

Abstract
TREM-2 is an immunoglobulin-like cell surface receptor associated with DAP12/KARAP that activates monocyte-derived dendritic cells (DCs) in vitro. Recently, it has been shown that genetic defects of human DAP12/KARAP and TREM-2 result in a rare syndrome characterized by bone cysts and presenile dementia called Nasu-Hakola disease. This observation suggests that TREM-2 may function in myeloid cells other than DCs, most probably osteoclasts (OCs) and microglial cells, which are involved in bone modeling and brain function. Consistent with this prediction, here we show that OC differentiation is dramatically arrested in TREM-2–deficient patients, resulting in large aggregates of immature OCs that exhibit impaired bone resorptive activity. These results demonstrate a critical role for TREM-2 in the differentiation of mononuclear myeloid precursors into functional multinucleated OCs.
Keywords: osteoclast, bone resorption, differentiation, dendritic cell, TREM
Introduction
Triggering receptors expressed on myeloid cells (TREM) are members of the immunoglobulin superfamily that promote myeloid cell activation by associating with the transmembrane adaptor protein DAP12/KARAP (1–4). DAP12 contains an immunoreceptor tyrosine-based activation motif that functions as docking site for the protein tyrosine kinases Syk and Zap70 (5, 6). These kinases mediate a cascade of phosphorylation events that ultimately activate the cell. Within the TREM receptor family, TREM-2 is selectively expressed on monocyte-derived DCs and promotes DC migration and activation in vitro, suggesting that it may have a role in antigen presentation and T cell stimulation in vivo (2). Rare TREM-2– and DAP12-deficient individuals have been recently identified (7, 8). Remarkably, these individuals are affected by a genetic syndrome characterized by bone cysts and presenile dementia, called Nasu-Hakola disease or polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (9). This observation suggests that the TREM-2–DAP12 complex may regulate the function of an unexpectedly vast array of myeloid cells, including not only DCs, but also osteoclasts (OCs) and microglial cells, which are critical for bone modeling and brain function, respectively. In this study, we examined TREM-2 function in human OCs.
Materials and Methods
In Vitro Generation of OCs and DCs.
Monocytes were isolated from PBMCs of Nasu-Hakola patients and healthy controls using anti-CD14 microbeads (Miltenyi Biotec) and cultured in RPMI supplemented with 10% FCS (Hyclone), nonessential amino acids, Na pyruvate, Glutamax, kanamycin (GIBCO BRL), 50 ng/ml GM-CSF (Leukine; Immunex), and 1 ng/ml IL-4 (R&D Systems) or 10 ng/ml M-CSF (R&D Systems) for 48 h. To generate OCs, cells were seeded in 24-well plates (3 × 105/well) or in glass chamber slides (1.5 × 105/well; Lab-Tek; Nalgene) with GM-CSF and IL-4. After 48 h the culture medium was supplemented with 300 ng/ml RANK-L, 10 ng/ml M-CSF, and the anti–class II mAb 3.8B1 (1:10 dilution of culture supernatant). Under these conditions, mature OC formation was observed after 7–10 d of culture. Generation of monocyte-derived DCs was performed as previously described (10).
Immunoblot Analysis.
TREM-2–deficient or control DCs were lysed at day 6 of culture. Cell lysates were run in 10% Tris-Glycine gel (Novex) with MES running buffer (2 × 105 cells were loaded in each lane). Gels were blotted onto nitrocellulose membranes and incubated with anti–TREM-2 antibodies (clones 21E10 and 20G3) or rabbit anti–DAP12 antiserum as previously described (2).
Flow Cytometry.
Monocytes were stained with anti–TREM-1 (mouse IgG1, clone 9E2), CD16 (mouse IgM, Dako), and CD14 (mouse IgG2b, TIB228). Monocyte-derived DCs and OC precursors were stained with anti–TREM-2 (mouse IgG1, clone 21E10), CD14, and CD16. PE- and FITC-labeled (Fab′)2 isotype-specific goat anti–mouse antibodies (Southern Biotechnology Associates, Inc.) were used as second step antibodies. LPS-activated DCs were stained with PE-labeled CD1a, CD86, and FITC-labeled CD83 and CD16 (Immunotech).
OC Phenotype.
OCs were stained in 24-well plates for tartrate-resistant acid phosphatase (TRAP) activity (Acid Phosphatase Kit; Sigma-Aldrich). To examine F-actin distribution, cells were fixed for 10 min in 0.1% glutaraldehyde and 3.7% formaldehyde, permeabilized for 5 min in 0.2% Triton X-100, and stained with Phalloidin-Oregon Green (Molecular Probes). A MRC-600/Biorad confocal microscope was used for analysis. Expression of calcitonin receptor and vitronectin receptor was assessed by RT-PCR. For this assay, OC RNA was prepared by lysing cells in situ with TRIzol (GIBCO BRL) at the end of the culture. Synthesis of cDNA and PCR were performed by standard techniques. Primers for vitronectin receptor were: 5′-GTTGGGAGATTAGACAGAGG-3′ and 5′-CTAGTGGGTCAAGATGTAGC-3′. Primers for calcitonin receptors were: 5′-ACTGCTGGCTGAGTGTGGAAA-3′, 5′-GAAGCAGTAGATGGTCGAAAC-3′ (11). PCR products were run on a 1.5% agarose gel and stained with ethidium bromide.
Resorption Assays.
Purified peripheral blood monocytes from TREM-2–deficient patients and healthy donors were seeded on either BD Biocoat™ Osteologic™ Multitest Slides (BD Biosciences) or dentin slices (Alpco Diagnostics). Cells were seeded at 0.6–1.5 × 105/well and cultured with GM-CSF and IL-4. After 48 h, culture medium was supplemented with receptor activator of NF-κB ligand (RANKL), mAb 3.8B1, and M-CSF. After 10–12 d, dentin slices were treated with 2 N sodium hydroxide to remove cells, washed with distilled water, stained with hematoxylin, and examined by light microscopy. Cells were removed from Biocoat™ slides with 5% sodium hypochlorite. Slides were washed with distilled water, air-dried, examined by light microscopy, and reexamined after Von Kossa staining, as described by the manufacturer (BD Biosciences).
Online Supplemental Material.
TREM-2–deficient immature osteoclasts degrade thin calcium phosphate layers. Monocytes from TREM-2–deficient patients (a–c) or healthy individuals (d–f) were cultured on thin films of calcium phosphate coated onto quartz slides. At day 10, cultures were inspected by light microscopy (a, b, d, and e) before (a and d) and after (b and e) cell removal. In b and e, lacunae were directly visualized by light microscopy (×2); in c and f, lacunae were detected after Von Kossa staining of slides (×10). Resorption areas appear as clear spots with a contrasting brown/black background. The exaggerated ability of TREM-2–deficient immature osteoclasts to degrade thin calcium phosphate layers suggests active phagocytic activity and ability to acidify the medium. Fig. S1 is available at http://www.jem.org/cgi/content/full/jem.20022220/DC1.
Results
TREM-2 Is Expressed on OC Precursors.
OCs are polykaryons of the monocyte/macrophage lineage that specialize in bone resorption (12, 13). They derive from the fusion of mononuclear myeloid precursors (14, 15) in the presence of two key cytokines: M-CSF and a TNF-related protein, RANKL (also known as OPGL, TRANCE, or ODF; 12). Osteoclastogenesis is also promoted by inflammatory cytokines, such as TNF-α and IL-1, which activate the transcription factor NF-κB (12), and by fusion regulatory proteins, such as CD98 (16). To generate human OCs in vitro, we used peripheral blood monocytes as precursors and cultured them with GM-CSF and IL-4 for the first 48–72 h. Cell culture medium was then supplemented with M-CSF, RANKL, and the anti–HLA-DR mAb 3.8B1, which delivers a potent myeloid cell activation signal by stimulating protein tyrosine phosphorylation (10). In these culture conditions, monocytes clustered in large aggregates and fused to form multinucleated cells with typical OC features. In control cultures, monocytes were maintained in GM-CSF and IL-4 to generate monocyte-derived DCs. Both culture conditions induced expression of TREM-2 on monocytic precursors, as did M-CSF, GM-CSF, and IL-4 alone (Fig. 1) . Within 12 h after the addition of mAb 3.8B1 and RANKL, differentiating progenitors reduced cell surface expression of TREM-2 to low but detectable levels (not depicted).
Figure 1.

Monocytic precursors from normal donors express TREM-2 at early times of culture with M-CSF or GM-CSF and IL-4. TREM-2 is expressed within 48–72 h of culture with M-CSF or GM-CSF and IL-4, reaching a maximum level at days 3–4. It is worth noting that IL-4 alone is sufficient to induce TREM-2 expression, consistent with a possible role of Th2 cytokines in regulation of TREM-2.
Incomplete Differentiation of TREM-2–deficient Monocytes into OCs.
To define the role of TREM-2 in OC function, we examined OCs from two TREM-2–deficient patients. Patients A and B have been previously shown to carry a homozygous mutation of the splice donor consensus site of intron 3 of the TREM-2 gene (8). This mutation results in the skipping of exon 3 (and perhaps additional exons) from the TREM-2 mature mRNA, most likely impairing cell surface expression of TREM-2. Indeed, OC precursors and monocyte-derived DCs from patients A and B expressed very little TREM-2 on the cell surface in comparison to those cultured from normal donors (Fig. 2 , a and b). Complete lack of expression was also observed in a patient carrying a mutation of DAP12 that has been recently characterized (Fig. 2 b; reference 17). To confirm that patients A and B have a selective defect of TREM-2, we analyzed another DAP12-associated receptor, TREM-1, which is expressed on peripheral blood monocytes and granulocytes. As shown in Fig. 2 c, TREM-1 was expressed on the monocytes of patient A and B, but not on DAP12-deficient monocytes. Immunoblot analysis confirmed that lack of TREM-2 in patients A and B is associated with normal DAP12 expression (Fig. 2 d).
Figure 2.
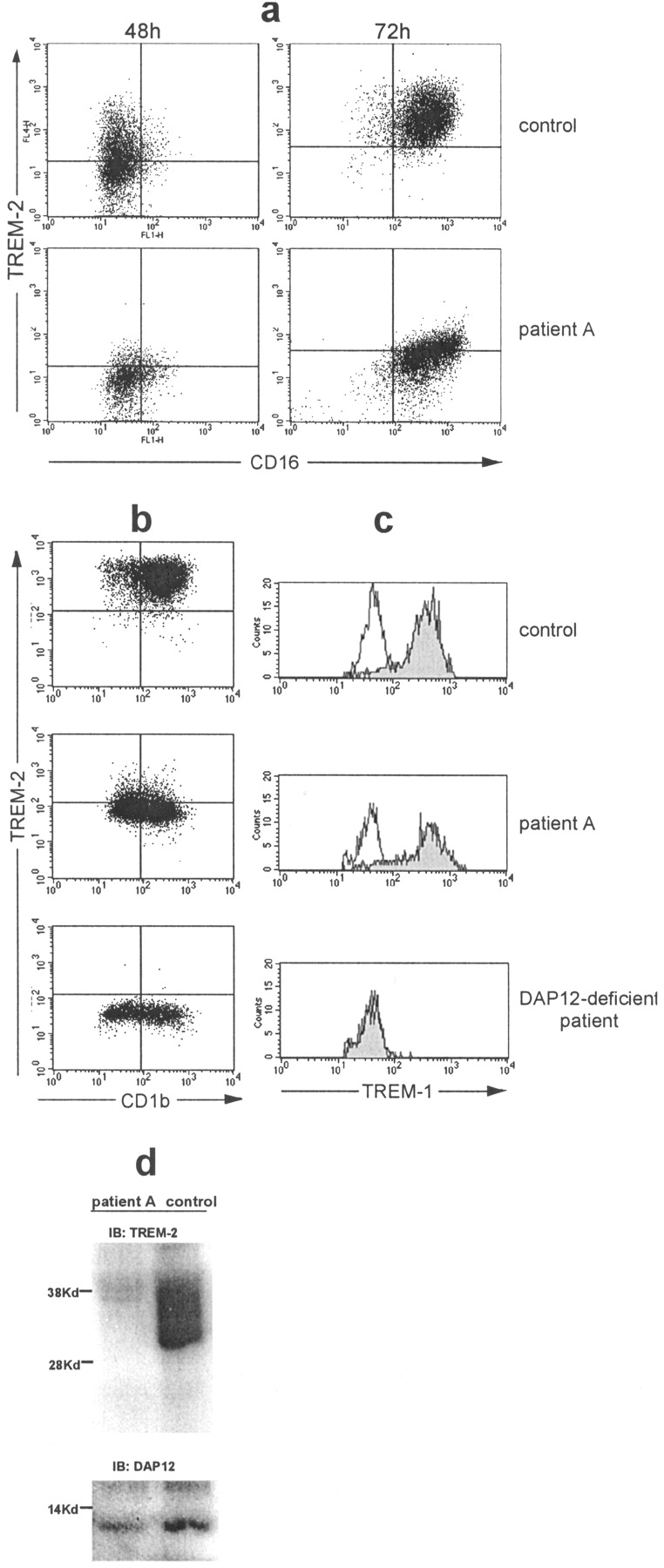
Patient A exhibits a selective lack of TREM-2 expression in monocytes cultured with M-CSF or GM-CSF and IL-4. (a) TREM-2 and CD16 expression in monocytes cultured for 48 or 72 h in the presence of M-CSF from normal or TREM-2–deficient patients. (b) TREM-2 and CD1b expression on control, TREM-2–deficient, and DAP12-deficient monocytes cultured for 4 d in GM-CSF and IL-4. (c) TREM-1 expression on freshly isolated monocytes from normal, TREM-2–deficient, and DAP12-deficient individuals. (d) Expression of TREM-2 and DAP12 proteins in normal and TREM-2–deficient monocyte-derived DCs at day 6 of culture assessed by immunoblot. Similar results were obtained with monocytes derived from patient B (not depicted).
OCs were generated in vitro from peripheral blood monocytes of patients and normal donors as described above. Within 7–10 d, monocytes from normal donors differentiated into multinucleated OCs that spread out and tightly attached to culture dishes or slides and expressed the intracellular TRAP (Fig. 3 , a and c). In striking contrast, TREM-2–deficient monocytes formed large aggregates of mononuclear cells that although intensely positive for TRAP, did not fuse to generate polykaryons, were unable to spread, and remained loosely adherent to tissue culture slides (Fig. 3, b and d). To quantify the defect of OC development, we counted cells with three or more nuclei generated after seeding 60,000 monocytic precursors in each of three wells of a 96-well plate. Normal monocytes yielded 180 ± 20 multinucleated cells/well, most of which displayed eight or more nuclei. TREM-2–deficient monocytes generated 4 ± 1 cells/well with no more than three nuclei. Thus, OC differentiation is obviously arrested in TREM-2–deficient patients. This implies that TREM-2 expression is required during osteoclastogenesis to generate a signal that promotes activation and fusion of precursors into mature OCs.
Figure 3.

Arrest of differentiation, defect of actin polymerization, reduced levels of vitronectin receptor (VNR), and lack expression of calcitonin receptor (CTR) in OCs from TREM-2–deficient patients. (a–d) Images of OC cultures derived from normal (a and c) and TREM-2–deficient individuals (b and d). Images were taken on a Nikon phase contrast microscope with a 10DL objective. In c and d, cells were stained for intracellular TRAP. Upon stimulation with M-CSF, RANKL, and mAb 3.8B1, TREM-2–deficient monocytic precursors do not fuse, but form large aggregates of cells intensely positive for the TRAP reaction. Multinucleated cells with up to three nuclei were rarely detected in the OC cultures from TREM-2–deficient patients (red arrow). (e–h) Actin cytoskeleton of OCs from normal (e and g) and TREM-2–deficient individuals (f and h) generated in glass chamber slides, stained on day 10 of culture with Phalloidin-Oregon Green and analyzed by confocal microscopy (×20). (i) Content of F actin/cell was determined in control and TREM-2–deficient cells by flow cytometry 48 h after stimulation of monocytes with M-CSF, RANKL, and the 3.8B1 mAb. At this early time point immature OCs could still be recovered from culture wells. (j) Three-fold dilutions of cDNAs obtained from control OC cultures, control monocytes, or TREM-2–deficient OC cultures were amplified by RT-PCR with oligonucleotide pairs specific for vitronectin receptor (VNR), calcitonin receptor (CTR), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). PCR products were separated by agarose gel electrophoresis and visualized with ethidium bromide. Molecular weight markers are indicated.
To further document the defect in osteoclastogenesis in TREM-2–deficient patients, we analyzed additional OC features. To resorb bone, OCs adhere tightly to the bone matrix, establishing a sealing zone (12, 13). This process involves rearrangement of the actin cytoskeleton to form an “actin ring.” The membrane enclosed by the actin ring forms a ruffled border, which is specialized for secretion of protons and lysosomal proteases that mediate bone resorption (13). Accordingly, OCs from normal donors exhibited a well-developed actin cytoskeleton with actin rings and podosomes (Fig. 3, e and g). In contrast, the actin cytoskeleton was barely detectable under the cell membrane in TREM-2–deficient immature OCs (Fig. 3, f and h). To quantify these differences in actin polymerization, OC precursors were stained with phalloidin, which binds to polymerized actin (F actin), and analyzed by flow cytometry. As shown in Fig. 3 i, the amount of F actin was significantly higher in normal than TREM-2–deficient OCs. This lack of adequate actin polymerization and cytoskeleton organization is consistent with the inability of TREM-2–deficient OCs to spread and adhere normally in tissue culture and suggests that their capacity to adhere tightly to bone matrix might be impaired in vivo. This hypothesis was further corroborated by analysis of vitronectin receptor expression, which has been implicated in OC adhesion and migration as well as bone resorption (18–20). As shown in Fig. 3 j, expression of the vitronectin receptor transcript was clearly reduced in TREM-2–deficient OCs as compared with normal OCs.
The receptor for calcitonin is another typical OC marker, as it distinguishes OCs from multinucleated giant cell macrophages (21). Therefore, we analyzed calcitonin receptor transcripts by RT-PCR and detected no expression in TREM-2–deficient OC, whereas this receptor was expressed in normal mature OCs (Fig. 3 j). Together with the lack of actin polymerization and cell polarization, this observation confirms the defect in osteoclastogenesis in TREM-2–deficient patients.
Impaired Resorptive Function of TREM-2–deficient OCs.
To test the impact of defective osteoclastogenesis on resorptive function, OCs from normal donors and TREM-2–deficient patients were cultured on dentin slices and the lacunae resulting from resorption were analyzed by light microscopy (Fig. 4) . In parallel experiments, resorption was also analyzed on thin films of calcium phosphate coated onto quartz slides (see Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20022220/DC1). Normal OCs created limited and well-defined excavations in the dentin slices and calcium phosphate surface that corresponded to the polykaryons (Fig. 4 and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20022220/DC1). In striking contrast, TREM-2–deficient immature OCs were incapable of excavating resorptive pits on dentin slices (Fig. 4). Only a few pits were detected that corresponded in number to the few multinucleated cells generated (Fig. 4). Unexpectedly, TREM-2–deficient immature OCs excavated numerous and large resorptive areas on calcium phosphate film that appeared to correspond to the large cellular aggregates formed in culture by immature OCs (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20022220/DC1). Thus, immature OCs derived from TREM-2–deficient individuals resorb bone matrix very inefficiently. However, they actively degrade thin calcium phosphate layers, possibly through phagocytic and acidification mechanisms.
Figure 4.

Impaired resorptive function of TREM-2–deficient immature OCs. Resorptive function of normal and TREM-2–deficient OCs on dentin slices (a–f). Monocytic precursors from healthy individuals (a and b) and TREM-2–deficient patients (c–f) were cultured on dentin slices. At day 10, dentin slices were stained with hematoxylin and inspected by light microscopy before (a, c, and e) and after (b, d, and f) cell removal. Resorption pits appear as dark spots stained with hematoxylin after cell removal. Normal monocytes differentiated into multinucleated OCs (a) that generated resorption pits (b). In general, TREM-2–deficient mononuclear immature OCs (c) did not excavate resorptive pits (d). Only a few pits were detected (f) that corresponded in number to the few multinucleated cells generated (e).
TREM-2–deficient Monocytes Reluctantly Differentiate into DCs.
In parallel with our examination of OC differentiation, we analyzed differentiation of TREM-2–deficient monocytes into DCs (22). Monocytes from normal donors maintained in GM-CSF/IL-4 for 4–6 d differentiated into DCs expressing high levels of CD1a. Remarkably, TREM-2–deficient monocytes differentiated into two distinct cell subsets: a small subset expressing the DC marker CD1a and a larger population expressing the macrophage marker CD16 but lacking CD1a (Fig. 5) . Only after 7 or more days of culture in GM-CSF and IL-4 did TREM-2–deficient monocytes acquire the CD1a+ CD16− phenotype typical of monocyte-derived DCs (not depicted). Upon stimulation with LPS, TREM-2–deficient DCs did up-regulate the costimulatory molecules CD86 and CD83, showing normal responsiveness to activation stimuli (Fig. 5). Thus, TREM-2–deficient monocytes favor a macrophage differentiation pathway. Only a prolonged treatment with GM-CSF and IL-4 overcomes this bias, eventually driving differentiation toward a DC pathway.
Figure 5.

A subset of TREM-2–deficient DCs expresses the macro-phage marker CD16. Immature DCs from normal (top panels) and TREM-2–deficient individuals (bottom panels) were generated by culturing peripheral blood monocytes with GM-CSF and IL-4. At day 4, DCs were analyzed for expression of CD1a, CD16, CD83, and CD86. A population of CD1a− CD16+ macrophage-like cells was detected in the cultures of TREM-2–deficient patient A but not in control cultures. Identical results were obtained from DC cultures of patient B (not depicted). DC activation was monitored 24 h after stimulation with LPS. CD86 and CD83 were up-regulated on LPS-activated DCs in both TREM-2–deficient and control DC.
Discussion
Our study shows that TREM-2–deficient individuals have a striking defect in OC development, which results in impaired bone resorption in vitro. TREM-2–deficient OC precursors fail to fuse into multinucleated cells, do not develop extensive rearrangements of actin cytoskeleton, and show reduced or no expression of typical OC markers such as vitronectin and calcitonin receptors. TREM-2–deficient immature OCs do express TRAP; however, this molecule is also expressed in other bone marrow–derived cells. This block in OC development leads to impaired performance in dentin resorption assays, potentially reflecting inefficient bone resorption in vivo. It will be important to determine how this defect contributes to the development of cysts and spontaneous fractures in bones of Nasu-Hakola disease patients (9). Consistent with our results, defective development and resorptive function of OCs have been recently observed in DAP12-deficient mice (23). Thus, TREM-2/DAP12 is an essential element in a unique pathway of bone remodeling and repair, which cooperates with previously defined OC differentiation pathways (12).
More generally, TREM-2 appears to play a major role in regulating myeloid lineage development. Our data are consistent with the hypothesis that TREM-2, by interacting with a yet unknown ligand on myeloid precursors or stromal cells, generates a positive signal that promotes precursor cell differentiation into OCs in the presence of M-CSF and RANKL or into DCs in the presence of GM-CSF and IL-4. In contrast, when TREM-2 is absent, myeloid cell differentiation is biased toward the macrophage lineage. Corroborating this, TREM-2–deficient monocytes are reluctant to differentiate into DCs and, if not driven by exogenous factors, favor the macrophage pathway. The exaggerated ability of TREM-2–deficient immature OCs to degrade thin calcium phosphate layers is also consistent with the retention of a phagocytic activity. This model suggests an inherent flexibility in the myeloid development program that is dependent on existing needs. Under homeostatic conditions, engagement of TREM-2 may favor OC differentiation, whereas inflammatory conditions may drive myeloid differentiation toward the macrophage lineage through engagement of different receptors. Accordingly, it has been recently shown that engagement of Toll-like receptors by bacterial products or CpG oligonucleotides directs OC differentiation toward a macrophage pathway (28, 29). TREM-2 may guide myeloid cell differentiation by modulating responsiveness to certain cytokines or reinforcing signaling pathways triggered by cytokine receptors or integrins (19, 24–27). The TREM-2/DAP12 signaling pathway may also play an important role in promoting actin polymerization, organization of the cytoskeleton, and ruffled border trafficking via recruitment of protein tyrosine kinases, activation of phospholipase Cγ1, and calcium mobilization (30). These functions are critical for promoting cell fusion as well as transforming OC precursors into a polarized cell type capable of resorbing bone in a highly regulated fashion (13).
Acknowledgments
We would like to thank Susan Gilfillan for reading the manuscript, Chris A. Nelson and Daved Fremont for recombinant RANKL, Michael G. Veith for scanning electron microscopy analysis, and Patricia and Philip A. Osdoby for helpful suggestions.
The online version of this article contains supplemental material.
References
- 1.Bouchon, A., F. Facchetti, M.A. Weigand, and M. Colonna. 2001. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 410:1103–1107. [DOI] [PubMed] [Google Scholar]
- 2.Bouchon, A., C. Hernandez-Munain, M. Cella, and M. Colonna. 2001. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 194:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daws, M.R., L.L. Lanier, W.E. Seaman, and J.C. Ryan. 2001. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 31:783–791. [DOI] [PubMed] [Google Scholar]
- 4.Chung, D.H., W.E. Seaman, and M.R. Daws. 2002. Characterization of TREM-3, an activating receptor on mouse macrophages: definition of a family of single Ig domain receptors on mouse chromosome 17. Eur. J. Immunol. 32:59–66. [DOI] [PubMed] [Google Scholar]
- 5.Lanier, L.L., and A.B. Bakker. 2000. The ITAM-bearing transmembrane adaptor DAP12 in lymphoid and myeloid cell function. Immunol. Today. 21:611–614. [DOI] [PubMed] [Google Scholar]
- 6.Tomasello, E., L. Olcese, F. Vely, C. Geourgeon, M. Blery, A. Moqrich, D. Gautheret, M. Djabali, M.G. Mattei, and E. Vivier. 1998. Gene structure, expression pattern, and biological activity of mouse killer cell activating receptor-associated protein (KARAP)/DAP-12. J. Biol. Chem. 273:34115–34119. [DOI] [PubMed] [Google Scholar]
- 7.Paloneva, J., M. Kestila, J. Wu, A. Salminen, T. Bohling, V. Ruotsalainen, P. Hakola, A.B. Bakker, J.H. Phillips, P. Pekkarinen, et al. 2000. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25:357–361. [DOI] [PubMed] [Google Scholar]
- 8.Paloneva, J., T. Manninen, G. Christman, K. Hovanes, J. Mandelin, R. Adolfsson, M. Bianchin, T. Bird, R. Miranda, A. Salmaggi, et al. 2002. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hakola, H.P. 1972. Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr. Scand. Suppl. 232:1–173. [PubMed] [Google Scholar]
- 10.Cella, M., C. Dohring, J. Samaridis, M. Dessing, M. Brockhaus, A. Lanzavecchia, and M. Colonna. 1997. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J. Exp. Med. 185:1743–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higuchi, Y., M. Ito, M. Tajima, S. Higuchi, N. Miyamoto, M. Nishio, M. Kawano, S. Kusagawa, M. Tsurudome, A. Sudo, et al. 1999. Gene expression during osteoclast-like cell formation induced by antifusion regulatory protein-1/CD98/4F2 monoclonal antibodies (MAbs): c-src is selectively induced by anti-FRP-1 MAb. Bone. 25:17–24. [DOI] [PubMed] [Google Scholar]
- 12.Teitelbaum, S.L. 2000. Bone resorption by osteoclasts. Science. 289:1504–1508. [DOI] [PubMed] [Google Scholar]
- 13.Vaananen, H.K., H. Zhao, M. Mulari, and J.M. Halleen. 2000. The cell biology of osteoclast function. J. Cell Sci. 113:377–381. [DOI] [PubMed] [Google Scholar]
- 14.Udagawa, N., N. Takahashi, T. Akatsu, H. Tanaka, T. Sasaki, T. Nishihara, T. Koga, T.J. Martin, and T. Suda. 1990. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA. 87:7260–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matayoshi, A., C. Brown, J.F. DiPersio, J. Haug, Y. Abu-Amer, H. Liapis, R. Kuestner, and R. Pacifici. 1996. Human blood-mobilized hematopoietic precursors differentiate into osteoclasts in the absence of stromal cells. Proc. Natl. Acad. Sci. USA. 93:10785–10790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsurudome, M., and Y. Ito. 2000. Function of fusion regulatory proteins (FRPs) in immune cells and virus-infected cells. Crit. Rev. Immunol. 20:167–196. [PubMed] [Google Scholar]
- 17.Kondo, T., K. Takahashi, N. Kohara, Y. Takahashi, S. Hayashi, H. Takahashi, H. Matsuo, M. Yamazaki, K. Inoue, K. Miyamoto, et al. 2002. Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): three genetic forms. Neurology. 59:1105–1107. [DOI] [PubMed] [Google Scholar]
- 18.McHugh, K.P., K. Hodivala-Dilke, M.H. Zheng, N. Namba, J. Lam, D. Novack, X. Feng, F.P. Ross, R.O. Hynes, and S.L. Teitelbaum. 2000. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Invest. 105:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura, I., M.F. Pilkington, P.T. Lakkakorpi, L. Lipfert, S.M. Sims, S.J. Dixon, G.A. Rodan, and L.T. Duong. 1999. Role of alpha(v)beta(3) integrin in osteoclast migration and formation of the sealing zone. J. Cell Sci. 112:3985–3993. [DOI] [PubMed] [Google Scholar]
- 20.Lakkakorpi, P.T., M.A. Horton, M.H. Helfrich, E.K. Karhukorpi, and H.K. Vaananen. 1991. Vitronectin receptor has a role in bone resorption but does not mediate tight sealing zone attachment of osteoclasts to the bone surface. J. Cell Biol. 115:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson, J.M. 2000. Multinucleated giant cells. Curr. Opin. Hematol. 7:40–47. [DOI] [PubMed] [Google Scholar]
- 22.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 23.Kaifu, T., J. Nakahara, M. Inui, K. Mishima, T. Momiyama, M. Kaji, A. Sugahara, H. Koito, A. Ujike-Asai, A. Nakamura, et al. 2003. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J. Clin. Invest. 111:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura, I., E. Jimi, L.T. Duong, T. Sasaki, N. Takahashi, G.A. Rodan, and T. Suda. 1998. Tyrosine phosphorylation of p130Cas is involved in actin organization in osteoclasts. J. Biol. Chem. 273:11144–11149. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura, I., L. Lipfert, G.A. Rodan, and T.D. Le. 2001. Convergence of alpha(v)beta(3) integrin- and macrophage colony stimulating factor–mediated signals on phospholipase Cgamma in prefusion osteoclasts. J. Cell Biol. 152:361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duong, L.T., P.T. Lakkakorpi, I. Nakamura, M. Machwate, R.M. Nagy, and G.A. Rodan. 1998. PYK2 in osteoclasts is an adhesion kinase, localized in the sealing zone, activated by ligation of alpha(v)beta3 integrin, and phosphorylated by src kinase. J. Clin. Invest. 102:881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanjay, A., A. Houghton, L. Neff, E. DiDomenico, C. Bardelay, E. Antoine, J. Levy, J. Gailit, D. Bowtell, W.C. Horne, et al. 2001. Cbl associates with Pyk2 and Src to regulate Src kinase activity, alpha(v)beta(3) integrin-mediated signaling, cell adhesion, and osteoclast motility. J. Cell Biol. 152:181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takami, M., N. Kim, J. Rho, and Y. Choi. 2002. Stimulation by toll-like receptors inhibits osteoclast differentiation. J. Immunol. 169:1516–1523. [DOI] [PubMed] [Google Scholar]
- 29.Zou, W., H. Schwartz, S. Endres, G. Hartmann, and Z. Bar-Shavit. 2002. CpG oligonucleotides: novel regulators of osteoclast differentiation. FASEB J. 16:274–282. [DOI] [PubMed] [Google Scholar]
- 30.McVicar, D.W., L.S. Taylor, P. Gosselin, J. Willette-Brown, A.I. Mikhael, R.L. Geahlen, M.C. Nakamura, P. Linnemeyer, W.E. Seaman, S.K. Anderson, et al. 1998. DAP12-mediated signal transduction in natural killer cells. A dominant role for the Syk protein-tyrosine kinase. J. Biol. Chem. 273:32934–32942. [DOI] [PubMed] [Google Scholar]