

Abstract
Two important Ras guanine nucleotide exchange factors, Son of sevenless (Sos) and Ras guanine nucleotide releasing protein (RasGRP), have been implicated in controlling Ras activation when cell surface receptors are stimulated. To address the specificity or redundancy of these exchange factors, we have generated Sos1/Sos2 double- or RasGRP3-deficient B cell lines and determined their ability to mediate Ras activation upon B cell receptor (BCR) stimulation. The BCR requires RasGRP3; in contrast, epidermal growth factor receptor is dependent on Sos1 and Sos2. Furthermore, we show that BCR-induced recruitment of RasGRP3 to the membrane and the subsequent Ras activation are significantly attenuated in phospholipase C-γ2–deficient B cells. This defective Ras activation is suppressed by the expression of RasGRP3 as a membrane-attached form, suggesting that phospholipase C-γ2 regulates RasGRP3 localization and thereby Ras activation.
Keywords: DAG, PLC-γ2, RasGRP3, ERK, B cell activation
Introduction
In B and T lymphocytes, activation of the small guanosine triphosphatase (GTPase) Ras is a crucial event that takes place when antigen or some lymphokine receptors are stimulated (1–7). By associating with different effector proteins, including Raf/MEK/ERK, Ras acts as a branch point to initiate multiple downstream signaling pathways and, hence, influence gene expression in these cells (8). Indeed, the Ras pathway has been implicated in supporting survival and differentiation of pre–B cells as well as mature B cells. Introduction of a constitutive active form of Ras into a Rag-null background can cause progression of pro–B cells to pre–B and subsequent mature B cells (9, 10). Conversely, expression of a dominant negative form of Ras markedly reduces the number of pre–B cells and immature B cells (11, 12). These findings, given the importance of pre–B cell receptor (BCR) and BCR in B cell survival and differentiation (13–17), suggest a crucial role for Ras in pre-BCR– and BCR-mediated cell fate decision.
Ras activity cycles between a GDP-bound off and a GTP-bound on conformation proximal to the inner surface of the plasma membrane. Ras is inactivated by its own intrinsic capability to induce GTP hydrolysis to GDP, and this GTPase activity can be accelerated by GTPase-activating proteins (GAPs). Conversely, guanine nucleotide exchange factors (GEFs) control the activation of Ras by catalyzing GDP release from Ras and, thereby, facilitating its association with GTP (18–20). Thus, Ras activity is controlled by the balanced activities of GEFs and GAPs. Hence, two alternative, but not necessarily mutually exclusive, models are thought to account for Ras activation upon antigen receptor stimulation as follows: (a) inhibition of GAP function; and (b) activation of GEF activity. With regard to the first possibility, the importance of association of p62DOK with p120RasGAP has been proposed in B cells (21, 22); this interaction is thought either to allosterically inhibit GAP functions, or to sequester this negative regulator, thereby physically preventing its interaction with Ras. However, the molecular details of GAP regulation are still fragmentary.
The other side of Ras regulation, namely by GEF activity, has been thought to be mediated by at least two types of Ras GEFs: (a) the well-characterized Son of sevenless (Sos) family; and (b) the recently identified Ras guanine nucleotide releasing protein (RasGRP) family (19, 23–25). Two lines of recent evidence favor the idea that the RasGRP family, rather than the Sos family, could play a more dominant role in Ras activation after antigen receptor ligation in the T cell system. First, RasGRPs contain a diacylglycerol (DAG)-binding C1 domain, possibly explaining the striking activation of Ras in peripheral T cells after phorbol ester stimulation (26–28). Second, RasGRP1−/− thymocytes fail to activate Ras and phosphorylate Erk when challenged with phorbol ester or when their TCRs are stimulated (26).
Despite striking T cell developmental defects in Ras GRP1−/− mice, B cell development appears to be apparently normal (26). Hence, in the case of B cells, considering the existence of four isoforms of the RasGRP family, one possibility is that another RasGRP family might play an important role in BCR-mediated Ras activation. Alternatively, in contrast to T cells, BCR might dominantly utilize the Sos family for Ras activation. Here, we report the consequences of disruption of either of these GEFs and demonstrate the mechanism by which BCR activates Ras.
Materials and Methods
Cells, Mice, Abs, and Reagents.
Wild-type and various mutant DT40 cells were cultured in RPMI 1640 (GIBCO™; Invitrogen) supplemented with 10% FCS, 1% chicken serum, 50 μM 2-ME (Sigma-Aldrich), 4 mM l-glutamine, and antibiotics. Establishment of phospholipase C (PLC)-γ2–deficient DT40 cells was described previously (29). Wild-type C57BL/6J mice were purchased from Clea Japan. Anti-Sos1 Ab, anti-Sos2 Ab, anti-RasGRP1 Ab, and anti-RasGRP3 Ab were obtained by immunizing rabbits with bacterially expressed glutathione S-transferase (GST) fusion proteins containing amino acid sequences coding chicken Sos1 (amino acids 1241–1336), chicken Sos2 (amino acids 1200–1297), chicken RasGRP1 (amino acids 690–787), and chicken RasGRP3 (amino acids 401–691), respectively. Anti–chicken IgM mAb, M4 (30) was used for stimulation of BCR and Western blotting. Anti-phospho–p44/p42 mitogen-activated protein kinase (MAPK) polyclonal Ab, anti-phospho–p44/p42 MAPK mAb (E10), and anti-p44/p42 MAPK polyclonal Ab were purchased from Cell Signaling Technology Inc. Anti–pan-Ras mAb (Ab-3) was purchased from Oncogene Research Products. Anti–H-Ras Ab (C-20) and anti–K-Ras Ab (F234) were purchased from Santa Cruz Biotechnology, Inc. The pEGFP-C2 vector was purchased from CLONTECH Laboratories, Inc. EGF was purchased from TOYOBO.
Expression Constructs and Transfection.
Chicken RasGRP3 cDNA was cloned into the pApuro expression vector. Using QuikChange™ (Stratagene), the arginine residue (218) is substituted to glutamate to create a GEF mutant construct (mGEFRasGRP3), and C1 domain is deleted to create a C1 domain deletion mutant construct (ΔC1RasGRP3). Wild-type RasGRP3 and ΔC1RasGRP3 were fused at its COOH terminus to enhanced green fluorescent protein (EGFP) by the PCR method, resulting in generation of RasGRP3–green fluorescent protein (GFP) and ΔC1RasGRP3-GFP. For construction of ΔC1-pbRasGRP3 and ΔC1-ΔpbRasGRP3, the modified pApuro vector was made to insert oligonucleotides encoding a polybasic domain or its mutant at its 3′ cloning site. Thus, the resulting polybasic domain and polybasic domain defective vector encode the following amino acids of the COOH terminus of chicken K-Ras: xRKHKEKMSKDGKKKKKKTKTKCIIM# or xRKHKEKMSKDG#, where x is the site of insertion of cDNA and # is the stop codon. ΔC1RasGRP3 was inserted into these vectors, resulting in in-frame fusion of cDNA to the polybasic domain. These constructs were transfected into each mutant DT40 cells by electroporation (550 V, 25 μF), and selected in the presence of 0.5 μg/ml puromycin. Human epidermal growth factor receptor (EGFR) cDNA was transfected and selected as described previously (31). Chicken H-Ras and K-Ras cDNAs were cloned into the pApuro expression vector and were transfected into 293T cells by the calcium phosphate coprecipitation method. Expression of transfected cDNAs was confirmed by Western blot analysis.
Generation of Various Deficient DT40 Cells.
Based on published sequence of murine Sos1, Sos2, RasGRP1, and human RasGRP3, we searched each chicken homologue using the expressed sequence tag (EST) database and obtained each chicken cDNA by RT-PCR with RNA from DT40 B cells. Genomic clones of Sos1, Sos2, RasGRP1, and RasGRP3 were obtained by PCR using oligonucleotides designed from each cDNA sequence and genomic DNA as a template. The targeting vector, pSos1-bleo, pSos1-hygro, or pSos1-puro was constructed by replacing the genomic fragment containing exons that correspond to murine Sos1 GEF domain, amino acid residues 740–947, with bleo, hygro, or puro cassette. The targeting constructs for Sos2 were designed for neo and hisD cassettes to replace the genomic fragment corresponding to chicken Sos2 GEF domain, amino acid residues 814–908. The hisD and zeo targeting constructs for RasGRP1 were made by replacing the genomic fragment containing chicken RasGRP1 GEF domain, amino acid residues 169–316, with hisD and zeo cassettes. The targeting constructs for RasGRP3 were designed for neo and bsr cassettes to replace the genomic fragment containing exons corresponding to chicken RasGRP3 GEF domain, amino acid residues 124–361. These targeting vectors were sequentially transfected into various mutant DT40 cells, resulting in generation of Sos1-, Sos2-, RasGRP1-, RasGRP3-, Sos1/Sos2 double-, and RasGRP1/RasGRP3 double-deficient DT40 cells. Selection for each drug-resistant clone was performed by using 0.3 mg/ml phleomycin, 2 mg/ml hygromycin, 0.5 μg/ml puromycin, 1 mg/ml histidiol, 50 μg/ml blastcidin, and 2 mg/ml G418.
Northern Blot Analysis.
RNA was prepared from wild-type DT40 cells using the guanidium thiocyanate method. Total RNA (20 μg) was separated in 1.2% formaldehyde gel, transferred to Hybond-N+ membrane (Amersham Biosciences), and probed with 32P-labeled chicken cDNA. β-actin (32) was used for internal control.
Immunoprecipitation and Western Blot Analysis.
For immunoprecipitation, cells were solubilized in NP-40 lysis buffer supplemented with protease and phosphatase inhibitors as described previously (33), and precleared lysates were incubated with proper Abs and protein A–agarose. For Western blot analysis, immunoprecipitates or cleared-cell lysates were resolved on SDS-PAGE, transferred to polyvinyldifluoride membrane (Bio-Rad Laboratories), and detected by the indicated Abs using the enhanced chemoluminescence system (Amersham Biosciences).
Ras-GTP Assay.
Ras-GTP assay was performed as described previously (34, 35). In brief, bacterially expressed GST–Ras binding domain (amino acids 1–149 of human cRaf-1 fused to GST) prebounded glutathione-Sepharose beads was prepared. Each M4-stimulated cell lysate in Mg2+-containing lysis buffer was incubated with the beads for 30 min at 4°C. Bound proteins were eluted with SDS-PAGE sample buffer and resolved on 15% SDS-PAGE and subjected to Western blotting with anti–pan-Ras Ab.
Calcium Measurements.
Calcium mobilization was measured as described previously (34). In brief, cells were loaded in Fura-2 AM at 37°C for 45 min, and washed twice, adjusted to 106 cells/ml. Cells were stimulated with 1.5 μg/ml M4 mAb at 37°C. Continuous monitoring of fluorescence from cell suspension was performed using a fluorescence spectrophotometer (model F-2000; Hitachi).
IP3 Generation Assay.
2 × 106 cells were stimulated with 1.5 μg/ml M4 mAb at 37°C for indicated times. Production of inositol 1,4,5-triphosphate (IP3) was measured using a commercial IP3 assay system (Biotrak TRK 1000; Amersham Biosciences) following the manufacturer's protocol.
Lentiviral Transduction.
The cDNA encoding GEF mutant of murine RasGRP3 (amino acid 218; arginine to glutamate) was cloned into modified pLenti6/V5-DEST (Invitrogen) containing EGFP as an expression marker downstream of the internal ribosomal entry site. The empty vector was used as a negative control. Culture supernatants containing lentivirus were generated using 293FT cell line and ViraPower Lentiviral Support Kit (Invitrogen) according to the manufacturer's instruction. Collected supernatants were centrifuged at 60,000 g for 2 h, and the virus pellets were resuspended in culture medium at 100:1 volume. Preparation of murine splenic B cells was described previously (36), and isolated B cells were transduced with lentivirus as follows. Purified resting B cells (106) were suspended in 500 μl of growth medium consisted of RPMI 1640 supplemented with 10% FCS, 50 μM 2-ME, 2 mM l-glutamine, and 4 mM sodium pyruvate, and followed by the addition of 100 μl of lentiviral supernatants. Hexadimethrine bromide (Sigma-Aldrich) was added at the final concentration of 8 μg/ml. The culture plates were centrifuged at 30°C with 2,500 revolutions/min for 90 min. 3 d after transfection, cells were collected and subjected to Western blot analysis using phospho-p44/p42 MAPK mAb (E10) or RT-PCR.
RT-PCR.
Expression levels of murine RasGRP3 and its GEF mutant (mGEFRasGRP3) were analyzed by semi-quantitative RT-PCR. In brief, total RNA was extracted from 4 × 105 cells using TRIzol (Invitrogen). Obtained RNA was reverse transcribed using random hexamer primer (TAKARA BIO) with SuperScriptII (Invitrogen) according to the manufacturer's instruction. For amplification of wild-type RasGRP3, 5′-CAAACCAAGTGCGCAACAAAG-3′ and 5′-CTCCTCCATCCAGTCTGGGAA-3′ were used as forward and reverse primers, and cycled 45 times at 94°C for 30 s, 63.5°C for 30 s, and 72°C for 30 s. For amplification of mGEFRasGRP3, 5′-CAAACCAAGTGCGCAACAAGA-3′ was used as a forward primer, and the reverse primer was the same with wild type, and cycled 45 times at 94°C for 30 s, 65.5°C for 30 s, and 72°C for 30 s. GAPDH was amplified as an internal control using 5′-CCATCACCATCTTCCAGGAG-3′ and 5′-CCTGCTTCACCACCTTCTTG-3′ as forward and reverse primers. Each PCR was performed using AmpliTaq Gold™ (Applied Biosystems).
Subcellular Analysis of RasGRP3.
Subcellular fractionation was performed as described previously (34) with several modifications. In brief, cells were incubated on ice water for 10 min and further incubated in the presence or absence of 10 μg/ml M4 for 30 min. Cells were stimulated at 37°C for 10 min and resuspended in hypotonic lysis buffer. Proteins were separated by 7% SDS-PAGE and analyzed by Western blotting using anti-RasGRP3 Ab.
Visualization of Membrane Translocation of RasGRP3-GFP.
Wild-type DT40 cells expressing RasGRP3-GFP or its C1 domain deletion mutant (ΔC1RasGRP3-GFP) were incubated with 5 μg/ml anti–chicken IgM mAb (M4) on ice for 20 min. PLC-γ2–deficient DT40 cells expressing RasGRP3-GFP were also incubated with M4 under the same condition. The cells were washed with ice-cold PBS and incubated at 37°C for 0 and 10 min followed by fixation with 0.4% paraformaldehyde (Sigma-Aldrich). Membrane translocation of RasGRP3-GFP fusion protein was visualized by a laser scanning confocal microscope (model LSM 510; Carl Zeiss MicroImaging, Inc.).
Flow Cytometric Analysis.
Cell surface expression of BCR or EGFR on various mutant DT40 cells was analyzed by FACSCalibur™ (Becton Dickinson) using FITC-conjugated anti–chicken IgM Ab (Bethyl) or mouse anti–human EGFR mAb and FITC-labeled anti–mouse IgG (Cappel). Expression level of EGFP was also monitored by FACSCalibur™.
Results
Requirement for PLC-γ2 in BCR-mediated Ras Activation.
Given the previous evidence that BCR-mediated Erk activation is impaired in PLC-γ2–deficient DT40 B cells (37), we first examined whether Ras activation is also perturbed in the mutant cells. As shown in Fig. 1 A, when compared with wild-type DT40 cells, BCR-mediated Ras activation was decreased by ∼50% in PLC-γ2–deficient cells. The anti-pan Ras Ab recognized two species of Ras in chicken DT40 B cells, presumably representing K-Ras (Fig. 1 B, top band) and H-Ras (Fig. 1 B, bottom band). Indeed, expression of the chicken K-Ras or H-Ras cDNA in 293T cells gave rise to a protein that comigrated with K-Ras or H-Ras in DT40 B cells, respectively (Fig. 1 B). Thus, PLC-γ2 is required for activation of both isoforms of Ras, which, in turn, contributes to Erk activation in BCR signaling context.
Figure 1.

Requirement for PLC-γ2 in BCR-mediated Ras activation. (A) BCR-mediated Ras and ERK activation in wild-type, PLC-γ2–deficient DT40 B cells. For Ras activation, 107 cells/lane were stimulated with 5 μg/ml M4, and cell lysates were mixed with beads containing the Ras binding domain of Raf. Elutes of the beads (top) or cell lysates (bottom) were subjected to Western blot analysis with anti–pan-Ras mAb. AP, affinity precipitation. For ERK activation, 107 cells were stimulated with 5 μg/ml M4, and cell lysates (2 × 106 cells/lane) were analyzed by Western blotting with anti-phospho–p44/p42 MAPK polyclonal Ab (top) and anti-p44/p42 MAPK polyclonal Ab (bottom). (B) Protein expression analysis of chicken H-Ras and K-Ras. Total cell lysates of DT40 cells (107 cells/lane) and 293T cells transfected with chicken H-Ras or K-Ras cDNA were prepared and analyzed by Western blotting using anti–pan-Ras, anti–H-Ras, and anti–K-Ras Abs. Although the bottom band was the major product of transfected chicken H-Ras in 293T cells, the faint top band, which has the same electrophoretic mobility as K-Ras, was detected when overexposed. Thus, the minor portion of the top band observed in DT40 B cells is likely to be composed of H-Ras. (C) Complementation of Ras activation after costimulation of BCR with PMA or ionomycin. PLC-γ2–deficient DT40 B cells were stimulated and analyzed as in A with 50 ng/ml PMA or 100 nM ionomycin. This concentration of ionomycin can induce calcium mobilization to the same extent as induced by M4 stimulation on wild-type DT40 B cells.
When temporal kinetics between Ras and Erk activation in wild-type DT40 cells were compared, the Erk activation was still sustained after 10 min of BCR stimulation despite a significant decline of Ras activation (Fig. 1 A). Therefore, in addition to regulation by Ras, another Ras-independent pathway is likely to operate on sustaining ERK activation. This Ras-independent pathway appears to lie downstream of PLC-γ2, because the sustained Erk activation at 10 min was abolished in PLC-γ2–deficient DT40 B cells.
Once PLC-γ2 is activated upon BCR engagement, the activated PLC-γ2 converts phosphatidylinositol 4,5-bisphosphate into the two second messengers, DAG and inositol 1,4,5-trisphosphate (IP3), which result in protein kinase C activation and calcium increases, respectively (38–41). To examine the relative importance of DAG and IP3 in BCR-mediated Ras activation, complementation experiments were performed with either PMA or ionomycin. In contrast to PMA treatment, addition of ionomycin to BCR cross-linking on PLC-γ2–deficient DT40 B cells barely stimulated Ras activation (Fig. 1 C), suggesting that DAG plays a more important role in Ras activation.
Targeted Disruption of Sos and RasGRP Family.
Next, we focused on the connection between PLC-γ2 and Ras activation. Because RasGRP family, but not Sos family, has a DAG-binding C1 domain, we reasoned that RasGRP family, rather than Sos family, might function as a downstream molecule of PLC-γ2, thereby contributing to BCR-mediated Ras activation. To directly address the necessity or redundancy of these molecules in BCR-mediated Ras activation, a genetic strategy was taken using DT40 B cells. Two members of Sos (Sos1 and Sos2) and four members of RasGRP (RasGRP1–RasGRP4; references 42–47) are known to be expressed with their combinations in a given cell type. RNA blot analysis of DT40 cells revealed that Sos1, Sos2, RasGRP1, and RasGRP3 were expressed in this cell line (Fig. 2 A). Hence, we established B cell lines lacking each gene encoding these proteins, either individually or in combination. Among these lines, in this analysis we particularly concentrated on analysis of DT40 B cell lines deleting Sos1/Sos2 doubly, RasGRP1/RasGRP3 doubly, and RasGRP3 alone. The lack of these proteins was verified by Western blot analysis (Fig. 2 B). The level of cell surface expression of BCR on these deficient lines was essentially the same as that on parental DT40 cells (Fig. 2 C). In addition to BCR, transfected EGFR was used as another prototype of receptors with potentially different signaling requirements.
Figure 2.
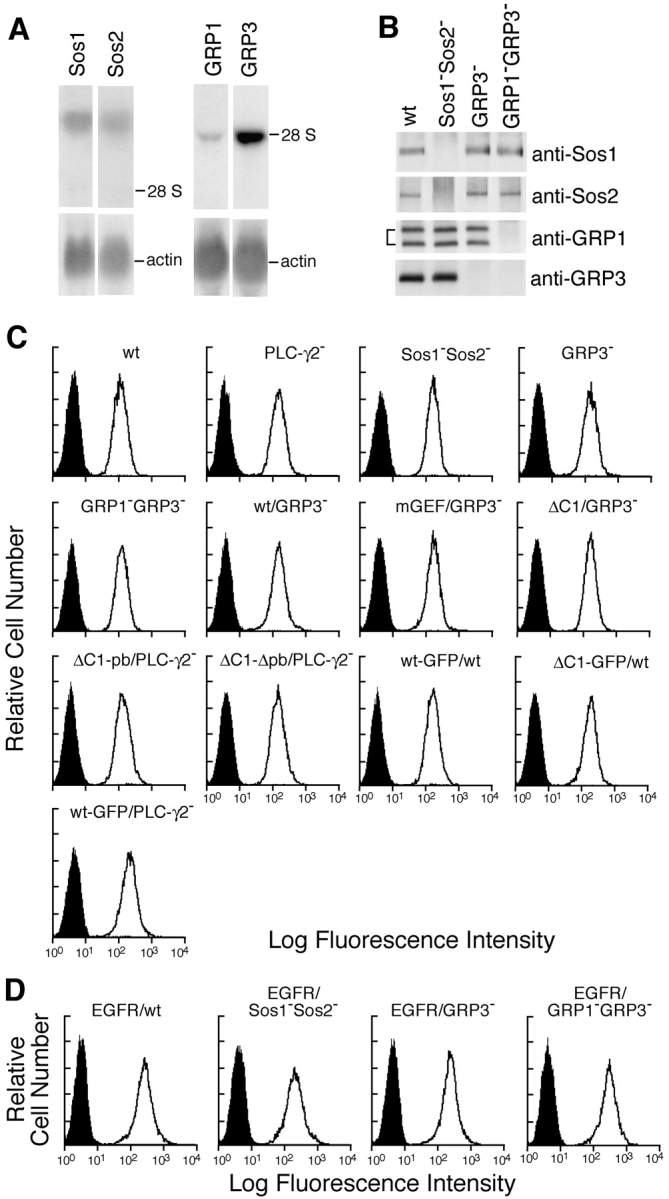
Generation of Sos1/Sos2 double-, RasGRP1/RasGRP3 double-, and RasGRP3-deficient DT40 B cells. (A) RNA expression of Sos1, Sos2, RasGRP1, and RasGRP3 was analyzed by Northern blot analysis using each chicken cDNA probe (top) or β-actin (bottom). The specific radio activity of each probe was roughly the same. Positions of 28S rRNA are shown. (B) Sos1, Sos2, RasGRP1, and RasGRP3 protein expression in wild-type and various mutant DT40 cells. Each protein was immunoprecipitated and detected by Western blotting with each specific Abs. Sos1−Sos2−, Sos1/Sos2 double-deficient DT40 cells. GRP3−, RasGRP3-deficient DT40 cells. GRP1−GRP3−, RasGRP1/RasGRP3 double-deficient DT40 cells. (C) Cell surface expression of BCR on wild-type and various DT40 mutants (see Fig. 6 A for designation). (D) Cell surface expression of transfected EGFR on wild-type and mutant DT40 cells.
RasGRP3 Is Necessary for BCR-mediated Ras Activation, Whereas Sos Family Is Necessary for EGFR-mediated Ras Activation.
As shown in Fig. 3 A, BCR-mediated Ras activation was unaffected in Sos1/Sos2 double-deficient DT40 cells, but was decreased by ∼60% in RasGRP3-deficient cells. This defective Ras activation was exacerbated to a small degree by double knockouts of RasGRP3 and RasGRP1, although RasGRP1 single-deficient DT40 cells exhibited apparently normal BCR-mediated Ras activation (unpublished data). Hence, we conclude that BCR-mediated Ras activation requires RasGRP3, but not the Sos family, in DT40 B cells. In addition, RasGRP1 appears to function as a redundant molecule of RasGRP3 and the relative importance of RasGRP3 in DT40 B cells is presumably attributable to dominant expression of RasGRP3 rather than RasGRP1 particularly in this cell line (Fig. 2 A). Consistent with the defective Ras activation in RasGRP3- or RasGRP1/RasGRP3 double-deficient DT40 cells, Erk activation was decreased in these mutants. However, the kinetics of Erk activation in these mutants was distinct from that of Ras, being marked at 1 min. These data imply that a Ras-independent pathway may compensate for the defective Erk activation in an attempt to cope with the deficit, particularly at the later time point.
Figure 3.

Differential requirement for RasGRP and Sos family in BCR- or EGFR-mediated Ras activation. (A) BCR-mediated Ras and ERK activation in wild-type, Sos1/Sos2 double-, RasGRP3-, and RasGRP1/RasGRP3 double-deficient DT40 cells. Ras and ERK activation was measured as described in Fig. 1. (B) EGFR-mediated Ras and ERK activation in each deficient DT40 cell. Cells were stimulated by 50 ng/ml EGF and analyzed as in Fig. 1. These experiments were performed at least three times and the representative results are shown.
In contrast to BCR, EGFR-mediated activation of both Ras and Erk was substantially reduced in Sos1/Sos2 double-deficient DT40 B cells and slightly perturbed in RasGRP3- or RasGRP1/RasGRP3 double-deficient cells (Fig. 3 B). This decrease cannot be ascribed to decreased expression of EGFR on DT40 B cells, because the level of cell surface expression of EGFR was similar among these clones (Fig. 2 D). Collectively, these data reveal that two types of receptors utilize distinct types of Ras GEFs to activate Ras in a given cell type.
To determine the importance of RasGRP family, particularly RasGRP3, in primary B cells as well, we took an approach of introducing a dominant-negative form of RasGRP3 into primary B cells by using a lentivirus system. Based on the data that overexpression of the GEF mutant of RasGRP3 in wild-type DT40 B cells inhibited BCR-mediated Ras activation (unpublished data), the similar mutant using the mouse RasGRP3 cDNA was subcloned into the HIV-based lentiviral vector containing a bicistronic mRNA encoding GFP. Because the Erk assay, rather than the Ras assay, needs a relatively small amount of infected B cells, we monitored BCR-mediated Erk activity in infected primary B cells. As shown in Fig. 4 B, primary B cells infected with a dominant negative form of RasGRP3 manifested a decrease in BCR-mediated Erk activation, compared with control viruses.
Figure 4.

Requirement for RasGRP3 in BCR-mediated ERK activation in primary B cells. (A) Expression of mGEFRasGRP3 in infected B cells. Splenic B cells from C57BL/6J mice were transduced with a GEF mutant of mouse RasGRP3 (mGEFRasGRP3) by lentivirus. Transduction efficiencies were monitored by EGFP expression. Fluorescence intensities of untransduced cells are shown in dashed lines (top). Expression levels of endogenous RasGRP3 and infected mGEFRasGRP3 were analyzed by RT-PCR as described in the Materials and Methods (bottom). (B) Attenuation of Erk activation by the expression of mGEFRasGRP3. Infected cells were stimulated with 12 μg/ml anti–mouse IgM and ERK activation was measured using anti-phospho–p44/p42 MAPK mAb (E10) (top) and anti-p44/p42 MAPK Ab (bottom).
To formally demonstrate that PLC-γ2 acts upstream to RasGRP3 in the context of BCR signaling, we examined the effect of disruption of RasGRP3 on PLC-γ2 activation. As demonstrated in Fig. 5 , BCR-mediated IP3 generation and subsequent calcium mobilization were unaffected in RasGRP3-deficient DT40 cells, supporting the idea that PLC-γ2 lies upstream of RasGRP3 in BCR signaling.
Figure 5.

BCR-mediated PLC-γ2 activation is not affected by loss of RasGRP3. Cells were stimulated by 1.5 μg/ml M4 for indicated time periods and subjected to the following analyses. (A) Calcium mobilization. Intracellular-free calcium levels in Fura-2–loaded cells were monitored by a fluorescence spectrophotometer. (B) IP3 generation. Soluble IP3 was extracted from 2 × 106 cells, and IP3 generation was measured by a Biotrak competitive binding assay system. The results were shown by mean ± standard error of three independent experiments.
The Catalytic Activity and C1 Domain of RasGRP3 Are Required for BCR-mediated Ras Activation.
The preceding results suggest that RasGRP3, when associated with the membrane by its interaction with DAG, gains access to its substrate Ras, converting it to the GTP-bound active form. This hypothesis predicts requirement for both catalytic activity and C1 domain of RasGRP3 in BCR-mediated Ras activation. These data are presented in Fig. 6 . Introduction of wild-type RasGRP3, but neither its GEF nor C1 deletion mutant, into RasGRP3-deficient DT40 B cells restored BCR-mediated Ras activation.
Figure 6.

Both catalytic activity and C1 domain of RasGRP3 are required in BCR-mediated Ras activation. (A) Schematic diagram of various mutant RasGRP3 constructs. (B) Protein expression analyses of various mutant DT40 cell lines. Wild-type, RasGRP3-, or PLC-γ2–deficient DT40 B cells expressing various RasGRP3 mutants are shown. Endogenous RasGRP3 is also shown in wild-type or PLC-γ2–deficient DT40 cells. Whole cell lysates prepared from 2 × 106 cells were analyzed by Western blotting using anti-RasGRP3 Ab or anti-BLNK Ab. (C) Analysis of BCR-mediated Ras activation in RasGRP3-deficient cells expressing wild-type RasGRP3 and its mutants was performed as described in Fig. 1. Each experiment was repeated at least three times.
Expression of RasGRP3 as a Membrane-attached Form in PLC-γ2–deficient Cells Restores Ras Activation.
By subcellular fractionation in wild-type DT40 B cells, RasGRP3 was localized in the membrane fraction before BCR stimulation to some extent. When stimulated, the stoichiometry of membrane-associated RasGRP3 was reproducibly, albeit to a small degree, enhanced. To demonstrate the importance of the interaction between DAG and the C1 domain of RasGRP3 in its translocation to the membrane, we examined the RasGRP3 status in PLC-γ2–deficient DT40 B cells. As expected, this BCR-mediated increase could not be observed in the mutant cells. Furthermore, the C1 deletion mutant of RasGRP3 (ΔC1RasGRP3) failed to move to the membrane fraction after BCR engagement (Fig. 7 A).
Figure 7.
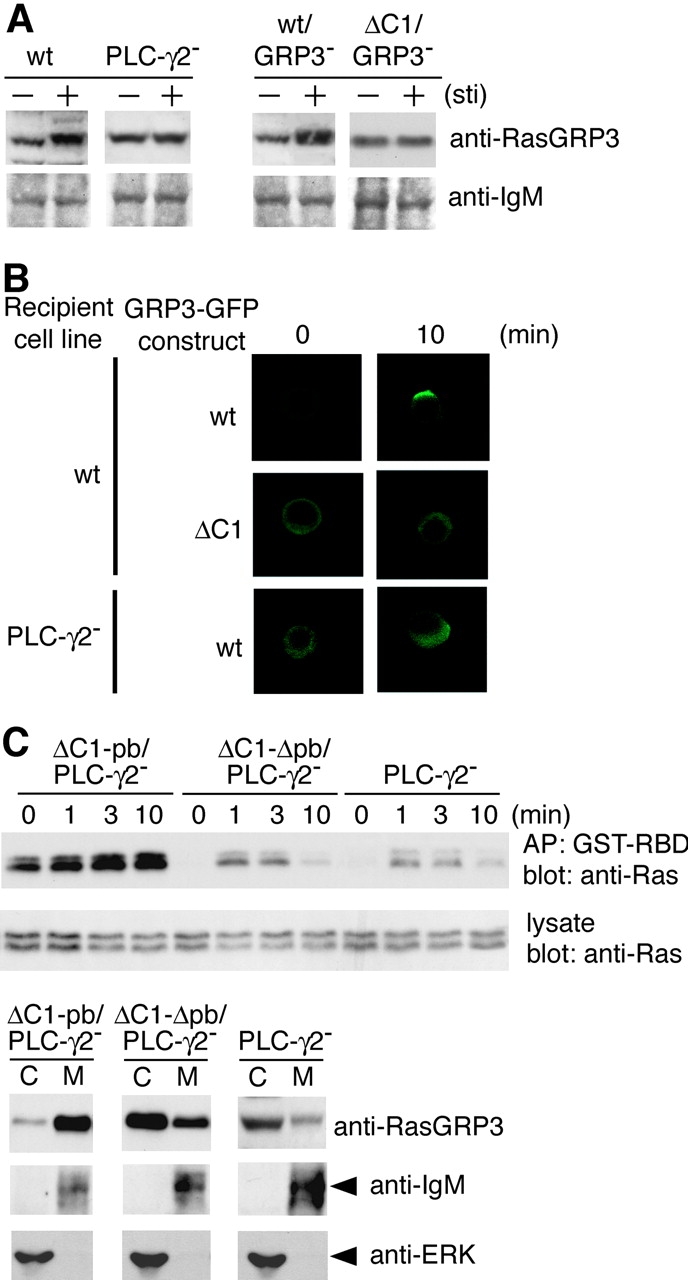
Membrane localization of RasGRP3 is necessary for Ras activation after BCR ligation. (A) Subcellular analysis of RasGRP3. Wild-type, PLC-γ2–deficient, wt/RasGRP3−, and ΔC1/RasGRP3− DT40 cells were stimulated by M4 for 10 min (+) and left unstimulated (−). Membrane fractions were resolved by SDS-PAGE and immunoblotted with anti-RasGRP3 Ab or anti-IgM Ab. (B) Impaired membrane translocation of RasGRP3 in PLC-γ2–deficient DT40 cells. Wild-type DT40 cells expressing RasGRP3-GFP (top) or its C1 deletion mutant (middle, ΔC1RasGRP3-GFP) were stimulated with anti–chicken IgM (M4) for indicated periods. PLC-γ2–deficient DT40 cells expressing RasGRP3-GFP were stimulated with M4 under the same condition (bottom). After the stimulation, cells were fixed and subcellular localization of RasGRP3-GFP fusion protein was visualized by confocal microscope as described in Materials and Methods. Among 100 cells counted for membrane localization, the numbers that showed clear membrane localization were 68 ± 16 for wild-type DT40 expressing RasGRP3-GFP, 8 ± 1 for wild-type DT40 cells expressing ΔC1RasGRP3-GFP, and 7 ± 1 for PLC-γ2–deficient DT40 cells expressing RasGRP3-GFP from three independent experiments. The representative results are shown. (C) Restoration of Ras activation by expression of a membrane-attached form of RasGRP3 in PLC-γ2–deficient DT40 cells. PLC-γ2–deficient DT40 cells expressing chimeras are shown as in Fig. 6 A. ΔC1RasGRP3 harboring the polybasic carboxyl terminus of K-Ras (ΔC1-pbRasGRP3/PLC-γ2−) or its polybasic amino acid–defective mutant (ΔC1-ΔpbRasGRP3/PLC-γ2−) were stimulated and analyzed with Ras activation as in Fig. 1 (top, GTP-bound Ras; middle, total Ras). Subcellular analyses of transfected RasGRP3 and endogenous RasGRP3 were also carried out before BCR stimulation. Samples were immunoblotted with anti-RasGRP3. Anti-IgM that recognized a μ heavy chain was used as a control for membrane fraction, and anti-ERK was used as a control for cytosolic fraction (bottom). C, cytosolic fraction. M, membrane fraction.
To further substantiate our conclusion, we performed microscopic analysis using RasGRP3 harboring GFP. As demonstrated in Fig. 7 B, wild-type RasGRP3-GFP, but not ΔC1RasGRP3-GFP, began to move to the membrane after 1 min of stimulation of BCR, and this membrane association persisted until at least 10 min after stimulation. After 10 min of stimulation, some DT40 cells exhibited RasGRP3-GFP at perinuclear region in addition to the membrane. In contrast to wild-type DT40 cells, in a PLC-γ2–deficient background, wild-type RasGRP3-GFP could not move to the membrane.
To address whether the defect in translocation of RasGRP3 in PLC-γ2–deficient DT40 B cells causes insufficient Ras activation, a chimera including ΔC1RasGRP3 and a polybasic domain of K-Ras (ΔC1-pbRasGRP3) and its defective mutant (ΔC1-ΔpbRasGRP3; reference 48) were expressed in PLC-γ2–deficient DT40 B cells (Fig. 6 A). As expected, before BCR stimulation, ΔC1-pbRas GRP3 was partitioned more into the membrane than wild-type RasGRP3 (Fig. 7 C). Ras activation status at resting level was increased by expression of ΔC1-pbRasGRP3 in PLC-γ2–deficient cells. When ligated by BCR, these cells exhibited a significant increase in Ras activation to the extent observed in wild-type DT40 B cells. These data suggest that membrane targeting of RasGRP3 can overcome the need for association of its C1 domain with DAG. However, a decline in active Ras after 10 min of BCR stimulation in wild-type DT40 cells (Fig. 1 A) was not recapitulated by expression of ΔC1-pbRasGRP3 (Fig. 7 C).
It has been shown that H-Ras and K-Ras are differentially localized in membrane rafts and outside the rafts, respectively, by the difference of their COOH-terminal membrane targeting sequence (49). Both H- and K-Ras are prenylated, but H-Ras is additionally modified by palmitoylation, whereas K-Ras contains a polybasic sequence (50, 51). Hence, activation of both H-Ras and K-Ras by the chimera containing the polybasic domain of K-Ras (ΔC1-pbRasGRP3) was somewhat unexpected. But, considering redistribution of H-Ras from the rafts into the bulk plasma membrane during receptor activation (49, 52, 53), this movement could provide H-Ras the possibility to gain access to ΔC1-pbRasGRP3. Alternatively, a small amount of ΔC1-pbRasGRP3 might be located in rafts under our experimental conditions, which, in turn, is responsible for H-Ras activation.
Discussion
Our previous work has pointed to a role for PLC-γ2 in Erk activation in response to BCR stimulation (37). The data presented here demonstrate that RasGRP3 couples PLC-γ2 to Ras, accounting at least partly for the defective BCR-mediated Erk activation in PLC-γ2–deficient B cells. In addition, our results show that BCR and EGFR utilize distinct types of RasGEFs, the RasGRP family and the Sos family, respectively, to activate Ras in the same cellular context.
Because the two src homology (SH) 3 domains of Grb2 bind to proline-rich residues near the C terminus of Sos, Grb2 is thought to mediate the translocation of Sos to the plasma membrane, thereby allowing Sos to activate membrane-bound Ras (54–58). In the case of EGFR, a requirement for Sos1 in EGFR-mediated Erk activation has been demonstrated already by using Sos1−/− fibroblasts, but this Erk activation is only partially blocked in this mutant cell (59, 60), therefore, subsequently raising the question of whether the remaining Erk activation is accounted for by residual Sos2 or by other GEF families. Hence, an almost complete block of EGER-mediated Ras and subsequent Erk activation in Sos1/Sos2 double-deficient DT40 B cells (Fig. 3 B) supports the former possibility. Furthermore, this result, together with our previous evidence that Grb2 is required for EGFR-mediated Ras activation in DT40 B cells (31), provides genetic evidence that the aforementioned mechanism indeed operates in B cells as well, at least in the case of transmembrane receptor tyrosine kinase signaling.
As in T cells, treatment of DAG analogues such as phorbol esters (PMAs) can induce the drastic activation of Ras in B cells (3), suggesting the existence of other Ras activation modes, in addition to the Grb2–Sos mechanism. This PMA-dependent Ras activation was initially thought to be mediated by protein kinase C isoforms containing single or double C1 domains. However, recent identification of RasGRPs as Ras activators possessing the C1 domain has inspired the possibility that RasGRPs are additional potential candidates to connect between PMA and Ras (23). Indeed, like a complete block of PMA-induced Ras activation in RasGRP1−/− thymocytes (26), RasGRP1/RasGRP3 double-deficient DT40 cells manifested almost complete block of PMA-induced Ras activation (unpublished data).
In fact, RasGRPs function in the BCR signaling context, because our data demonstrate the inhibition of BCR-mediated Ras activation in RasGRP1/RasGRP3 double-deficient DT40 B cells (Fig. 3 A). Despite a significant decrease in BCR-mediated Ras activation, this mutant cell line still exhibits Ras activation, which could be explained by the following possibilities. First, as there exists three Ras GEF families (RasGRF, RasGRP, and Sos family; reference 23), the full BCR-mediated Ras activation might require other GEF families, Sos and/or RasGRF, in addition to RasGRP. Based on our data using Sos1/Sos2 double-deficient DT40 B cells, dominant involvement of the Sos family would be unlikely. Nevertheless, considering that a gene knockout approach sometimes causes functional up-regulation of other molecules in an attempt to cope with the deficit, it is still possible that the Sos family plays a role, albeit small, in BCR-mediated Ras activation. This possibility might explain the fact that Ras activation takes place, at least partly, independent of PLC-γ2 (Fig. 1 A), as Sos activation does not require PLC-γ. Second, because ablation of both RasGRP1 and RasGRP3 results in a slightly more severe defect in Ras activation than in single RasGRP3 deletion (Fig. 3 A), another RasGRP member (RasGRP2 or RasGRP4) could be a candidate for mediating the residual BCR-mediated Ras activation. Third, assuming that BCR activates Ras through operating two mechanisms concomitantly, by activating Ras GEFs and by inhibiting Ras GAPs, the residual Ras activation in RasGRP1/RasGRP3 double-deficient cells could be due to ongoing inhibition of Ras GAPs during the BCR signal (8). Additional studies are underway to define which of the preceding possibilities is the most likely in DT40 B cells.
Because both Grb2–Sos and PLC-γ2–RasGRP pathways coexist in B cells, a question arises about how their selective engagement, depending on receptor types such as BCR or EGFR, takes place for Ras activation. Both receptors use Grb2 and PLC-γ2 in their signal transduction, because BCR-mediated Rac1 activation or EGFR-mediated calcium response is inhibited in Grb2- or PLC-γ2–deficient DT40 B cells, respectively (reference 61; unpublished data). Thus, why would Grb2 or PLC-γ2 be used more preferentially for EGFR- or BCR-mediated Ras activation, respectively? As the NH2-terminal Grb2 SH3 domain is able to bind to proline-rich residues in the B cell linker (BLNK) as well as Sos (62), this Grb2-dependent association may be skewing more toward the BLNK when BCRs are stimulated. Indeed, this type of induced interaction between a proline-rich sequence in CD3ɛ and an SH3 domain of adaptor molecule Nck has been reported in TCR-stimulated T cells (63). Similarly, the association between Grb2 and the BLNK might be induced or stabilized upon BCR engagement, possibly sequestering Sos from Grb2. Hence, the selection of binding partners for Grb2, depending on receptor types, may underlie the basis for differential outcomes through Grb2.
In contrast to BCR, EGFR did not essentially require PLC-γ2 for Ras activation in DT40 B cells (unpublished data). This result is consistent with previous evidence that EGFR-mediated Erk activation occurs normally in PLC-γ1−/− fibroblasts (64). These observations might simply suggest that DAG, a product of PLC-γ2 action, is necessary, but not sufficient, for activation of RasGRP3. If so, BCR, but not EGFR, could provide the second signal, in addition to generating DAG, both of which are required for activation of RasGRP3. Alternatively, given the relatively small activation of PLC-γ2 upon EGFR engagement, compared with the BCR (unpublished data), the differential requirement for PLC-γ2 might reflect the quantitative differences; low levels of PLC-γ2 activity are not sufficient for activation of RasGRP3.
Two lines of our evidence shown here support the proposed model that PLC-γ, after being activated upon antigen receptor engagement, generates DAG in the plasma membrane, thereby facilitating membrane recruitment of RasGRPs and its subsequent interaction with Ras (27). First, the membrane recruitment of RasGRP3 was severely affected by loss of PLC-γ2, whereas absence of RasGRP3 did not affect PLC-γ2 activation. Moreover, this Ras GRP3 recruitment required its C1 domain. Second, the necessity of membrane localization of RasGRP3 for its activation was demonstrated by experiments using its membrane-attached construct (Fig. 7 C, ΔC1-pbRasGRP3). In line with this model, the defective membrane recruitment of RasGRP1 in Vav3-deficient DT40 cells is likely accounted for by the insufficient PLC-γ2 activation in this mutant line (65). Recent results indicate that RasGRP1 may be involved not only in the stimulation of Ras at the plasma membrane but also at the Golgi apparatus in COS cells and Jurkat cells (66). Indeed, consistent with a previous report (67), in addition to the plasma membrane localization of RasGRP3 in DT40 cells, we also observed that RasGRP3 could localize at the perinuclear region, presumably Golgi in some DT40 cells after 10 min of BCR stimulation (Kashiwagi, K., and N. Saito, personal communication). Hence, it is possible that translocation of RasGRP3 to the perinuclear region might participate in the sustained Ras activation.
Constitutive localization of ΔC1-pbRasGRP3 enhanced activation status of Ras before BCR stimulation, but the Ras status was further activated upon BCR ligation, simply suggesting that additional BCR-mediated event might be required for optimal Ras activation. In this regard, consistent with the previous data (68), we also detected the BCR-mediated electrophoretic mobility shift of RasGRP3 in DT40 and primary B cells (unpublished data), presumably reflecting phosphorylation status. Therefore, this phosphorylation-dependent modification could be a potential additional mechanism.
Although initiation of Ras activation in BCR signaling requires membrane localization of RasGRP3, the subsequent inactivation of Ras at 10 min after BCR stimulation (Fig. 1 A) appears not to be simply explained by dissociation of RasGRP3 from the membrane. Indeed, our biochemical and microscopic analyses indicated that RasGRP3 was still located in the membrane after 10 min of BCR stimulation. Hence, inactivation of RasGRP3, independently of membrane localization, might take place at this time point, or RasGAPs might be activated, thereby leading to a decrease in active Ras.
Despite the common ability of RasGRP and Sos to activate Ras, the pathways to which these GEFs are coupled seem not to be simply overlapping or redundant. Given that various receptors such as c-kit and cytokine receptors stimulate Ras and subsequent Erk, these pathways could provide the B cells with the ability to discriminate between these stimuli and possibly modulate the subsequent biological responses.
Acknowledgments
We would like to thank our colleagues M. Kurosaki for expert technical assistance, and Y. Mori and M. Nishida for advice on the microscopic analysis.
This work was supported by grants to T.K. from the Ministry of Education, Science, Sports, and Culture in Japan and from the Uehara Memorial Foundation.
The present address of A. Hashimoto is Osaka Bioscience Institute, Osaka 565-0847, Japan.
Abbreviations used in this paper: BCR, B cell receptor; BLNK, B cell linker; DAG, diacylglycerol; EGFP, enhanced green fluorescent protein; EGFR, epidermal growth factor receptor; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; GFP, green fluorescent protein; GST, glutathione S-transferase; GTPase, guanosine triphosphatase; MAPK, mitogen-activated protein kinase; PLC, phospholipase C; RasGRP, Ras guanine nucleotide releasing protein; SH, src homology; Sos, Son of sevenless.
References
- 1.Downward, J., J.D. Graves, P.H. Warne, S. Rayter, and D.A. Cantrell. 1990. Stimulation of p21ras upon T-cell activation. Nature. 346:719–723. [DOI] [PubMed] [Google Scholar]
- 2.Graves, J.D., J. Downward, M. Izquierdo-Pastor, S. Rayter, P.H. Warne, and D.A. Cantrell. 1992. The growth factor IL-2 activates p21ras proteins in normal human T lymphocytes. J. Immunol. 148:2417–2422. [PubMed] [Google Scholar]
- 3.Harwood, A.E., and J.C. Cambier. 1993. B cell antigen receptor cross-linking triggers rapid protein kinase C independent activation of p21ras1. J. Immunol. 151:4513–4522. [PubMed] [Google Scholar]
- 4.Izquierdo, M., J. Downward, J.D. Graves, and D.A. Cantrell. 1992. Role of protein kinase C in T-cell antigen receptor regulation of p21ras: evidence that two p21ras regulatory pathways coexist in T cells. Mol. Cell. Biol. 12:3305–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazarus, A.H., K. Kawauchi, M.J. Rapoport, and T.L. Delovitch. 1993. Antigen-induced B lymphocyte activation involves the p21ras and ras.GAP signaling pathway. J. Exp. Med. 178:1765–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxton, T.M., I. van Oostveen, D. Bowtell, R. Aebersold, and M.R. Gold. 1994. B cell antigen receptor cross-linking induces phosphorylation of the p21ras oncoprotein activators SHC and mSOS1 as well as assembly of complexes containing SHC, GRB-2, mSOS1, and a 145-kDa tyrosine-phosphorylated protein. J. Immunol. 153:623–636. [PubMed] [Google Scholar]
- 7.Smit, L., G. van der Horst, and J. Borst. 1996. Sos, Vav, and C3G participate in B cell receptor-induced signaling pathways and differentially associate with Shc-Grb2, Crk, and Crk-L adaptors. J. Biol. Chem. 271:8564–8569. [DOI] [PubMed] [Google Scholar]
- 8.Genot, E., and D.A. Cantrell. 2000. Ras regulation and function in lymphocytes. Curr. Opin. Immunol. 12:289–294. [DOI] [PubMed] [Google Scholar]
- 9.Shaw, A.C., W. Swat, R. Ferrini, L. Davidson, and F.W. Alt. 1999. Activated Ras signals developmental progression of recombinase-activating gene (RAG)-deficient pro–B lymphocytes. J. Exp. Med. 189:123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw, A.C., W. Swat, L. Davidson, and F.W. Alt. 1999. Induction of Ig light chain gene rearrangement in heavy chain-deficient B cells by activated Ras. Proc. Natl. Acad. Sci. USA. 96:2239–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iritani, B.M., K.A. Forbush, M.A. Farrar, and R.M. Perlmutter. 1997. Control of B cell development by Ras-mediated activation of Raf. EMBO J. 16:7019–7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagaoka, H., Y. Takahashi, R. Hayashi, T. Nakamura, K. Ishii, J. Matsuda, A. Ogura, Y. Shirakata, H. Karasuyama, T. Sudo, et al. 2000. Ras mediates effector pathways responsible for pre–B cell survival, which is essential for the developmental progression to the late pre–B cell stage. J. Exp. Med. 192:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurosaki, T. 2002. Regulation of B cell fates by BCR signaling components. Curr. Opin. Immunol. 14:341–347. [DOI] [PubMed] [Google Scholar]
- 14.Niiro, H., and E.A. Clark. 2002. Regulation of B-cell fate by antigen-receptor signals. Nat. Rev. Immunol. 2:945–956. [DOI] [PubMed] [Google Scholar]
- 15.Reth, M. 2001. Oligomeric antigen receptors: a new view on signaling for the selection of lymphocytes. Trends Immunol. 22:356–360. [DOI] [PubMed] [Google Scholar]
- 16.Rajewsky, K. 1996. Clonal selection and learning in the antibody system. Nature. 381:751–758. [DOI] [PubMed] [Google Scholar]
- 17.Meffre, E., R. Casellas, and M.C. Nussenzweig. 2000. Antibody regulation of B cell development. Nat. Immunol. 1:379–385. [DOI] [PubMed] [Google Scholar]
- 18.Bar-Sagi, D., and A. Hall. 2000. Ras and Rho GTPases: a family reunion. Cell. 103:227–238. [DOI] [PubMed] [Google Scholar]
- 19.Campbell, S.L., R. Khosravi-Far, K.L. Rossman, G.J. Clark, and C.J. Der. 1998. Increasing complexity of Ras signaling. Oncogene. 17:1395–1413. [DOI] [PubMed] [Google Scholar]
- 20.Reuther, G.W., and C.J. Der. 2000. The Ras branch of small GTPases: Ras family members don't fall far from the tree. Curr. Opin. Cell Biol. 12:157–165. [DOI] [PubMed] [Google Scholar]
- 21.Tamir, I., J.C. Stolpa, C.D. Helgason, K. Nakamura, P. Bruhns, M. Daeron, and J.C. Cambier. 2000. The RasGAP-binding protein p62dok is a mediator of inhibitory FcγRIIB signals in B cells. Immunity. 12:347–358. [DOI] [PubMed] [Google Scholar]
- 22.Yamanashi, Y., T. Tamura, T. Kanamori, H. Yamane, H. Nariuchi, T. Yamamoto, and D. Baltimore. 2000. Role of the rasGAP-associated docking protein p62dok in negative regulation of B cell receptor-mediated signaling. Genes Dev. 14:11–16. [PMC free article] [PubMed] [Google Scholar]
- 23.Quilliam, L.A., J.F. Rebhun, and A.F. Castro. 2002. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog. Nucleic Acid Res. Mol. Biol. 71:391–444. [DOI] [PubMed] [Google Scholar]
- 24.Cullen, P.J., and P.J. Lockyer. 2002. Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell Biol. 3:339–348. [DOI] [PubMed] [Google Scholar]
- 25.Ehrhardt, A., G.R. Ehrhardt, X. Guo, and J.W. Schrader. 2002. Ras and relatives--job sharing and networking keep an old family together. Exp. Hematol. 30:1089–1106. [DOI] [PubMed] [Google Scholar]
- 26.Dower, N.A., S.L. Stang, D.A. Bottorff, J.O. Ebinu, P. Dickie, H.L. Ostergaard, and J.C. Stone. 2000. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat. Immunol. 1:317–321. [DOI] [PubMed] [Google Scholar]
- 27.Ebinu, J.O., S.L. Stang, C. Teixeira, D.A. Bottorff, J. Hooton, P.M. Blumberg, M. Barry, R.C. Bleakley, H.L. Ostergaard, and J.C. Stone. 2000. RasGRP links T-cell receptor signaling to Ras. Blood. 95:3199–3203. [PubMed] [Google Scholar]
- 28.Roose, J., and A. Weiss. 2000. T cells: getting a GRP on Ras. Nat. Immunol. 1:275–276. [DOI] [PubMed] [Google Scholar]
- 29.Takata, M., Y. Homma, and T. Kurosaki. 1995. Requirement of phospholipase C-γ2 activation in surface immunoglobulin M-induced B cell apoptosis. J. Exp. Med. 182:907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen, C.L., J.E. Lehmeyer, and M.D. Cooper. 1982. Evidence for an IgD homologue on chicken lymphocytes. J. Immunol. 129:2580–2585. [PubMed] [Google Scholar]
- 31.Hashimoto, A., M. Kurosaki, N. Gotoh, M. Shibuya, and T. Kurosaki. 1999. Shc regulates epidermal growth factor-induced activation of the JNK signaling pathway. J. Biol. Chem. 274:20139–20143. [DOI] [PubMed] [Google Scholar]
- 32.Kost, T.A., N. Theodorakis, and S.H. Hughes. 1983. The nucleotide sequence of the chick cytoplasmic β-actin gene. Nucleic Acids Res. 11:8287–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inabe, K., M. Ishiai, A.M. Scharenberg, N. Freshney, J. Downward, and T. Kurosaki. 2002. Vav3 modulates B cell receptor responses by regulating phosphoinositide 3-kinase activation. J. Exp. Med. 195:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishiai, M., M. Kurosaki, R. Pappu, K. Okawa, I. Ronko, C. Fu, M. Shibata, A. Iwamatsu, A.C. Chan, and T. Kurosaki. 1999. BLNK required for coupling Syk to PLCγ2 and Rac1-JNK in B cells. Immunity. 10:117–125. [DOI] [PubMed] [Google Scholar]
- 35.Taylor, S.J., and D. Shalloway. 1996. Cell cycle-dependent activation of Ras. Curr. Biol. 6:1621–1627. [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto, A., K. Takeda, M. Inaba, M. Sekimata, T. Kaisho, S. Ikehara, Y. Homma, S. Akira, and T. Kurosaki. 2000. Cutting edge: essential role of phospholipase C-γ2 in B cell development and function. J. Immunol. 165:1738–1742. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto, A., H. Okada, A. Jiang, M. Kurosaki, S. Greenberg, E.A. Clark, and T. Kurosaki. 1998. Involvement of guanosine triphosphatases and phospholipase C-γ2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J. Exp. Med. 188:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurosaki, T., A. Maeda, M. Ishiai, A. Hashimoto, K. Inabe, and M. Takata. 2000. Regulation of the phospholipase C-γ2 pathway in B cells. Immunol. Rev. 176:19–29. [DOI] [PubMed] [Google Scholar]
- 39.DeFranco, A.L. 1997. The complexity of signaling pathways activated by the BCR. Curr. Opin. Immunol. 9:296–308. [DOI] [PubMed] [Google Scholar]
- 40.Reth, M., and J. Wienands. 1997. Initiation and processing of signals from the B cell antigen receptor. Annu. Rev. Immunol. 15:453–479. [DOI] [PubMed] [Google Scholar]
- 41.Pleiman, C.M., D. D'Ambrosio, and J.C. Cambier. 1994. The B-cell antigen receptor complex: structure and signal transduction. Immunol. Today. 15:393–399. [DOI] [PubMed] [Google Scholar]
- 42.Clyde-Smith, J., G. Silins, M. Gartside, S. Grimmond, M. Etheridge, A. Apolloni, N. Hayward, and J.F. Hancock. 2000. Characterization of RasGRP2, a plasma membrane-targeted, dual specificity Ras/Rap exchange factor. J. Biol. Chem. 275:32260–32267. [DOI] [PubMed] [Google Scholar]
- 43.Ebinu, J.O., D.A. Bottorff, E.Y. Chan, S.L. Stang, R.J. Dunn, and J.C. Stone. 1998. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science. 280:1082–1086. [DOI] [PubMed] [Google Scholar]
- 44.Kawasaki, H., G.M. Springett, S. Toki, J.J. Canales, P. Harlan, J.P. Blumenstiel, E.J. Chen, I.A. Bany, N. Mochizuki, A. Ashbacher, et al. 1998. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. USA. 95:13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reuther, G.W., Q.T. Lambert, J.F. Rebhun, M.A. Caligiuri, L.A. Quilliam, and C.J. Der. 2002. RasGRP4 is a novel Ras activator isolated from acute myeloid leukemia. J. Biol. Chem. 277:30508–30514. [DOI] [PubMed] [Google Scholar]
- 46.Yamashita, S., N. Mochizuki, Y. Ohba, M. Tobiume, Y. Okada, H. Sawa, K. Nagashima, and M. Matsuda. 2000. CalDAG-GEFIII activation of Ras, R-ras, and Rap1. J. Biol. Chem. 275:25488–25493. [DOI] [PubMed] [Google Scholar]
- 47.Yang, Y., L. Li, G.W. Wong, S.A. Krilis, M.S. Madhusudhan, A. Sali, and R.L. Stevens. 2002. RasGRP4, a new mast cell-restricted Ras guanine nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Identification of defective variants of this signaling protein in asthma, mastocytosis, and mast cell leukemia patients and demonstration of the importance of RasGRP4 in mast cell development and function. J. Biol. Chem. 277:25756–25774. [DOI] [PubMed] [Google Scholar]
- 48.Tognon, C.E., H.E. Kirk, L.A. Passmore, I.P. Whitehead, C.J. Der, and R.J. Kay. 1998. Regulation of RasGRP via a phorbol ester-responsive C1 domain. Mol. Cell. Biol. 18:6995–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prior, I.A., A. Harding, J. Yan, J. Sluimer, R.G. Parton, and J.F. Hancock. 2001. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 3:368–375. [DOI] [PubMed] [Google Scholar]
- 50.Hancock, J.F., A.I. Magee, J.E. Childs, and C.J. Marshall. 1989. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 57:1167–1177. [DOI] [PubMed] [Google Scholar]
- 51.Hancock, J.F., H. Paterson, and C.J. Marshall. 1990. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 63:133–139. [DOI] [PubMed] [Google Scholar]
- 52.Niv, H., O. Gutman, Y. Kloog, and Y.I. Henis. 2002. Activated K-Ras and H-Ras display different interactions with saturable nonraft sites at the surface of live cells. J. Cell Biol. 157:865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hancock, J.F. 2003. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 4:373–384. [DOI] [PubMed] [Google Scholar]
- 54.Schlessinger, J. 1993. How receptor tyrosine kinases activate Ras. Trends Biochem. Sci. 18:273–275. [DOI] [PubMed] [Google Scholar]
- 55.Buday, L., and J. Downward. 1993. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell. 73:611–620. [DOI] [PubMed] [Google Scholar]
- 56.Egan, S.E., B.W. Giddings, M.W. Brooks, L. Buday, A.M. Sizeland, and R.A. Weinberg. 1993. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 363:45–51. [DOI] [PubMed] [Google Scholar]
- 57.Goudreau, N., F. Cornille, M. Duchesne, F. Parker, B. Tocque, C. Garbay, and B.P. Roques. 1994. NMR structure of the N-terminal SH3 domain of GRB2 and its complex with a proline-rich peptide from Sos. Nat. Struct. Biol. 1:898–907. [DOI] [PubMed] [Google Scholar]
- 58.Li, N., A. Batzer, R. Daly, V. Yajnik, E. Skolnik, P. Chardin, D. Bar-Sagi, B. Margolis, and J. Schlessinger. 1993. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature. 363:85–88. [DOI] [PubMed] [Google Scholar]
- 59.Wang, D.Z., V.E. Hammond, H.E. Abud, I. Bertoncello, J.W. McAvoy, and D.D. Bowtell. 1997. Mutation in Sos1 dominantly enhances a weak allele of the EGFR, demonstrating a requirement for Sos1 in EGFR signaling and development. Genes Dev. 11:309–320. [DOI] [PubMed] [Google Scholar]
- 60.Qian, X., L. Esteban, W.C. Vass, C. Upadhyaya, A.G. Papageorge, K. Yienger, J.M. Ward, D.R. Lowy, and E. Santos. 2000. The Sos1 and Sos2 Ras-specific exchange factors: differences in placental expression and signaling properties. EMBO J. 19:642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johmura, S., M. Oh-hora, K. Inabe, Y. Nishikawa, K. Hayashi, E. Vigorito, D. Kitamura, M. Turner, K. Shingu, M. Hikida, and T. Kurosaki. 2003. Regulation of Vav Localization in membrane rafts by adaptor molecules Grb2 and BLNK. Immunity. 18:777–787. [DOI] [PubMed] [Google Scholar]
- 62.Wienands, J., J. Schweikert, B. Wollscheid, H. Jumaa, P.J. Nielsen, and M. Reth. 1998. SLP-65: a new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J. Exp. Med. 188:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gil, D., W.W. Schamel, M. Montoya, F. Sanchez-Madrid, and B. Alarcon. 2002. Recruitment of Nck by CD3ɛ reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 109:901–912. [DOI] [PubMed] [Google Scholar]
- 64.Ji, Q.S., S. Ermini, J. Baulida, F.L. Sun, and G. Carpenter. 1998. Epidermal growth factor signaling and mitogenesis in Plcg1 null mouse embryonic fibroblasts. Mol. Biol. Cell. 9:749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caloca, M.J., J.L. Zugaza, D. Matallanas, P. Crespo, and X.R. Bustelo. 2003. Vav mediates Ras stimulation by direct activation of the GDP/GTP exchange factor Ras GRP1. EMBO J. 22:3326–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bivona, T.G., I. Perez De Castro, I.M. Ahearn, T.M. Grana, V.K. Chiu, P.J. Lockyer, P.J. Cullen, A. Pellicer, A.D. Cox, and M.R. Philips. 2003. Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nature. 424:694–698. [DOI] [PubMed] [Google Scholar]
- 67.Caloca, M.J., J.L. Zugaza, and X.R. Bustelo. 2003. Exchange factors of the RasGRP family mediate Ras activation in the Golgi. J. Biol. Chem. 278:33465–33473. [DOI] [PubMed] [Google Scholar]
- 68.Teixeira, C., S.L. Stang, Y. Zheng, N.S. Beswick, and J.C. Stone. 2003. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood. 102:1414–1420. [DOI] [PubMed] [Google Scholar]