicated that the reduction of IGF-IR expression may consequently affect the IGF-I–IGF-1R signal on development of the mammary gland, including retarded ductal development, fewer Cap cells in the TEB, and reduced BrdU staining and cyclin D1 expression (Figs. 1 and 3). The aforementioned observations concerning the prepubertal AR −/− mammary glands suggest a tight association of AR and growth factor signal in the mammary gland. Together, our results provide the first in vivo evidence showing that AR plays a significant role in the prepubertal as well as adult mammary gland development.
Reduced ER Activity in AR−/− Mammary Glands.
Early analyses indicated that MAPK could also influence ER function (25), and that cyclin D1 could also be a downstream target gene for ER (26). Defects in MAPK and cyclin D1 may suggest that ER signals could also be impaired in the female AR −/− mice. Therefore, we ovariectomized 4-wk-old mice, treated them with E2 for 2 d, and harvested the mammary glands for the comparison of the ER activity by examining ER target gene expression between AR −/− and AR +/+ mice. We found that E2-induced Efp (27) and HGF (28) were down-regulated in female AR −/− mice as compared with AR +/+ mice (Fig. 3 D). Early works also indicated that both Efp and HGF were important factors for breast cell growth (20, 29). Interestingly, we found that PR expression in mammary glands was similar between AR −/− and AR +/+ (Fig. 3 D). This finding is consistent with a previous paper showing that PR expression is E2/ER-independent in 5-wk-old or younger mice (30). Nevertheless, we found that the serum levels of PR's ligand, P, was reduced in 12- to 16-wk-old adult female AR −/− mice (Fig. 3 E), which may result in the reduction of P/PR activity in mature mice. As the P/PR signal pathway plays important roles for the tertiary ductal branching and alveolar development (19), the lower P/PR activity in AR −/− mice may contribute to the retarded branching and lobuloalveolar formation in the development of mature stage mammary glands.
The AR−/− MCF7 Cells Exhibit Severe Defects in Growth and Colony Formation.
To further dissect the mechanisms of AR roles in breast tissue at molecular and cellular levels, we applied the homologous recombination by using a targeting vector carrying a promoterless neomycin cassette to generate AR-deficient (AR −/−) MCF7 cells (Fig. 4 A). Two AR −/− MCF7 clones have been successfully obtained, and the targeted loci were confirmed by Southern blot analysis (Fig. 4 B). In these two homologous clones, the expression and the ligand-activated transcriptional activity of AR were indeed abrogated (Fig. 4, C and D). We found that AR −/− MCF7 cells exhibit a severe impairment in proliferation when cultured in media containing normal, steroid-deprived or 10−10 M E2-treated serum (Fig. 4 E). The soft-agar colony formation assay also showed that the colony number of AR +/+ MCF7 cells was increased in response to 10−10 M E2 or 100 ng/ml HRG-α, an activator for the HER2/HER3/HER4 family, whereas the colony formation of AR −/− MCF7 cells was largely reduced (Fig. 4 F). To determine whether abrogating AR expression in AR +/+ MCF7 cells can also reduce the cell growth capacity, we used an AR siRNA expression vector that showed an efficient knock down of AR in cells transfected with np-AR, a natural AR promoter-driven AR expression plasmid (Fig. 4 G). We found that the mRNA levels of Ki67 and c-myc were reduced by 42 and 81%, respectively, in AR +/+ MCF7 cells transfected with AR siRNA, compared with the cells transfected with vector alone (Fig. 4 H). Overall, Fig. 4 implies that AR plays an important role for the growth of MCF7 breast cancer cells.
Figure 4.

Targeted deletion of AR gene in MCF7 cells results in severe defects in cell proliferation and colony formation. (A) Schematic diagram of the strategy of targeting AR genes in MCF7 cells. (B) Genotyping by Southern blot analysis. Genomic DNA extracted from neomycin-resistant clones was digested with XbaI. The untargeted and targeted loci produced ∼9.0-kb and 3.5-kb bands, respectively. (C) The AR protein was ablated in AR −/− MCF7 cells. (D) The ligand-activated transcriptional activity of AR was reduced in AR +/− MCF7 cells and abrogated in AR −/− MCF7 cells, compared with AR +/+ MCF7 cells. (E) The proliferation of AR −/− MCF7 cells was reduced in media containing 10% FBS (left) or 10% CDS-FBS with ethanol (e) or 10−10 M E2 (right), compared with AR +/+ MCF7 cells, using the MTT proliferation assay. (F) The soft-agar colony formation capacity of AR −/− MCF7 cells was largely reduced compared with AR +/+ MCF7 cells. (G) AR siRNA efficiently knocked down AR in the cells transfected with np-AR. Full length of the 110-kD AR was detected by anti-AR antibody (NH27) using Western blotting assays. Cells were cotransfected with pEGFP-C1 vector for normalization of transfection efficiency. GFP expression was detected with anti-GFP antibody. (H) The mRNA expression of Ki67 and c-myc, but not Bcl-2, was reduced in AR +/+ MCF7 cells transfected with AR siRNA using electroporation, compared with the cells transfected with vector alone. Electroporation was performed using 0.4-cm cuvettes and Gene Pulser II set at 280 V and 950 μF.
The Growth Factor–mediated Proliferation and MAPK Activation Is Impaired in AR−/− MCF7.
Next, we examined whether the loss of AR impairs the growth factor–mediated proliferation and MAPK activation in AR −/− MCF7 cells. Treatment of AR +/+ MCF7 cells with IGF-I, EGF, or HRG-α could stimulate cell proliferation and activate GAL4-Elk1, a direct target of MAPK, in a low serum-containing medium (Fig. 5, A and B) . In contrast, the growth factor–stimulated cell proliferation and MAPK activation were largely impaired in the AR −/− MCF7 cells (Fig. 5, A and B). Using another strategy by transfection of AR siRNA into AR +/+ MCF7 cells, we found that suppression of AR expression could also diminish IGF-I/EGF/HRG-α–induced MAPK activation (Fig. 5 B). Moreover, the reduced transcriptional activity of GAL4-Elk1 in AR −/− MCF7 cells could be rescued by transfection of np-AR (Fig. 5 C). Interestingly, adding EGF with np-AR could synergistically enhance the transactivation of GAL4-Elk1 in AR −/− MCF7 cells compared with the cells treated with EGF alone (Fig. 5 C), suggesting a significant involvement of AR in the growth factor signaling pathway. Furthermore, the AR-activated GAL4-Elk1 activity could be diminished by MAPK phosphatase-1 (CL-100) or a specific inhibitor U0126, as well as Ras-DN or Raf-DN (Fig. 5 D; reference 14). Also, the reduction of MAPK activation by AR siRNA could be recovered by MEK-CA, Ras-CA, or Raf-CA, but not by Rac-CA (15) or PI3K (Fig. 5 E, p110 catalytic subunit). These results indicate that AR is an important upstream regulator of the Ras/Raf/MAPK cascade.
Figure 5.

AR was essential for growth factor and estrogen signaling pathway. (A) Growth factor–induced cell proliferation was impaired in AR −/− MCF7 cells, compared with AR +/+ MCF7 cells. Cultures were incubated with RPMI 1640 media containing 0.2% serum treated with or without growth factors for 8 d. (B) The steady-state level of the active form of MAPK was lower in AR −/− MCF7 cells than that in AR +/+ MCF7 cells, when cells were cultured in the media containing 1% HI-FBS for 5 d (left). Growth factor–induced transcriptional activity of GAL4-Elk1 was diminished in AR −/− MCF7 cells (middle) and in AR +/+ MCF7 cells transfected with AR siRNA (right). (C) The reduced MAPK activity was restored by np-AR, which expresses full-length AR and is driven by natural AR promoter. np-AR synergistically enhanced EGF-induced GAL4-Elk1 transactivation. (D) The AR-FL–activated GAL4-Elk1 transactivation can be inhibited by a MAPK phosphatase (CL-100), a specific inhibitor U0126, dominant-negative Ras (Ras-DN), or Raf (Raf-DN). AR-FL, full-length WT AR. (E) Constitutively activated MEK1 (MEK-CA), Ras (Ras-CA), or Raf (Raf-CA), but not Rac (Rac-CA) or PI3K (p110 catalytic subunit), can block the suppressive effect of AR siRNA on the activity of GAL4-Elk1. (F) The transcriptional activity of ER is reduced in AR −/− MCF7 cells, compared with AR +/+ MCF7 cells. (G) The reduced ER activity in AR −/− MCF7 cells was restored by np-AR. pG5-Luc and ERE-Luc were the reporters for GAL4-Elk1 and ER, respectively. pRL-TK was used as an internal control. Transfections were performed using SuperFect according to the manufacturer's instructions. Values shown are the mean ± SD from at least four independent experiments.
The Transcriptional Activity of ER Is Defective in AR−/− MCF7 Cells.
Using an ERE-Luc reporter, we further compared the ER activity in AR +/+ and AR −/− MCF7 cells. The transcriptional activities of ER were reduced by 58.8, 53.8, and 55.0% in AR −/− MCF7 cells in the presence of E2 at 10−12, 10−10, and 10−8 M, respectively (Fig. 5 F). The reduced transcriptional activity of ER in AR −/− MCF7 cells could be restored by transfection of np-AR (Fig. 5 G). These results are consistent with the data in Fig. 3 C showing that the ER target gene expression is reduced in AR −/− mouse breasts.
The NH2 Terminus/DNA-binding Domain (DBD) of AR Is Required for MAPK Activation.
To further determine which functional domain of AR is required to restore the normal MAPK activity in AR −/− MCF7 cells, various AR fragments were reintroduced into AR −/− MCF7 cells (Fig. 6 A). We found that the NH2 terminus together with DBD (but not ligand-binding domain [LBD]), LBD with deletion of helix 12 domain (LBD-dH12), DBD alone, or NH2 terminus alone can restore the MAPK activation (Fig. 6 A). Next, because an AR mutant (AR-R608K) has been suggested to be associated with male breast cancer (31), we were interested to know its effect on the MAPK activation. We found that in AR −/− MCF7 cells, AR-R608K had a higher induction fold on MAPK activation than AR-FL (Fig. 6 B), suggesting that the contribution of AR-R608K to breast cancer incidence may involve the excessive activation of MAPK. Next, the requirement of AR NH2 terminus/DBD, but not LBD, to restore the MAPK activation was further confirmed by using a double mutation AR (AR-R614H-dprm) with a deletion of the proline-rich motif (dprm) at the NH2-terminal region (32) and a point mutation on the second zinc-finger motif (R614H) at the DBD (33). Although AR with a single mutation, AR-R614H or AR-dprm, still partially retains the ability to activate MAPK, the double mutation AR-R614H-dprm almost loses the whole capacity to restore the MAPK activity, even though these AR mutants contain intact LBDs (Fig. 6 B). Together, these restoration assays indicate that AR, through its NH2 terminus/DBD domains, plays an important role for the MAPK activation in MCF7 cells.
Figure 6.

The NH2 terminus/DBD of AR were required for normal MAPK activation, and an AR mutant (R608K) was associated with the excessive activation of MAPK. (A) Ectopical expression of AR restored the defective MAPK activity in AR −/− MCF7 cells. The NH2 terminus together with the DBD, but not N, DBD, LBD, or LBD-dH12 alone, were required for activating MAPK, using a transient transfection assay (middle) and a Western blot (bottom) with anti–phospho-MAPK and anti-MAPK antibodies. All of the sequence constructs were FLAG-tagged and inserted into pCDNA3 vector. V, vector alone. dH4–12, AR with deletion from helix 4 to helix 12. (B) The AR-R608K–induced GAL4-Elk1 transactivation was higher than AR-FL. AR-R614H-dprm, containing a point mutation (R614H) and a dprm, lost the ability to activate MAPK, whereas AR-R614H or AR-dprm still partially retained MAPK activation capacity. pG5-Luc was the reporter for GAL4-Elk1, and 5 ng/well pRL-TK was used for internal control. Values shown are mean ± SD from at least four independent experiments. (C) The proposed molecular mechanisms. The AR abrogation in mammary glands or mammary cancer cells retards the growth or development via the impairments of the growth factor and ER signaling pathways. The reduced ER activity, as demonstrated by the decreased target gene expression (Efp and HGF), may partly result from the impairment of the growth factors/MAPK signaling pathway. The reduced PR activity may be due to the reduction of ER activity and/or the serum level of P after puberty but not before puberty (asterisk). The decreased cyclin D1 expression may be caused by the impairments of both the growth factor/MAPK and ER signaling pathways. Together, these impaired signals may contribute to the developmental defects of mouse mammary glands in AR −/− mice and growth inhibition in breast cancer cells lacking AR.
Discussion
Using three different approaches (Cre-lox conditional KO, siRNA, and homologous recombination) to abrogate the AR function in female mice and MCF7 breast cancer cells, we demonstrate that AR may go through interruption of MAPK activity and ER signaling to exert its important roles for the normal mouse mammary gland development and MCF7 breast cancer cell growth. These results are in agreement with early papers showing that both MAPK and ER are essential factors for mammary gland development and breast cancer growth. For example, female ER −/− mice exhibit undeveloped mammary glands similar to those of newborn mice, indicating the essential roles of ER in the ductal growth (21), and the loss of ER can significantly delay the onset of tumor induction in MMTV-Neu (Neu/ER −/−) or –Wnt-1 (Wnt-1/ER −/−) transgenic mice lacking functional ER (36, 37). The MAPK inhibitor PD098059 could inhibit both the mammary gland alveolar morphogenesis (20) and Her2/Neu-, H-Ras– or C-myc–initiated mammary tumor growth (38).
To dissect how AR influences MAPK activity, we found that loss of AR could disrupt the IGF-I–, EGF-, and HRG-α–induced MAPK activity (Fig. 4 B) and reduced IGF-IR expression in AR −/− mice (Fig. 3 C). Bonnette et al. (23) found that mice with defective IGF-IR have less branching and decreased cellular proliferation of TEBs in developing mammary glands, and these defects could be only partly restored during pregnancy. Similar phenomena also occurred in AR −/− mice (Figs. 1 and 2), suggesting that the suppression of IGF-IR by the AR abrogation may contribute to the retarded mammary gland development in AR −/− mice. Consistent with data (Fig. 4 F and Fig. 5 B) showing that the loss of AR interrupts HRG-α–induced anchorage-independent cell growth and MAPK activation, Watson et al. (39) found that in transgenic rats with overexpression of HER2–Neu in the mammary gland, normal males, but not castrated males, developed mammary tumors. These results may suggest the potential cross-talk between androgen–AR and HER2–Neu signaling pathways in mammary tumor progression.
Further mechanism dissection studies indicate that AR-induced MAPK activation is via the Ras/Raf/MAPK cascade (Fig. 5 D), although the detailed mechanisms of how AR influences Ras-Raf activity remain unclear. Migliaccio et al. (32) reported that AR could activate MAPK via the interaction between the proline-rich motif of AR and the SH3 domain of c-Src. They demonstrated that this interaction occurred quickly and could be initiated in a short time by androgen or estrogen treatment. In contrast, Fig. 6 A demonstrates that AR NH2 terminus/DBD, without the LBD, can induce MAPK activity, suggesting that AR, but not androgen, is the major factor to activate MAPK activity. This raises an interesting question: what is the role of AR in the breast cancer development? Early analyses of androgen–AR roles in breast cancer mainly focused on the effect of androgen treatment on the breast cancer showing that androgen could either promote or suppress breast cancer growth (5, 6, 40). On the other hand, the effect of AR protein itself may also possibly go through interaction with other proteins resulting in nongenomic and/or nonandrogenic activities. It is likely that AR signals may use multiple pathways, including the classic androgen/AR→AR target genes of genomic actions as well as AR→AR interaction proteins of nongenomic action to exert its roles in the breast cancer progression. This is in agreement with early papers showing that ER could also cross-talk to MAPK in breast cancer cells (25, 41). In addition to estrogens, ER could be activated via phosphorylation at Ser118 by MAPK to induce its target gene expression (25). In return, ER could also induce the Ras/Raf/MAPK cascade via nongenomic action (32). Therefore, our results showing that AR could influence both MAPK and ER signals suggest that the reduction of ER activity could be due to the reduced MAPK activity, and the reduced MAPK activity may be due to the reduced ER activity in female mice and MCF7 cells lacking AR.
In conclusion, this paper provides the first in vivo evidence showing that AR may go through growth factors, MAPK, and ER/PR signals (summary in Fig. 6 C) to control the normal breast development, and modulate the breast cancer cell proliferation, especially in the conditions of absence of or reduced E2 (Fig. 4 E). Whether this would imply there is a positive correlation between AR expression and breast cancer incidence/progression in women with lower circulating E2, such as with the postmenopausal stages, remains an interesting question for future study. Epidemiological papers support this correlation suggesting that AR expression is more significantly associated with breast cancer in postmenopausal women than premenopausal women (9, 13, 42), and up to 50% of the AR-positive breast cancers are ER- and/or PR-negative (13, 43). Finally, as AR NH2 terminus/DBD, but not LBD, may play essential roles to modulate the growth factor signaling pathways in MCF7 breast cancer cells, targeting the function of AR NH2 terminus/DBD may represent a new potential therapeutic approach to the battle against breast cancer.
Acknowledgments
We thank J. Xu, H. Land, P. Keng, D.A. Pearce, P.A. di Sant'Agnese, K.-L. Guan, and J. Chernoff for helpful discussions and their reagents. We also thank K. Wolf for proofreading.
This work was supported by National Institutes of Health grants DK60905, Army DAMD17-02-1-0557, NYSDOH-C017947, and the George H. Whipple Professorship Endowment.
S. Yeh and Y.-C. Hu contributed equally to this paper.
Abbreviations used in this paper: AR, androgen receptor; BrdU, bromodeoxyuridine; CDS, charcoal dextran–stripped; DBD, DNA-binding domain; dprm, deletion of the proline-rich motif; E2, 17β-estradiol; Efp, estrogen-responsive finger protein; EGF, epidermal growth factor; ER, estrogen receptor; GFP, green fluorescent protein; HGF, hepatocyte growth factor; HI, heat-inactivated; HRG-α, heregulin-α; IGF-I, insulin-like growth factor I; LBD, ligand-binding domain; Luc, luciferase; MAPK, mitogen-activated protein kinase; np, natural promoter–driven; P, progesterone; PR, P receptor; siRNA, small interfering RNA; TBS, Tris-buffered saline; TEB, terminal end bud; UTR, untranslated region.
References
- 1.Chang, C.S., J. Kokontis, and S.T. Liao. 1988. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 240:324–326. [DOI] [PubMed] [Google Scholar]
- 2.McKenna, N.J., and B.W. O'Malley. 2002. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 108:465–474. [DOI] [PubMed] [Google Scholar]
- 3.Secreto, G., and B. Zumoff. 1994. Abnormal production of androgens in women with breast cancer. Anticancer Res. 14:2113–2117. [PubMed] [Google Scholar]
- 4.Berrino, F., P. Muti, A. Micheli, G. Bolelli, V. Krogh, R. Sciajno, P. Pisani, S. Panico, and G. Secreto. 1996. Serum sex hormone levels after menopause and subsequent breast cancer. J. Natl. Cancer Inst. 88:291–296. [DOI] [PubMed] [Google Scholar]
- 5.Dimitrakakis, C., J. Zhou, and C.A. Bondy. 2002. Androgens and mammary growth and neoplasia. Fertil. Steril. 77(Suppl.):26–33. [DOI] [PubMed] [Google Scholar]
- 6.Birrell, S.N., J.M. Bentel, T.E. Hickey, C. Ricciardelli, M.A. Weger, D.J. Horsfall, and W.D. Tilley. 1995. Androgens induce divergent proliferative responses in human breast cancer cell lines. J. Steroid Biochem. Mol. Biol. 52:459–467. [DOI] [PubMed] [Google Scholar]
- 7.Szelei, J., J. Jimenez, A.M. Soto, M.F. Luizzi, and C. Sonnenschein. 1997. Androgen-induced inhibition of proliferation in human breast cancer MCF7 cells transfected with androgen receptor. Endocrinology. 138:1406–1412. [DOI] [PubMed] [Google Scholar]
- 8.Wilson, C.M., and M.J. McPhaul. 1996. A and B forms of the androgen receptor are expressed in a variety of human tissues. Mol. Cell. Endocrinol. 120:51–57. [DOI] [PubMed] [Google Scholar]
- 9.Lea, O.A., S. Kvinnsland, and T. Thorsen. 1989. Improved measurement of androgen receptors in human breast cancer. Cancer Res. 49:7162–7167. [PubMed] [Google Scholar]
- 10.Kuenen-Boumeester, V., T.H. Van der Kwast, W.L. van Putten, C. Claassen, B. van Ooijen, and S.C. Henzen-Logmans. 1992. Immunohistochemical determination of androgen receptors in relation to oestrogen and progesterone receptors in female breast cancer. Int. J. Cancer. 52:581–584. [DOI] [PubMed] [Google Scholar]
- 11.Bayer-Garner, I.B., and B. Smoller. 2000. Androgen receptors: a marker to increase sensitivity for identifying breast cancer in skin metastasis of unknown primary site. Mod. Pathol. 13:119–122. [DOI] [PubMed] [Google Scholar]
- 12.Yeh, S., M.Y. Tsai, Q. Xu, X.M. Mu, H. Lardy, K.E. Huang, H. Lin, S.D. Yeh, S. Altuwaijri, X. Zhou, et al. 2002. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc. Natl. Acad. Sci. USA. 99:13498–13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bieche, I., B. Parfait, S. Tozlu, R. Lidereau, and M. Vidaud. 2001. Quantitation of androgen receptor gene expression in sporadic breast tumors by real-time RT-PCR: evidence that MYC is an AR-regulated gene. Carcinogenesis. 22:1521–1526. [DOI] [PubMed] [Google Scholar]
- 14.Sugimoto, T., S. Stewart, M. Han, and K.L. Guan. 1998. The kinase suppressor of Ras (KSR) modulates growth factor and Ras signaling by uncoupling Elk-1 phosphorylation from MAP kinase activation. EMBO J. 17:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sells, M.A., U.G. Knaus, S. Bagrodia, D.M. Ambrose, G.M. Bokoch, and J. Chernoff. 1997. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7:202–210. [DOI] [PubMed] [Google Scholar]
- 16.Hu, Y.C., C.R. Shyr, W. Che, X.M. Mu, E. Kim, and C. Chang. 2002. Suppression of estrogen receptor-mediated transcription and cell growth by interaction with TR2 orphan receptor. J. Biol. Chem. 277:33571–33579. [DOI] [PubMed] [Google Scholar]
- 17.Sui, G., C. Soohoo, B. Affar el, F. Gay, Y. Shi, W.C. Forrester, and Y. Shi. 2002. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl. Acad. Sci. USA. 99:5515–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silberstein, G.B. 2001. Postnatal mammary gland morphogenesis. Microsc. Res. Tech. 52:155–162. [DOI] [PubMed] [Google Scholar]
- 19.Lydon, J.P., F.J. DeMayo, C.R. Funk, S.K. Mani, A.R. Hughes, C.A. Montgomery, Jr., G. Shyamala, O.M. Conneely, and B.W. O'Malley. 1995. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 9:2266–2278. [DOI] [PubMed] [Google Scholar]
- 20.Niemann, C., V. Brinkmann, E. Spitzer, G. Hartmann, M. Sachs, H. Naundorf, and W. Birchmeier. 1998. Reconstitution of mammary gland development in vitro: requirement of c-met and c-erbB2 signaling for branching and alveolar morphogenesis. J. Cell Biol. 143:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couse, J.F., and K.S. Korach. 1999. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr. Rev. 20:358–417. [DOI] [PubMed] [Google Scholar]
- 22.Kleinberg, D.L., M. Feldman, and W. Ruan. 2000. IGF-I: an essential factor in terminal end bud formation and ductal morphogenesis. J. Mammary Gland Biol. Neoplasia. 5:7–17. [DOI] [PubMed] [Google Scholar]
- 23.Bonnette, S.G., and D.L. Hadsell. 2001. Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology. 142:4937–4945. [DOI] [PubMed] [Google Scholar]
- 24.Brisken, C., A. Ayyannan, C. Nguyen, A. Heineman, F. Reinhardt, J. Tan, S.K. Dey, G.P. Dotto, R.A. Weinberg, and T. Jan. 2002. IGF-2 is a mediator of prolactin-induced morphogenesis in the breast. Dev. Cell. 3:877–887. [DOI] [PubMed] [Google Scholar]
- 25.Kato, S., H. Endoh, Y. Masuhiro, T. Kitamoto, S. Uchiyama, H. Sasaki, S. Masushige, Y. Gotoh, E. Nishida, H. Kawashima, et al. 1995. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 270:1491–1494. [DOI] [PubMed] [Google Scholar]
- 26.Said, T.K., O.M. Conneely, D. Medina, B.W. O'Malley, and J.P. Lydon. 1997. Progesterone, in addition to estrogen, induces cyclin D1 expression in the murine mammary epithelial cell, in vivo. Endocrinology. 138:3933–3939. [DOI] [PubMed] [Google Scholar]
- 27.Inoue, S., A. Orimo, T. Hosoi, S. Kondo, H. Toyoshima, T. Kondo, A. Ikegami, Y. Ouchi, H. Orimo, and M. Muramatsu. 1993. Genomic binding-site cloning reveals an estrogen-responsive gene that encodes a RING finger protein. Proc. Natl. Acad. Sci. USA. 90:11117–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang, J.G., A. Bell, Y. Liu, and R. Zarnegar. 1997. Transcriptional regulation of the hepatocyte growth factor gene by the nuclear receptors chicken ovalbumin upstream promoter transcription factor and estrogen receptor. J. Biol. Chem. 272:3928–3934. [DOI] [PubMed] [Google Scholar]
- 29.Urano, T., T. Saito, T. Tsukui, M. Fujita, T. Hosoi, M. Muramatsu, Y. Ouchi, and S. Inoue. 2002. Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature. 417:871–875. [DOI] [PubMed] [Google Scholar]
- 30.Haslam, S.Z. 1988. Acquisition of estrogen-dependent progesterone receptors by normal mouse mammary gland. Ontogeny of mammary progesterone receptors. J. Steroid Biochem. 31:9–13. [DOI] [PubMed] [Google Scholar]
- 31.Lobaccaro, J.M., S. Lumbroso, C. Belon, F. Galtier-Dereure, J. Bringer, T. Lesimple, M. Namer, B.F. Cutuli, H. Pujol, and C. Sultan. 1993. Androgen receptor gene mutation in male breast cancer. Hum. Mol. Genet. 2:1799–1802. [DOI] [PubMed] [Google Scholar]
- 32.Migliaccio, A., G. Castoria, M. Di Domenico, A. de Falco, A. Bilancio, M. Lombardi, M.V. Barone, D. Ametrano, M.S. Zannini, C. Abbondanza, and F. Auricchio. 2000. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 19:5406–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beitel, L.K., L. Prior, D.M. Vasiliou, B. Gottlieb, M. Kaufman, R. Lumbroso, C. Alvarado, B. McGillivray, M. Trifiro, and L. Pinsky. 1994. Complete androgen insensitivity due to mutations in the probable alpha-helical segments of the DNA-binding domain in the human androgen receptor. Hum. Mol. Genet. 3:21–27. [DOI] [PubMed] [Google Scholar]
- 34.Deleted in proof.
- 35.Deleted in proof.
- 36.Hewitt, S.C., W.P. Bocchinfuso, J. Zhai, C. Harrell, L. Koonce, J. Clark, P. Myers, and K.S. Korach. 2002. Lack of ductal development in the absence of functional estrogen receptor alpha delays mammary tumor formation induced by transgenic expression of ErbB2/neu. Cancer Res. 62:2798–2805. [PubMed] [Google Scholar]
- 37.Bocchinfuso, W.P., W.P. Hively, J.F. Couse, H.E. Varmus, and K.S. Korach. 1999. A mouse mammary tumor virus-Wnt-1 transgene induces mammary gland hyperplasia and tumorigenesis in mice lacking estrogen receptor-alpha. Cancer Res. 59:1869–1876. [PubMed] [Google Scholar]
- 38.Amundadottir, L.T., and P. Leder. 1998. Signal transduction pathways activated and required for mammary carcinogenesis in response to specific oncogenes. Oncogene. 16:737–746. [DOI] [PubMed] [Google Scholar]
- 39.Watson, P.A., K. Kim, K.S. Chen, and M.N. Gould. 2002. Androgen-dependent mammary carcinogenesis in rats transgenic for the Neu proto-oncogene. Cancer Cell. 2:67–79. [DOI] [PubMed] [Google Scholar]
- 40.Xie, B., S.W. Tsao, and Y.C. Wong. 1999. Sex hormone-induced mammary carcinogenesis in female noble rats: the role of androgens. Carcinogenesis. 20:1597–1606. [DOI] [PubMed] [Google Scholar]
- 41.Greene, G.L. 2003. In vivo imaging reveals estrogen receptor's hidden personality. Nat. Med. 9:22–23. [DOI] [PubMed] [Google Scholar]
- 42.Honma, N., G. Sakamoto, F. Akiyama, Y. Esaki, M. Sawabe, T. Arai, T. Hosoi, N. Harada, M. Younes, and K. Takubo. 2003. Breast carcinoma in women over the age of 85: distinct histological pattern and androgen, oestrogen, and progesterone receptor status. Histopathology. 42:120–127. [DOI] [PubMed] [Google Scholar]
- 43.Brys, M., M. Wojcik, H. Romanowicz-Makowska, and W.M. Krajewska. 2002. Androgen receptor status in female breast cancer: RT-PCR and Western blot studies. J. Cancer Res. Clin. Oncol. 128:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]