

Abstract
Antigen-pulsed dendritic cells (DCs) are used as natural adjuvants for vaccination, but the factors that influence the efficacy of this treatment are poorly understood. We investigated the parameters that affect the migration of subcutaneously injected mouse-mature DCs to the draining lymph node. We found that the efficiency of DC migration varied with the number of injected DCs and that CCR7+/+ DCs migrating to the draining lymph node, but not CCR7−/− DCs that failed to do so, efficiently induced a rapid increase in lymph node cellularity, which was observed before the onset of T cell proliferation. We also report that DC migration could be increased up to 10-fold by preinjection of inflammatory cytokines that increased the expression of the CCR7 ligand CCL21 in lymphatic endothelial cells. The magnitude and quality of CD4+ T cell response was proportional to the number of antigen-carrying DCs that reached the lymph node and could be boosted up to 40-fold by preinjection of tumor necrosis factor that conditioned the tissue for increased DC migration. These results indicate that DC number and tissue inflammation are critical parameters for DC-based vaccination.
Keywords: dendritic cell, T cell priming, CCL21, migration, dendritic cell–based vaccination
Introduction
Tissue-resident DCs and DC precursors, such as monocytes or immature DCs recruited from the blood, are stimulated by microbial products and inflammatory cytokines to undergo a coordinately regulated process of maturation and migration (1). Immature DCs express receptors for inflammatory chemokines and, upon induction of maturation, up-regulate CCR7, a chemokine receptor that drives their migration to the lymphatics (2, 3). CCR7 has two ligands, CCL21, which is expressed constitutively by endothelial cells of lymphatic vessels and of high endothelial venules (HEV) and by stromal cells present in the T cell zone, and CCL19, which is produced by stromal cells and mature DCs in the T cell zone (4–7). These chemokines attract CCR7+ mature DCs into lymphatics and promote extravasation of CCR7+ naive and central memory T cells through HEV, thus orchestrating their encounter in the T cell area of antigen-stimulated lymph nodes.
Lack of CCR7 or its ligands leads to dramatic changes in DC and lymphocyte traffic and abnormal lymph node architecture (8, 9). In CCR7-deficient mice, Langerhans cells and dermal DCs do not migrate to draining lymph nodes and the mice show delayed kinetics of antibody responses and lack contact sensitivity and delayed-type hypersensitivity (DTH) reactions (8). In plt/plt mice, which carry a spontaneous mutation that abolishes CCL19 expression and restricts CCL21 expression to the lymphatic endothelium, DCs and naive T cells fail to enter the T cell zone of secondary lymphoid organs (9, 10). Surprisingly, plt/plt mice can mount CD4+ T cell responses that are of slower kinetics but higher magnitude than wild-type mice and have normal antiviral CD8+ T cell responses (11, 12).
Besides chemokines, lipid mediators have been recently identified as modulators of DC migration and cell traffic through the lymph node. Responsiveness to CCL19 is increased by leukotrienes and by PGE2 through functional modulation or increased expression of CCR7 on DCs (13–15). Furthermore, sphingosine-1-phosphate (S1P), and its analogue derived by phosphorylation of the drug FTY720, prevents exit of lymphocytes from the lymph node (16)
Human in vitro–generated DCs are used as adjuvants for vaccination against cancer (17). Most protocols involve the in vitro loading of DCs with tumor antigen, followed by induction of maturation and s.c. injection. Although this treatment has been demonstrated to be effective in stimulating immune responses, a variety of factors are being considered to optimize DC vaccination. These include DC lineage, nature of the maturation stimulus, and route, frequency, and dosage of DC injection. The number of DCs injected, and ultimately the number of DCs that end up in the T cell zone, likely represents an important variable that impacts on T cell priming. On the one hand, high DC number increases the probability of DC–T cell encounter, delivers a sustained stimulation through monogamous or successive interactions and reduces competition among T cells (18, 19). On the other hand, low numbers of poorly stimulatory DCs induce abortive T cell proliferation and tolerance (20–22).
In this study we dissected the parameters that affect the migration of BM-derived mature DCs to the draining lymph nodes and analyzed the impact of DC migration on T cell priming.
Materials and Methods
Mice and Reagents.
BALB/c (H-2d) mice were from Charles River Laboratories and transgenic DO11.10 (H-2d) mice, expressing on 90% of CD4+ T cells a single T cell receptor specific for MHC class II and the OVA peptide 323–339, were from The Jackson Laboratory. CCR7−/− mice (8) were backcrossed on BALB/c for eight generations. Mice were treated in accordance with the Swiss Federal Veterinary Office guidelines. Recombinant mouse TNF and IL-1α were purchased from R&D Systems. CFA or IFA were from Sigma-Aldrich.
BM-derived DCs and Spleen Cells.
BM cells from femurs of CCR7+/+ or CCR7−/− mice were cultured in RPMI 1640 (GIBCO BRL) supplemented with 10% FCS (HyClone) and recombinant mouse GM-CSF (R&D Systems) according to current protocols. To induce maturation, 8–10 d BM-DC cultures were stimulated with 0.5 μg/ml LPS (Sigma-Aldrich) for 24 h. At that time point DCs were homogeneously CCR7+ as detected by staining with a CCL21–Ig fusion protein (provided by K. Karjalainen, Institute for Research in Biomedicine, Bellinzona, Switzerland). CD11c+ and CD11c− spleen cells were isolated using CD11c-coated magnetic beads (Miltenyi Biotec). Cells were labeled with 2.5 μM 5- and 6-carboxyfluorescein diacetate succinimidyl ester (CFSE) or 10 μM 5- and 6-(4-chloromethyl)benzoyl-amino-tetramethylrhodamine (CMTMR) intracellular fluorescent dyes according to the manufacturer's instructions (Molecular Probes). After labeling, cells were washed extensively in PBS and injected s.c. into the footpad or flank (for experiments involving use of CFA or IFA). No differences in the extent of DC migration to lymph nodes were found when CFSE or CMTMR was used.
Adoptive Transfer and Immunization.
Spleen and lymph node cells from DO11.10 mice were labeled with CFSE. 3 × 106 transgenic KJ1–26+ cells were injected into the tail vein of syngeneic BALB/c mice. For immunization, DCs were collected from culture and loaded in vitro with 20 μM OVA323–339 peptide (Invitrogen) at 37°C for 1 h. Cells were then washed extensively in PBS and injected s.c. into adoptively transferred mice. The DTH response was evaluated by challenging DO11.10-transferred, DC-OVA–primed mice with 10 μg whole OVA in PBS intradermally in the ear, 7 d after priming.
FACS® Analysis.
To analyze the number of migrating DCs, popliteal lymph nodes were minced into small fragments and treated with 1 mg/ml collagenase-D (Roche) at 37°C for 45 min. Cells were passed through a mesh, washed in PBS, and cell viability was determined with Trypan Blue (Sigma-Aldrich). Priming of CFSE-labeled KJ1–26+ transgenic T cells was evaluated by staining lymph node cells with PE-labeled anti-CD44 (BD Biosciences) and biotinylated anti-clonotype KJ1-26 (Caltag). Secondary reagent was streptavidin-APC (BD Biosciences). Cytokine production was evaluated by stimulating lymph node T cells with PMA and ionomycin for 4 h with the addition of Brefeldin A for the last 2 h (Sigma-Aldrich). Fluoresceinated antibodies against TNF and IFN-γ (BD Biosciences) were used after fixation in 2% paraformaldehyde and permeabilization with 0.5% saponin (Sigma-Aldrich). All samples were analyzed on a FACSCalibur™ (BD Biosciences).
In Vivo Skin Sensitization.
Green fluorescent CMFDA (5-chloromethyl-fluoresceindiacetate) cell tracker (Molecular Probes) was dissolved in a 50/50 (vol/vol) acetone-butyl phtalate mixture just before application. Mice were painted with 500 μl on the shaved abdomen. Draining lymph nodes were collected and cells were stained with APC-labeled CD11c antibody (BD Biosciences) and analyzed by flow cytometry using propidium iodide to exclude dead cells.
In Situ Hybridization and Immunohistochemistry.
Skin samples were collected, fixed in 4% formalin, and embedded in paraffin. Sense or antisense 35S-labeled riboprobes (831 bp) were generated by in vitro transcription (Boehringer) from the mouse SLC cDNA corresponding to positions 1–831 of the published sequence (10). Tissue sections were dewaxed, rehydrated, and subjected to in situ hybridization as previously described (23). Sections were dipped in Kodak photo emulsion NTB-2 and exposed in darkness for 2 wk at 4°C. Development and fixation were performed according to the manufacturer's instructions. Tissue cross sections were deparaffinized in xylene and rehydrated through graded alcohol solutions. Sections were boiled in target retrieval solution at pH 6.0 (DakoCytomation) and washed in 50 mM Tris and 0.15 M sodium chloride, pH 7.5 (Tris-buffered saline). After blocking, sections were incubated overnight at room temperature with goat polyclonal anti–mouse 6C-kine antibodies (1 μg/ml; R&D Systems), followed by incubation with biotinylated rabbit anti–goat immunoglobulin (1.3 μg/ml; DakoCytomation) and streptavidin-alkaline phosphatase (DakoCytomation). Sections were developed with New Fuchsin (DakoCytomation) according to the manufacturer's instructions.
Online Supplemental Material.
Fig. S1 shows the kinetics of tissue conditioning for increased DC migration after TNF injection. Fig. S2 shows migration of OVA-loaded DCs in naive and OVA-primed mice. Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20030448/DC1.
Results and Discussion
Cooperative Migration of Injected DCs to the Draining Lymph Node.
To investigate the capacity of in vitro–generated DCs to migrate to the draining lymph node we injected into the footpad of syngeneic mice increasing numbers of BM-derived, LPS-matured CCR7+ DCs labeled with the fluorescent dye CFSE. On day 2 draining lymph nodes were isolated and the total number of CFSE+ cells was measured by FACS® analysis. The number of DCs recovered in the lymph nodes increased as a function of the number of injected DCs (Fig. 1 A). However, the efficiency of DC migration, i.e., the percentage of injected DCs recovered in lymph nodes, varied with the number of cells injected (Fig. 1 B). Thus, migration efficiency was 3% (range 0.6 ÷ 4.7) when 2 × 106 DCs were injected, but dropped to 0.01% when 105 DCs were used. Finally, the efficiency of migration slightly decreased when >2 × 106 DCs were injected (Fig. 1 B). These results suggest that migration of injected DCs to the draining lymph node is cooperative at low DC inputs and reaches a saturation point when high DC numbers are injected.
Figure 1.
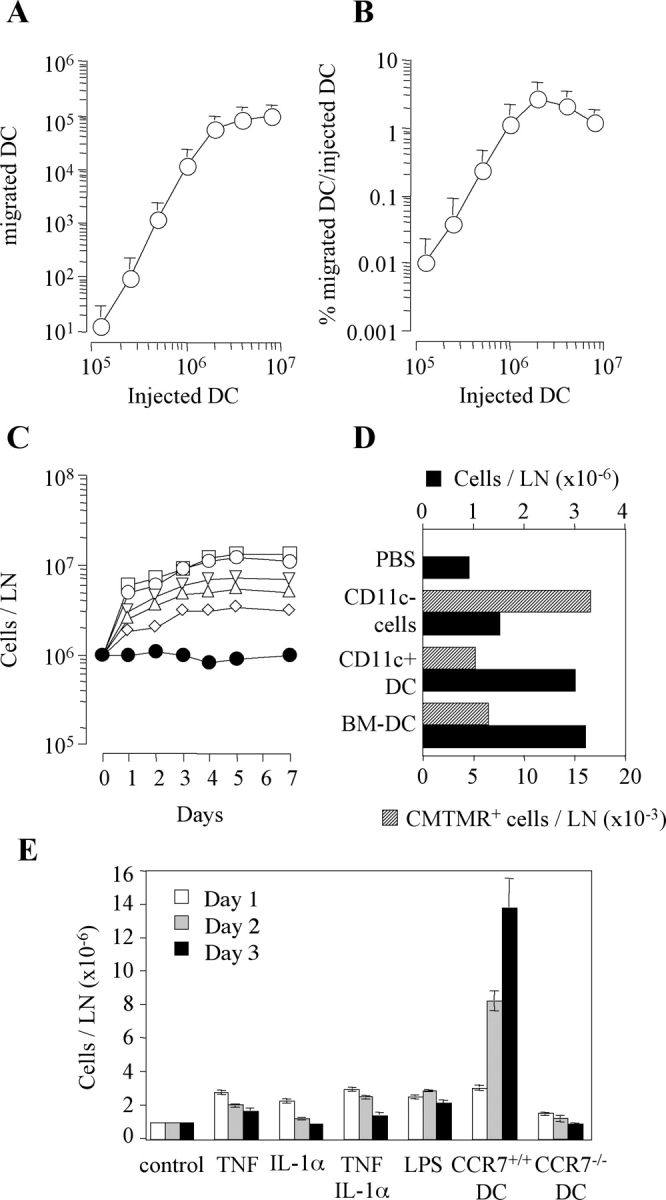
Variable efficiency of DC migration to draining lymph node and consequent lymph node congestion. Increasing numbers of CFSE-labeled mature BM-derived DCs were injected into the footpads of syngeneic mice. The total number of DCs recovered in the draining lymph node (LN; A) and the percentage of injected DCs recovered (B) on day 2 is shown. Mean ± SD of three independent experiments is shown, each performed using groups of two mice per condition. (C) Groups of two mice were injected s.c. with increasing numbers of mature DCs. Total cell number in the pooled draining lymph nodes (LN) was measured at different time points. The number of DCs injected was: 2 × 106 (□), 106 (○), 0.5 × 106 (▿), 0.25 × 106 (▵), and 0.125 × 106 (⋄). Mice injected with PBS were used as control (•). The experiment was repeated with comparable results. (D) Mice were injected s.c. with PBS, 106 BM-derived DC (BM-DC), 106 CD11c+ DCs isolated from the spleen or 10 × 106 CD11c− spleen cells that had been labeled with CMTMR. The total cell number in the lymph node (solid bars) and the number of CMTMR+ migrated cells (shaded bars) are shown. One out of three experiments performed is shown. (E) Groups of three mice per condition per time point were injected s.c. with PBS (control), 300 ng TNF, and/or IL-1α, 100 μg LPS, or 106 BM-derived syngeneic DCs from CCR7+/+ or CCR7−/− mice. The total cell number (means ± SD) in the draining lymph node was measured on days 1, 2, and 3.
Migrated DCs Rapidly Induce an Increase in Lymph Node Cellularity.
In the above experiments we observed that injection of DCs resulted in an increase in lymph node cellularity that was detectable on day 1 and reached a plateau by days 3 and 4 (Fig. 1 C). The extent of increase was dependent on the number of DCs injected. On day 1 as few as 2 × 105 DCs induced a twofold increase in lymph node cell number, whereas a maximum increase of ∼fivefold was reached upon injection of >106 DCs. Because cell proliferation was not detected before day 2 (unpublished data), the increase in cell number observed on day 1 must be the consequence of congestion of cell traffic due to either increased entry, decreased exit, or both.
Lymph node congestion was also induced by injection of 106 freshly isolated splenic CD11c+ DCs (Fig. 1 D). In contrast, a 10-fold higher number of CD11c− splenic cells failed to do so in spite of efficient migration to the draining lymph node (Fig. 1 D). An increase in lymph node total cell number was modestly induced by injection of inflammatory cytokines (TNF or IL-1, alone or in combination) or adjuvants (IFA, CFA, or LPS; Fig. 1 E and unpublished data), further suggesting a direct role for migrating DCs in this process. To understand whether DC migration to the lymph node was required, we compared DCs generated from BM cells of CCR7+/+ or CCR7−/− mice (8) for their capacity to induce lymph node congestion. Injection of CCR7−/− DCs, which produced comparable amounts of cytokines in vitro (unpublished data) but failed to migrate to the draining lymph node (8), did not induce cell accumulation in the draining lymph node (Fig. 1 E). We conclude that an increase in lymph node cellularity is rapidly induced and maintained by mature DCs that have reached the draining lymph node.
Subcutaneous Injection of DCs or Inflammatory Cytokines Conditions Tissue for Increased DC Migration.
The finding that DC migration to lymph nodes is cooperative prompted us to investigate whether DCs could promote the migration of other DCs. Therefore, we injected CMTMR-labeled DCs into footpads that were treated 24 h earlier with either CFSE-labeled DCs or PBS as control. Preinjection of CFSE-labeled DCs boosted up to 10-fold the efficiency of migration of subsequently injected CMTMR-labeled DCs (Fig. 2 A). Increased DC migration was also elicited by preinjection of TNF or IL-1α (Fig. 2 B), and was most prominent at low numbers of injected DCs (Fig. 2 C). To be effective, cytokines needed to be injected 6–8 h before DCs whereas no enhancement in DC migration was observed when cytokines were administered together with DCs (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20030448/DC1). Time course experiments indicated that DCs injected into tissues conditioned by a previous injection of DCs, TNF, or IL-1α reached the lymph nodes more rapidly and persisted at higher numbers compared with DCs injected in nonconditioned tissues (Fig. 2 D). Finally, preconditioning with TNF greatly enhanced the hapten-induced migration of endogenous CD11c+ DCs over a 3-d period (Fig. 2 E).
Figure 2.

The efficiency of DC migration to draining lymph node is increased by TNF, IL-1, and by DCs themselves. (A) PBS or 106 CFSE-labeled DCs were injected into the footpads of syngeneic mice. 24 h later, the mice received a second injection of 106 CMTMR-labeled DCs in the same footpad. Mice were killed 24 h after the second injection and the number of CMTMR+ DCs migrated to the draining lymph nodes was analyzed. (B) The experiment was performed as in A after the injection of PBS, unlabeled 106 DCs, TNF, or IL-1α. Mean of three experiments each performed with groups of two mice per condition is shown. (C) Footpads pretreated with either PBS (•) or TNF (○) were injected with increasing doses of CMTMR-labeled DCs and the number of migrated DCs recovered in the draining lymph node was measured 2 d later. (D) Footpads of mice pretreated with PBS (•), TNF (□), or 106 unlabeled DCs (○) were injected with 106 CMTMR DCs and the number of migrated DCs was evaluated daily. Values are from pooled lymph nodes of two mice per time point. (E) CMFDA was applied to the skin of mice that had been pretreated with either PBS (•) or TNF (○). The number of fluorescent endogenous CD11c+ DCs recovered in the draining lymph node is shown.
Taken together, the above results indicate that DC migration can be boosted in a paracrine fashion by TNF and by maturing DCs that are known to secrete TNF and other proinflammatory cytokines.
Proinflammatory Cytokines Up-regulate CCL21 Expression in Lymphatic Endothelium.
We investigated whether the increased DC migration observed after tissue conditioning by injection of DCs or TNF could be related to an increased expression of CCL21 or to the induction of alternative CCR7-independent migratory pathways. As shown in Fig. 3 , lymphatic endothelial cells constitutively expressed CCL21. However, CCL21 expression was dramatically up-regulated 8 h after DC or TNF injection both at mRNA and protein level. These results suggest that up-regulation of CCL21 might be one mechanism by which tissue inflammation increases DC recruitment into lymphatics.
Figure 3.
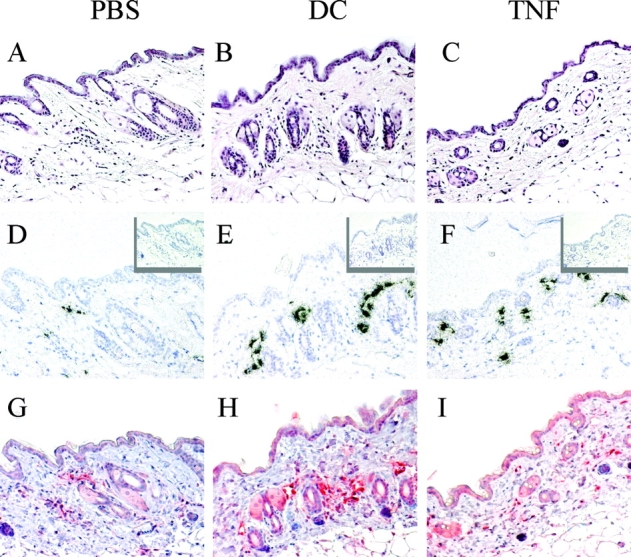
CCL21 is up-regulated on lymphatic endothelial cells after injection of mature DCs or TNF. Mice were injected intradermally with PBS, 105 syngeneic DCs, or 10 ng TNF. 8 h later, skin samples were collected and embedded in paraffin. Serial sections were stained with hematoxylin and eosin (A–C), or hybridized with antisense (D–F) or sense (inset) 35S-labeled CCL21 riboprobe or stained with a polyclonal antibody to mouse CCL21 (G–I). ×20.
To further investigate whether CCR7-independent mechanisms were involved in the enhanced DC migration observed in conditioned tissues, we tested whether in these conditions CCR7−/− DCs might be capable of migrating to the draining lymph node. As shown in Fig. 4 , CCR7−/− DCs injected into the footpad failed to migrate to the draining lymph node regardless of whether the tissue was conditioned by injection of TNF, CCR7+/+ DCs, or CCR7−/− DCs. However, subcutaneously injected CCR7−/− DCs efficiently promoted migration of CCR7+/+ DCs (Fig. 4), further supporting the notion that tissue conditioning by DCs is a local event. We conclude that the enhanced DC migration observed upon tissue conditioning is mediated mainly through up-regulation of CCL21 rather than through alternative migratory pathways.
Figure 4.

CCR7−/− DCs fail to migrate to the draining lymph node but efficiently condition the tissue for increased migration of CCR7+/+ DCs. Mice (two per condition) were injected with 106 CCR7+/+ or CCR7−/− syngeneic DCs, TNF, or PBS. 24 h later, all mice received a second injection at the same site of 106 CMTMR-labeled CCR7−/− or CCR7+/+ DCs. The total number of migrated CMTMR-labeled DCs was measured in the draining lymph node after an additional 24 h (gate). Live cells gated as propidium iodide (PI) negative are shown. One of two experiments performed is shown.
Impact of DC Migration and Tissue Conditioning on T Cell Priming.
To examine the impact of DC migration on T cell priming we transferred CFSE-labeled TCR transgenic CD4+ DO11.10 T cells into syngeneic mice and challenged the mice by injecting increasing numbers of LPS-matured DCs pulsed with optimal concentrations of the relevant OVA peptide (Fig. 5 A). When high numbers of DCs were injected, T cells were efficiently activated as determined by CFSE dilution. However, when the injected DCs decreased below 0.75 × 106, the efficiency of T cell priming dropped dramatically and dividing T cells failed to accumulate. Thus, a twofold decrease in DC number resulted in a much higher decrease (up to fourfold) in the number of activated T cell blasts recovered on day 3.
Figure 5.

Impact of DC migration and tissue conditioning on T cell response. (A) 3 × 106 KJ1–26+ T cells from DO11.10 mice were labeled with CFSE and adoptively transferred into syngeneic BALB/c mice. Control or conditioned mice were primed by injecting OVA323–339–pulsed syngeneic DCs in the footpad. CFSE profiles (day 3) of KJ1.26-gated cells and the absolute number of proliferating T cells after injection of increasing numbers of OVA-pulsed DCs. One representative experiment out of three is shown. (B) CFSE profiles (day 4) of KJ1-26–gated cells and total number of proliferating cells in mice conditioned with PBS, TNF, or 106 syngeneic DCs and primed by 105 OVA-pulsed DCs. Data are from pooled lymph nodes from two mice. (C) CFSE profile (day 4) of KJ1-26–gated T cells and total number of proliferating T cells in control or TNF-conditioned mice primed by 0.1 × 106 or 106 OVA-pulsed CMTMR-labeled DCs. The number of DCs recovered in the same lymph nodes was as follows: nonconditioned, 0.1 × 106 DCs injected, <10; TNF-conditioned, 0.1 × 106 DCs injected, 1,180; nonconditioned, 106 DCs injected, 1,600; TNF-conditioned, 106 DCs injected, 9,320. (D) TNF and IFN-γ production elicited by PMA and ionomycin in KJ1-26–gated cells recovered from draining (top panels) and nondraining (bottom panels) lymph nodes. The total number of cytokine-secreting cells was calculated from the percentage and the total number of cells and is indicated in the dot plot. (E) Mice were challenged by injection of 10 μg OVA in the ear pinna 7 d after priming. Controls include naive mice and mice adoptively transferred with naive DO11.10 T cells (unprimed). Ear thickness was measured 24 h after challenge. One out of two experiments performed is shown.
Next, we tested whether conditioning the injection site might enhance T cell response to limiting numbers of antigen-pulsed DCs. 105 OVA-pulsed DCs were injected into footpads that were conditioned either by unpulsed DCs, TNF, or PBS as control. Conditioning by DCs or TNF increased >10-fold the number of dividing D011.10 CD4+ transgenic T cells recovered in the draining lymph node 4 d after challenge (Fig. 5 B). The enhancing effect of TNF conditioning on T cell response was maximal at low DC input (105) but was also observed when relatively high DC numbers (106) were injected (Fig. 5 C). In all cases the T cell proliferative response was proportional to the number of DCs that reached the lymph node. Indeed, comparable numbers of migrated DCs and proliferating T cells were found after injection of either 106 DCs into nonconditioned tissue or 105 DCs into TNF-conditioned tissue. Migration of OVA-pulsed DCs under nonconditioning or conditioning regimes was comparable in naive or OVA-primed animals (Fig. S2, available at http://www.jem.org.cgi/content/full/jem.20030448/DC1), ruling out the possibility that memory or effector CD4+ T cells may affect DC migration to the lymph node. Furthermore, tissue conditioning led to a higher number of T cells producing TNF and IFN-γ in the draining and, especially, in the nondraining lymph nodes (Fig. 5 D), consistent with an increased differentiation and migration of effector cells. Tissue conditioning also enhanced effector function in peripheral tissues as measured by the DTH response to intradermal injection of OVA (Fig. 5 E).
Taken together, the above results indicate that conditioning the injection sites leads to T cell responses that are of higher magnitude and more effective that those elicited in the absence of tissue conditioning.
Concluding Remarks.
We have shown that migration of mature DCs to the draining lymph nodes is regulated at the level of entry into lymphatic vessels by inflammatory cytokines through up-regulation of CCL21. Mature DCs that reach the lymph node induce a rapid and sustained congestion of lymphocyte traffic and their number determines the magnitude of T cell proliferation and effector response.
Changes in the kinetics of lymphocyte traffic through antigen-stimulated lymph nodes have been known for a long time (24, 25). Recent studies suggest that increased cell influx might be mediated by increased availability of chemokines displayed on HEV, whereas decreased efflux through the lymphatic sinuses might be induced by production of S1P (16, 26). We have shown that injection of mature DCs leads to a rapid and sustained increase in the size of the draining lymph nodes. The increase in total lymph node cell number is observed before the onset of cell proliferation, is sustained for several days, and is proportional to the number of injected DCs. The fact that CCR7−/− DCs, which are incapable of migrating to the lymph node, do not cause lymph node congestion indicates that this phenomenon is mediated by mature DCs that have reached the lymph node through mechanisms that still need to be identified but might be related to release of chemokines or S1P.
The finding that DC migration can be boosted by inflammatory stimuli through up-regulation of CCL21 on lymphatic endothelial cells was unexpected, because CCL21 has been considered the prototype of constitutive chemokines (27). Therefore, our results delineate an additional level at which inflammation acts to increase T cell responses. Inflammatory stimuli not only promote recruitment of immature DCs into tissues and initiate the DC maturation process, but also boost recruitment of mature DCs into lymphatics. The increased DC migration that follows tissue conditioning can lead to up to a 40-fold increase in the number of IFN-γ–producing T cells generated, as well as in a stronger DTH reaction. Therefore, tissue conditioning might be considered a strategy to increase the efficiency of T cell priming by DC-based vaccines. It remains to be established whether this treatment in primed individuals may result in recruitment of effector cytotoxic T cells and killing of antigen-loaded DCs within the tissue (28, 29).
In conclusion, our results underline the impact of absolute DC numbers on T cell priming and indicate that optimal numbers of DCs have to be injected to fully exploit the T cell response potential.
Acknowledgments
We thank A. Gett for critical reading and comments.
This work has been supported in part by the Swiss National Science Foundation (grant no. 31-63885) and by the European Community (contract no. QLK2-CT-2001-01205). The Institute for Research in Biomedicine is supported by the Helmut Horten Foundation.
Abbreviations used in this paper: CFSE, 5- and 6-carboxyfluorescein diacetate succinimidyl ester; CMTMR, 5- and 6-(4-chloromethyl)benzoyl-amino-tetramethylrhodamine; DTH, delayed-type hypersensitivity; HEV, high endothelial venules; S1P, sphingosine-1-phosphate.
The online version of this article contains supplemental material.
References
- 1.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 2.Dieu, M.C., B. Vanbervliet, A. Vicari, J.M. Bridon, E. Oldham, S. Ait-Yahia, F. Briere, A. Zlotnik, S. Lebecque, and C. Caux. 1998. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 188:373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sallusto, F., P. Schaerli, P. Loetscher, C. Schaniel, D. Lenig, C.R. Mackay, S. Qin, and A. Lanzavecchia. 1998. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 28:2760–2769. [DOI] [PubMed] [Google Scholar]
- 4.Ngo, V.N., H.L. Tang, and J.G. Cyster. 1998. Epstein-Barr virus–induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J. Exp. Med. 188:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gunn, M.D., K. Tangemann, C. Tam, J.G. Cyster, S.D. Rosen, and L.T. Williams. 1998. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA. 95:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kriehuber, E., S. Breiteneder-Geleff, M. Groeger, A. Soleiman, S.F. Schoppmann, G. Stingl, D. Kerjaschki, and D. Maurer. 2001. Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J. Exp. Med. 194:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willimann, K., D.F. Legler, M. Loetscher, R.S. Roos, M.B. Delgado, I. Clark-Lewis, M. Baggiolini, and B. Moser. 1998. The chemokine SLC is expressed in T cell areas of lymph nodes and mucosal lymphoid tissues and attracts activated T cells via CCR7. Eur. J. Immunol. 28:2025–2034. [DOI] [PubMed] [Google Scholar]
- 8.Forster, R., A. Schubel, D. Breitfeld, E. Kremmer, I. Renner-Muller, E. Wolf, and M. Lipp. 1999. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 99:23–33. [DOI] [PubMed] [Google Scholar]
- 9.Gunn, M.D., S. Kyuwa, C. Tam, T. Kakiuchi, A. Matsuzawa, L.T. Williams, and H. Nakano. 1999. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 189:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakano, H., and M.D. Gunn. 2001. Gene duplications at the chemokine locus on mouse chromosome 4: multiple strain-specific haplotypes and the deletion of secondary lymphoid-organ chemokine and EBI-1 ligand chemokine genes in the plt mutation. J. Immunol. 166:361–369. [DOI] [PubMed] [Google Scholar]
- 11.Mori, S., H. Nakano, K. Aritomi, C.R. Wang, M.D. Gunn, and T. Kakiuchi. 2001. Mice lacking expression of the chemokines CCL21-ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J. Exp. Med. 193:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junt, T., H. Nakano, T. Dumrese, T. Kakiuchi, B. Odermatt, R.M. Zinkernagel, H. Hengartner, and B. Ludewig. 2002. Antiviral immune responses in the absence of organized lymphoid T cell zones in plt/plt mice. J. Immunol. 168:6032–6040. [DOI] [PubMed] [Google Scholar]
- 13.Robbiani, D.F., R.A. Finch, D. Jager, W.A. Muller, A.C. Sartorelli, and G.J. Randolph. 2000. The leukotriene C(4) transporter MRP1 regulates CCL19 (MIP-3beta, ELC)-dependent mobilization of dendritic cells to lymph nodes. Cell. 103:757–768. [DOI] [PubMed] [Google Scholar]
- 14.Scandella, E., Y. Men, S. Gillessen, R. Forster, and M. Groettrup. 2002. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood. 100:1354–1361. [DOI] [PubMed] [Google Scholar]
- 15.Luft, T., M. Jefford, P. Luetjens, T. Toy, H. Hochrein, K.A. Masterman, C. Maliszewski, K. Shortman, J. Cebon, and E. Maraskovsky. 2002. Functionally distinct dendritic cell (DC) populations induced by physiologic stimuli: prostaglandin E(2) regulates the migratory capacity of specific DC subsets. Blood. 100:1362–1372. [DOI] [PubMed] [Google Scholar]
- 16.Mandala, S., R. Hajdu, J. Bergstrom, E. Quackenbush, J. Xie, J. Milligan, R. Thornton, G.J. Shei, D. Card, C. Keohane, et al. 2002. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 296:346–349. [DOI] [PubMed] [Google Scholar]
- 17.Banchereau, J., B. Schuler-Thurner, A.K. Palucka, and G. Schuler. 2001. Dendritic cells as vectors for therapy. Cell. 106:271–274. [DOI] [PubMed] [Google Scholar]
- 18.Kedl, R.M., J.W. Kappler, and P. Marrack. 2003. Epitope dominance, competition and T cell affinity maturation. Curr. Opin. Immunol. 15:120–127. [DOI] [PubMed] [Google Scholar]
- 19.Smith, A.L., M.E. Wikstrom, and B. Fazekas de St Groth. 2000. Visualizing T cell competition for peptide/MHC complexes: a specific mechanism to minimize the effect of precursor frequency. Immunity. 13:783–794. [DOI] [PubMed] [Google Scholar]
- 20.Kurts, C., H. Kosaka, F.R. Carbone, J.F. Miller, and W.R. Heath. 1997. Class I–restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 186:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernandez, J., S. Aung, W.L. Redmond, and L.A. Sherman. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194:707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonifaz, L., D. Bonnyay, K. Mahnke, M. Rivera, M.C. Nussenzweig, and R.M. Steinman. 2002. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazzucchelli, L., A. Blaser, A. Kappeler, P. Scharli, J.A. Laissue, M. Baggiolini, and M. Uguccioni. 1999. BCA-1 is highly expressed in Helicobacter pylori-induced mucosa-associated lymphoid tissue and gastric lymphoma. J. Clin. Invest. 104:R49–R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cahill, R.N., H. Frost, and Z. Trnka. 1976. The effects of antigen on the migration of recirculating lymphocytes through single lymph nodes. J. Exp. Med. 143:870–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall, J.G., and B. Morris. 1965. The immediate effect of antigens on the cell output of a lymph node. Br. J. Exp. Pathol. 46:450–454. [PMC free article] [PubMed] [Google Scholar]
- 26.Stein, J.V., A. Rot, Y. Luo, M. Narasimhaswamy, H. Nakano, M.D. Gunn, A. Matsuzawa, E.J. Quackenbush, M.E. Dorf, and U.H. von Andrian. 2000. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function–associated antigen 1–mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J. Exp. Med. 191:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossi, D., and A. Zlotnik. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18:217–242. [DOI] [PubMed] [Google Scholar]
- 28.Hermans, I.F., D.S. Ritchie, J. Yang, J.M. Roberts, and F. Ronchese. 2000. CD8+ T cell-dependent elimination of dendritic cells in vivo limits the induction of antitumor immunity. J. Immunol. 164:3095–3101. [DOI] [PubMed] [Google Scholar]
- 29.Schrama, D., L.O. Pedersen, P. Keikavoussi, M.H. Andersen, P. Straten Pt, E.B. Brocker, E. Kampgen, and J.C. Becker. 2002. Aggregation of antigen-specific T cells at the inoculation site of mature dendritic cells. J. Invest. Dermatol. 119:1443–1448. [DOI] [PubMed] [Google Scholar]