

Abstract
PGD2, a lipid mediator released from mast cells, is known to participate in allergic reactions. However, the mechanism by which PGD2 contributes to such reactions remains unclear. We established a novel experimental model of asthma that permitted direct assessment of the role of PGD2 in airway inflammation. Antigen-sensitized mice were exposed to aerosolized prostaglandin D2 (PGD2) 1 d before challenge with low-dose aerosolized antigen. Not only the numbers of eosinophils, lymphocytes, and macrophages but also the levels of IL-4 and IL-5 in bronchoalveolar lavage fluid were higher in PGD2-pretreated mice than in control mice. The expression of macrophage-derived chemokine (MDC), a chemoattractant for Th2 cells, was greater in PGD2-pretreated mice than in control. Injection of anti-MDC antibody into PGD2-pretreated mice markedly inhibited inflammatory cell infiltration as well as Th2 cyto-kine production after antigen challenge. These results indicate that PGD2 accelerates Th2 type inflammation by induction of MDC. Our results suggest that this mechanism may play a key role in the development of human asthma and that MDC might be a target molecule for therapeutic intervention.
Keywords: bronchial asthma, chemokines, epithelial cells, prostanoids, Th2 cells
Introduction
Th2 cells play a critical role in the pathogenesis of bronchial asthma (1–4). Th2 cells produce cytokines such as IL-4, -5, -9, and -13, which induce IgE production as well as mast cell and eosinophil activation (5, 6). Recent analyses of mice lacking T1/ST2, a Th2-specific cell surface molecule, and those lacking signal transducer and activator of transcription–6, strongly support important roles of Th2 cells and the cytokines they secrete in the development of bronchial asthma (7, 8). Th2 cells are recruited into the airway mucosa (9–13) and are found in the bronchoalveolar lavage (BAL) fluid of patients with asthma (14–20).
Over the past few years, the ability of chemokines (chemotactic cytokines) to attract inflammatory cells to the lung in patients with asthma has received considerable attention. Recent papers suggest that bronchial epithelial cells may directly perpetuate Th2 type airway inflammation by producing certain chemokines, such as thymus- and activation-regulated chemokine (TARC) and macrophage-derived chemokine (MDC; references 21–23). These two chemokines, ligands for CC chemokine receptors (CCRs; reference 24) 4, are potent chemoattractants for Th2 cells (25–27) and have been implicated in Th2 type inflammation associated with the development of airway hyperresponsiveness (AHR; references 28–30). We have reported previously that IL-9, a Th2 type cytokine, is essentially involved in Th2 type pulmonary inflammation associated with AHR in a murine model of asthma, possibly via induction of MDC in bronchial epithelial cells (31). These results suggested that once Th2 cells are recruited into the lung, Th2 type inflammation might be exaggerated via the IL-9–MDC loop. Thus, identification of factors that initially induce MDC is critical to understanding the mechanism initiating Th2 type pulmonary inflammation.
Mast cells also have an important part in asthma. Activated mast cells contribute to asthmatic pulmonary inflammation by producing a variety of chemical mediators and cytokines. Prostaglandin D2 (PGD2), the major cyclooxygenase metabolite of arachidonic acid (32–34), is one chemical mediator released in large amounts by mast cells during asthmatic attacks in humans. PGD2 has been proposed as a marker of mast cell activation in asthma (35, 36). Recent papers have shown that mice lacking G protein–coupled receptors were resistant to experimentally induced allergic asthma, suggesting that PGD2 mediated by PGD receptors participates in the development of allergic asthma (37). Furthermore, Fujitani et al. reported recently that the levels of Th2 cytokines are elevated, accompanied by increased accumulation of eosinophils and lymphocytes, in the lungs of prostaglandin D synthase transgenic mice (38). These works suggested that PGD2 is involved in Th2 type pulmonary inflammation characterized by recruitment of eosinophils and Th2 cells. However, the mechanism by which PGD2 functions remains unclear.
We have developed a novel model of inducible allergic asthma by carefully titrating the dose of antigen. This model enabled us to directly assess the effect of inhaled PGD2. Using this model, we show here that PGD2 increased the recruitment of eosinophils and CD4+ T cells, especially Th2 cells, into the lung. We also found that PGD2 exerts its asthma-promoting activity via induction of MDC in pulmonary epithelial cells. Our results provide evidence that PGD2 acts via MDC, thought to function as a molecular link between PGD2 and Th2 type inflammation in asthma.
Materials and Methods
Animals.
Specific pathogen-free male BALB/c mice (6–8-wk-old; Japan SLC Inc.) were used in all experiments. The study protocol was reviewed and approved by the Dokkyo University School of Medicine Committee on Animal Care and complies with National Institutes of Health guidelines for animal care.
Sensitization and Antigen Challenge of Mice.
To investigate the roles of PGD2 in the development of Th2 type response, we analyzed pulmonary inflammation in mice sensitized and challenged with low doses of antigen. In preliminary studies, we determined the degree of antigen exposure that induced marginal pulmonary responses and inflammation, as compared with established murine models of asthma (Fig. 1, method 1). First, BALB/c mice were sensitized and challenged with antigen according to a conventional protocol (31, 39). In brief, mice were sensitized on days 0 and 5 by i.p. injections of OVA (Sigma-Aldrich) (8 μg/mouse) adsorbed on aluminum hydroxide (Wako Pure Chemical Industries). To induce experimental asthma, the sensitized mice were challenged with aerosolized 1% OVA (two 60-min sessions separated by an interval of 4 h) on day 17. Second, to evaluate the effects of PGD2 (Fig. 1, method 2), sensitized mice were exposed to aerosolized PGD2 (10−3 M) or saline on day 16, followed by one provocation with aerosolized 0.1% OVA for 30 min on day 17 (a low-dose antigen asthma model). In preliminary studies, we determined the amount of PGD2 that induced no obvious histological changes, such as inflammatory cell recruitment and tissue edema. Each mouse was killed at the indicated times before or after challenge (Fig. 1) .
Figure 1.

Protocols used in this work. Method (1), conventional standard asthma model; method (2), newly established asthma model for analysis of effects in PGD2-pretreated mice; and method (3), analysis of effects of anti-MDC antibody (αMDC) on PGD2-promoted Th2 type inflammation and AHR. *, Analysis of cytokines, cell populations, and histological findings before 0, 3, or 24 h after OVA challenge.
BAL.
BAL was performed immediately before and 3 and 24 h after the last aerosol challenge. At the time of lavage, the mice were killed with an overdose of 100 mg/kg body weight of sodium pentobarbitone (pentobarbital sodium). The trachea was exposed and cannulated with polyethylene tubing. The lungs were lavaged with three 0.5-ml aliquots of saline. The supernatant was stored at −70°C.
Total cell numbers were counted with a hemocytometer. Cytospin preparations of BAL cells were stained with Diff-Quik solution (Dade Behring Inc.) to determine the differential cell count, evaluated on the basis of at least 500 leukocytes.
Histological Examination of Lung.
The lungs were taken from the mice, fixed in neutralized buffered formalin, and embedded in paraffin. Sections 3-μm thick were stained with Luna solution for eosinophils and toluidine blue for mast cells.
OVA-specific T Cell and Bone Marrow Mast Cell (BMMC) Response In Vitro.
10 d after intraperitoneal immunization with OVA as described above in the Sensitization and Antigen Challenge of Mice section, mice spleen cells were isolated and cultured with 10−9–10−5 M PGD2 during stimulation with 10 μg/ml OVA for 24 h (31, 40). For BMMC culture, intact femurs and tibias were removed from mice, and bone marrow cells were harvested by repeated flushing with MEM. A cell culture was established at a density of 3 × 106 cells/ml in IMDM, supplemented with 10% FCS (inactivated at 56°C), 2 mM l-glutamine, 1 mM pyruvate, 100 U/ml penicillin, 100 mg/ml streptomycin, 20 U/ml mIL-3, and 50 U/ml mIL-4. Nonadherent cells were transferred to fresh culture plates every 2–3 d for a total of at least 21 d to remove adherent macrophages and fibroblasts (31, 41). Toluidine blue staining revealed that the resulting cell population consisted of >99% BMMCs. These cells were cultured with 0.25 μM ionomycin during stimulation with 10−9–10−5 M PGD2.
Detection of Pulmonary T1/ST2+ T Cells.
To examine the accumulation of Th2 cells in the lungs, we analyzed the expression of T1/ST2, a Th2 surface marker, on intrapulmonary CD3+ cells obtained from the PGD2-treated sensitized animals at the indicated times before and after challenge (Fig. 1, method 2). Lungs were perfused with PBS without magnesium or calcium to minimize contamination of the final lung cell population by cells from blood. The tissues were suspended in RPMI 1640 (HyClone Laboratories) medium containing 1 mg/ml type II collagenase (Worthington Biochemical Corp.) and incubated at 37°C for 30 min to permit digestion. The tissues were pressed through a 70-μm cell strainer (Becton Dickinson). Total lung cells were suspended in 40% isotonic Percoll solution (Amersham Biosciences), pelleted by centrifugation at 900 g for 20 min, and washed in the medium after hemolysis of the cells by suspension in lysing buffer (ACK; BioWhittaker Inc.). Cells gathered from the BLA fluid of five mice were used for analysis for each experimental condition because the absolute cell number per mouse was extremely small. The lung cells and BAL fluid cells were stained with CD3 and T1/ST2 and analyzed by flow cytometry. Phenotypic analysis of lung cells was performed with the use of PE-conjugated anti-CD3 (clone 145–2C11) and FITC-conjugated anti–mouse T1/ST2 (clone 3E10; Millennium Pharmaceuticals Inc.). All antibodies, except anti-T1/ST2, were purchased from BD Biosciences. As a control, cells incubated with an isotype-matched, directly conjugated antibody with irrelevant specificity were used. After incubation for 30 min at 4°C, the cells were washed, and immunofluorescence analysis was performed on a FACSCalibur™ flow cytometer (Becton Dickinson). The results were analyzed with CELLQuest™ software (Becton Dickinson).
Immunocytochemistry.
Paraffin sections of lung tissue were deparaffinized and hydrated by submersion in xylene followed by reagent-grade alcohol. The sections were rinsed for 5 min and incubated with 0.3% H2O2 for 30 min to quench endogenous peroxidase activity. After washing three times in Trizma buffer solution for 15 min, the sections were incubated with goat-anti–mouse MDC or TARC antibody or an isotype control antibody overnight. Next, the sections were washed three times in Trizma buffer solution for 15 min, and a rabbit anti–goat IgG secondary antibody was applied for 30 min. After washing, the sections were incubated with streptavidin peroxidase reagent for 30 min. The sections were washed again and stained with peroxidase substrate solution until the desired intensity was reached. After rinsing in running water, the sections were counterstained with hematoxylin. The used reagents were derived from commercially available streptavidin-biotin kits (DakoCytomation).
Culture of Primary Human Bronchial Epithelial Cells.
In these studies, we used primary human bronchial epithelial cells (Normal Human Cell System; Sanko Junyaku). Cells were maintained in bronchial epithelial growth media (SABM; Sanko Junyaku) and supplemented with 7.5 g/liter of bovine pituitary extract, 0.5 g/liter hydrocortisone, 0.5 mg/l of human recombinant epidermal growth factor, 0.5 g/liter epinephrine, 10 g/liter transferrin, 5 g/liter insulin, 0.1 mg/l retinoic acid, 6.5 mg/l 3,3′,5-triiodo-l-thryonine, 50 g/liter bovine serum albumin, and 0.1% GA-1000 (Sanko Junyaku) in 25-cm2 tissue plates at 37°C in 5% CO2. Media were replaced every other day. Cells reached confluency in 9–9.5 d, and nearly confluent cells were subcultured using trypsin-EDTA at a ratio of 1:8 at passage 3 for experimental treatment.
Preparation of mRNA and Reverse Transcriptase (RT)–PCR.
Total RNA was extracted from the primary human bronchial epithelial cells by Trisolv, a modified guanidine thiocyanate-phenol-chloroform method, as recommended by the manufacturer (Biotec Laboratories). RT-PCR was performed to determine the relative quantities of TARC and MDC mRNA, using a modified method as described previously (42). In brief, 1 μg RNA was reverse-transcribed using oligo (dT) primers; the cDNA underwent 30, 30, or 22 cycles of amplification with primers specific for TARC, MDC, and β-actin. The sequences of primers from the coding regions of human genes were as follows: MDC, 5′-TACAGACTGCACTCCTGGTTGTCC-3′ and 5′-TTCTGGCGGGGAGCAGCTATAATG-3′; TARC, 5′-CACGCAGCTCGAGGGACCAATGTG-3′ and 5′-TCAAGACCTCTCAAGGCTTTGCAGG-3′; β-actin, 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′ and 5′-CTAGAAGCATTGCGGTGGACGATGGAGGG-3′. PCR products in agarose gel were detected by ethidium bromide staining.
Stimulation of Primary Human Bronchial Epithelial Cells with PGD2.
To assess the effects of PGD2 on MDC and TARC gene expression, primary human bronchial epithelial cells were stimulated with various concentrations of PGD2 (10−9–10−5 M). Cultures were harvested after stimulation for 48 h and analyzed by RT-PCR.
Effect of Anti-MDC Antibody on PGD2-mediated Pulmonary Response.
For blocking experiments, intraperitoneally sensitized PGD2- or saline-treated mice were given 20 μg/mouse of neutralizing rabbit polyclonal antibody against murine MDC (anti-MDC antibody; R&D Systems). This antibody was administered i.v. 30 min before OVA challenge or saline. As control, OVA-challenged mice were given the same amount of rabbit Ig control Ig-G (DakoCytomation) by i.v. injection (Fig. 1, method 3). In these mice, AHR was assessed 24 h after challenge.
Determination of AHR after Allergen Challenge.
AHR to inhaled methacholine in conscious, spontaneously breathing mice was measured by barometric whole body plethysmography (Buxco Electronics) as described previously (31, 43–45). Mice were placed in the main chamber, and baseline readings were obtained and averaged for 3 min. Saline or increasing concentrations of 3–25 mg/ml methacholine was aerosolized through an inlet of the main chamber for 3 min, and readings were taken and averaged for 3 min after each challenge. Airway reactivity was expressed as enhanced pause (Penh) values for each concentration of methacholine.
Measurement of Cytokines and Chemokines.
Concentrations of cytokines (IL-2, IL-4, IL-5, IL-13, and IFN-γ) and chemokines (regulated upon activation normal T cell expressed and secreted [RANTES], eotaxin, and monocyte chemotactic protein [MCP]–1) in culture supernatants and BAL fluid supernatants were measured by ELISA kits (R&D Systems) according to the manufacturer's instructions.
Statistical Analysis.
Data are expressed as means ± SEM. The statistical significance of differences between groups was assessed by analysis of variance. P values of <0.05 were considered to indicate statistical significance.
Results
Description of the Model System.
Compared with the antigen doses usually used for challenge (31, 39), much smaller amounts of OVA were given to BALB/c mice to determine the antigen challenge required to optimally highlight the effect of PGD2 inhalation on pulmonary inflammation (Fig. 1, method 1). In preliminary studies, we found that 0.1% OVA inhalation for 30 min, instead of two rounds of inhalation of 1% OVA for 60 min (as used in conventional protocols), induced only marginal inflammation in the lung (unpublished data). We also confirmed that total cell numbers and cell populations in BAL fluid of sensitized mice were not altered 24 h after provocation with PGD2 alone (Fig. 2 A, 0 h) when no challenging antigens were given. This finding indicated that PGD2 alone had no chemoattractive effect in the lung. In contrast, 3 h after low-dose antigen (0.1% OVA) inhalation, total cell numbers in BAL fluid were significantly higher in PGD2-pretreated mice than in saline-pretreated control mice (Fig. 2 A). The infiltrated cells consisted predominantly of eosinophils, lymphocytes, and macrophages, with few neutrophils. Further increases in each cell type were seen 24 h after low-dose antigen challenge, as compared with the control (Fig. 2 B). The numbers of eosinophils and lymphocytes in BAL fluid did not differ significantly between the conventional asthma model challenged with 1% OVA and the PGD2-pretreated mice challenged with 0.1% OVA 24 h after antigen challenge. In contrast, the number of macrophages in the conventional asthma model was significantly greater than that in the PGD2-pretreated mice (Fig. 2 B). The results of histological examinations were consistent with the findings in BAL fluid (unpublished data). These data suggested that pretreatment with PGD2 augmented low-dose antigen-induced Th2 type pulmonary inflammation involving mainly eosinophils and lymphocytes.
Figure 2.

Effects of PGD2 on infiltration of inflammatory cells in BAL fluid. The number of inflammatory cells in BAL fluid was determined 0, 3 (A), and 24 h (B) after the last challenge on experimental day 17, as described in Materials and Methods. Data represent means ± SEM per group (n = 6). Differences were considered significant if P < 0.05. (A) *, P < 0.05, between PGD2-pretreated low-dose 0.1% OVA-challenged groups (black bars) and saline-pretreated, low-dose OVA-challenged groupsgroups (white bars). (B) *, P < 0.05, between PGD2-pretreated, low-dose 0.1% OVA-challenged groups (black bars) and saline-pretreated, low-dose OVA-challenged groups (white bars). Gray bars represent data obtained with the conventional asthma model. Groups of mice pretreated with PGD2 but challenged with saline are represented by diagonally striped bars. N.S., not significant; N.D., not detected.
Effect of PGD2 on Cytokine and Chemokine Concentrations in BAL Fluid.
First, the expression of the chemokines eotaxin, RANTES, and MCP-1 were measured in BAL fluid obtained from sensitized animals after OVA challenge (Fig. 1, methods 1 and 2). None of these chemokines were detected immediately before or 3 h after OVA challenge in either PGD2-pretreated animals or in conventional asthma models (unpublished data). Although increased levels of all three chemokines were found in conventional asthma models 24 h after 1% OVA challenge, only eotaxin was increased by low-dose OVA (0.1%) challenge to a level similar to that in conventional asthma models (Fig. 3) . We also analyzed Th1 (IL-2 and IFN-γ) and Th2 (IL-4 and IL-5) cytokines in BAL fluid. No Th1 cytokines were detected in any model, consistent with the exclusively Th2-dependent nature of these models. Moreover, there was no significant difference in the levels of Th2 cytokines in BAL fluid 24 h (Fig. 4 A, 0 h before OVA challenge) after PGD2 pretreatment. This result indicated that PGD2 alone had no significant effect on the induction of Th2 cytokines or on cell infiltration in the lung. Notably, 3 h after low-dose antigen (0.1% OVA) inhalation, Th2 cytokine levels were significantly higher in PGD2-pretreated mice than in the saline-pretreated control mice (Fig. 4 A). IL-5 levels were elevated similarly in PGD2-treated mice and in conventional asthma models 24 h after OVA challenge, whereas the IL-4 level was slightly lower in the former (Fig. 4 B). The levels of Th2 cytokines increased 24 h after challenge, as compared with those 3 h after challenge; no such increase was observed in saline-treated animals (Fig. 4 B). These results suggested that PGD2 pretreatment could lead to intrapulmonary accumulation of Th2 cytokines in sensitized animals exposed to low-dose antigen.
Figure 3.
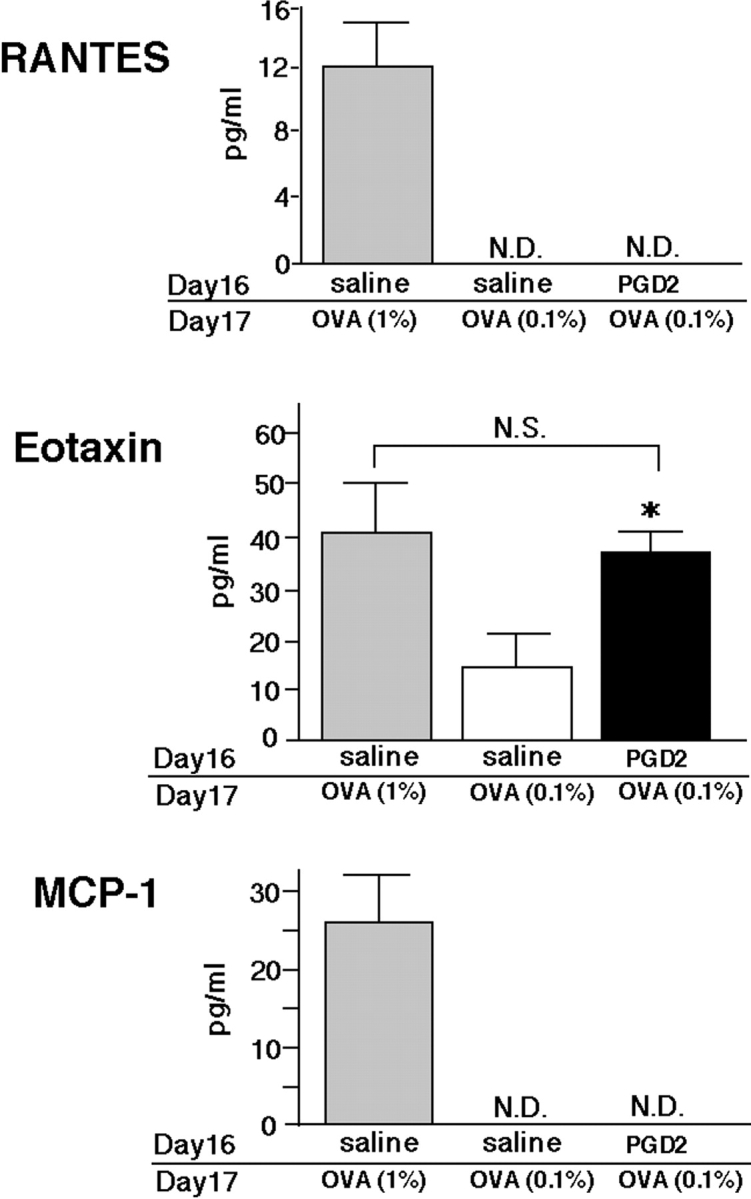
Effects of PGD2 on chemokine concentrations in BAL fluid. RANTES, eotaxin, and MCP-1 concentrations in BAL fluid were assessed 24 h after the last challenge. Gray and black bars represent the conventional asthma model groups and the PGD2-pretreated, low-dose OVA-challenged groups, respectively. Data are expressed as means ± SEM per group (n = 6). *, Significant differences (P < 0.05) between PGD2-pretreated, low-dose OVA-challenged groups (black bars) and saline-pretreated, low-dose OVA-challenged groups (white bars). N.S., not significant; N.D., not detected.
Figure 4.

Effects of PGD2 on Th1 (IL-2 and IFN-γ) and Th2 (IL-4 and IL-5) cytokine production in BAL fluid. Cytokine levels in BAL fluid were determined 0, 3 (A), and 24 h (B) after the last challenge on experimental day 17, as described in Materials and Methods. (A) Data are expressed as means ± SEM per group (n = 6). *, P < 0.05, between PGD2-pretreated, low-dose OVA-challenged (black bars) and saline-pretreated, low-dose OVA-challenged groups (white bars). (B) Gray bars represent the conventional asthma model groups. Data are expressed as means ± SEM per group (n = 6). *, P < 0.05, between PGD2-pretreated, low-dose OVA-challenged groups (black bars) and saline-pretreated, low-dose OVA-challenged groups (white bars) or PGD2-pretreated, saline-challenged groups (hatched bars). N.S., not significant difference. N.D., not detectable.
Effects of PGD2 on Cytokine Production by OVA-specific T Cells and BMMCs In Vitro.
To determine whether PGD2 stimulation directly influences Th2 cytokine production, we analyzed ovalbumin-specific T cell response in vitro. Addition of PGD2 did not affect ovalbumin-induced Th2 cytokine production by spleen T cells (Fig. 5) . In addition, PGD2 did not affect ionomycin-induced Th2 cytokine production by bone marrow–derived mast cells (unpublished data). These results indicate that PGD2 does not directly up-regulate the production of Th2 cytokines and suggest that PGD2 is involved in the recruitment of activated Th2 cells into the lung after antigen exposure.
Figure 5.

Effect of PGD2 on cytokine production by OVA-specific T cells in vitro. Spleen cells from sensitized and challenged mice were cultured for 24 h with 10−9–10−5 M PGD2 during 10 μg/ml OVA stimulation. Data are expressed as means ± SEM. These results are representative of three independent experiments.
Effects of PGD2 on the Expression of MDC mRNA and Protein In Vivo and In Vitro.
Next, we examined the role of MDC or TARC in Th2 cell recruitment into the lung in terms of the action of PGD2. First, we observed that MDC mRNA was expressed in primary human bronchial epithelial cells in a dose-dependent manner in response to 10−9–10−5 M PGD2 after a 48-h incubation (Fig. 6) . In contrast, TARC mRNA was constitutively expressed and not influenced by PGD2. We analyzed MDC expression immunohistochemically 3 and 24 h after OVA challenge in our murine asthma model (Fig. 1, method 2). The PGD2-pretreated mice showed strong staining for MDC in bronchial epithelial cells from 3 (Fig. 7 A) to 24 h (not depicted) after antigen (0.1% OVA) challenge. In the saline-pretreated mice, however, no MDC was detected after antigen (0.1% OVA) challenge (Fig. 7 B). When PGD2-pretreated mice inhaled saline, MDC staining was much less prominent as compared with that after antigen challenge (Fig. 7 C). No MDC staining was observed in saline-pretreated, saline-challenged control mice (Fig. 7 D).
Figure 6.

Effects of PGD2 on expression of MDC mRNA in vitro. MDC mRNA was expressed in primary human bronchial epithelial cells in a dose-dependent manner to 10−9–10−5 M PGD2 after 48-h incubation, whereas TARC mRNA was constitutively expressed and not influenced by PGD2. These results are representative of three independent experiments.
Figure 7.

Effects of PGD2 on expression of MDC protein in vivo. Lung sections were obtained from PGD2-pretreated, low-dose OVA-challenged group after 3 h (A), saline-pretreated, low-dose OVA-challenged group after 3 h (B), PGD2-pretreated, saline-inhalation group after 3 h (C), and saline-pretreated, saline-inhalation group after 3 h (D). The sections were stained with polyclonal antibodies against mouse MDC (original magnification, 200).
Inhibition of Th2 Cytokine Production and Recruitment of Inflammatory Cells by Anti-MDC Antibody in PGD2-treated Mice.
To examine if MDC expressed in the lung had a functional role in our asthma model, we injected anti-MDC antibody into the mice just before challenge (Fig. 1, method 3). As compared with control IgG, anti-MDC antibody significantly inhibited the accumulation of eosinophils, lymphocytes, and macrophages in BAL fluid in PGD2-pretreated mice 24 h after OVA challenge (Fig. 8 A). The levels of IL-4 and IL-5 in BAL fluid were also significantly reduced by treatment with anti-MDC antibody in the PGD2-pretreated mice (Fig. 8 B).
Figure 8.

Inhibition of Th2 cytokines and recruitment of inflammatory cells by anti-MDC antibody in PGD2-treated mice. The numbers of inflammatory cells (A) and levels of cytokines (B) in BAL fluid were determined 24 h after low-dose OVA challenge on experimental day 17, as described in Materials and Methods. Saline-pretreated, OVA-challenged groups are represented by gray bars. Data represent means ± SEM per group (n = 5). *, Significant differences (P < 0.05) between the PGD2-pretreated, OVA-challenged control-IgG treatment groups (white bars) and PGD2-pretreated, OVA-challenged, anti-MDC treatment groups (black bars).
Detection of Pulmonary T1/ST2+ T Cells in Sensitized Animals.
FACS® analysis revealed similar increases in accumulations of total CD3+ T cells and T1/ST2+ CD3+ T cells in the lungs of all sensitized mice, irrespective of antigen challenge or PGD2 exposure (Fig. 1, method 2; and Fig. 9 A). In contrast, the percentage of T1/ST2+CD3+ cells (Th2 cells) within CD3+ T cells increased sixfold, and accordingly, the absolute number of T1/ST2+CD3+ T cells increased threefold 24 h after OVA challenge in PGD2-pretreated mice, as compared with the respective values in saline-treated mice (Fig. 9 B). The migration of Th2 cells into the lung was strikingly inhibited by anti-MDC antibody treatment (Fig. 9 B). These results indicate that migration of Th2 cells from peripheral vessels to airway lumens in response to MDC expressed in bronchial epithelial cells resulted in increased accumulations of lymphocytes, including Th2 cells, and increased Th2 cytokine production in the lung.
Figure 9.

Detection of pulmonary T1/ST2+ CD3+ T cells. (A) The numbers of T1/ST2+ CD3+ T cells and CD3+ T cells in pulmonary tissues were determined by FACS® analysis 0, 3, and 24 h after low-dose OVA challenge. White and black bars represent the nonimmunized groups and immunized groups, respectively. Data represent means ± SEM per group (n = 5). *, P < 0.05, between nonimmunized groups and each immunized group. (B) The numbers of T1/ST2+ CD3+ T cells and CD3+ T cells in BAL fluid were determined 24 h after OVA challenge. Pretreatment with PGD2 followed by control-IgG treatment and anti-MDC antibody treatment are shown by white bars and black bars, respectively. Gray bars represent saline exposure without the antibody. These results are representative of three independent experiments.
Inhibition of Allergen-induced AHR by Treatment with Anti-MDC Antibody.
AHR, a prominent pathological alteration in murine models of asthma, was evaluated 24 h after OVA challenge (Fig. 1, method 3; and Fig. 10) . All PGD2-pretreated, OVA-challenged mice were more sensitive than saline-pretreated and saline-challenged mice in terms of AHR induction. However, AHR in PGD2-pretreated, OVA-challenged mice treated with anti-MDC antibody was significantly lower than that in control Ig–treated mice. The rate of change in Penh with increasing doses of methacholine was significantly lower after treatment with anti-MDC antibody than after treatment with control IgG. This finding implies that anti-MDC antibody significantly inhibited the development of AHR in PGD2-pretreated, OVA-challenged mice.
Figure 10.

Inhibition of PGD2 pretreatment and low-dose allergen-induced AHR by administration of anti-MDC antibody. Mice were first exposed to nebulized saline, followed by increasing doses (3–25 mg/ml) of nebulized methacholine for 3 min each. Breathing indices were read for 3 min after each nebulization, and Penh values were determined. AHR was analyzed 24 h after low-dose OVA challenge in each group. Data represent means ± SEM per group (n = 5). *, Significant differences (P < 0.05) between the PGD2-pretreated, OVA-challenged, control-IgG treatment groups (closed squares) and PGD2-pretreated, OVA-challenged, anti-MDC treatment groups (closed circles). Saline-pretreated, saline-challenged, control-IgG treatment groups are represented by open circles.
Discussion
We have described a new mouse model of asthma in which treatment with exogenous PGD2 was necessary for the development of marked pulmonary inflammation and accelerated AHR. This novel model of asthma was specifically established to analyze the mechanism by which PGD2 contributes to the pathogenesis of asthma. Antigens used for both sensitization and challenge were carefully titrated, and the optimal timing of PGD2 pretreatment was examined.
PGD2 is a major prostanoid produced by mast cells; however, its role in the pathogenesis of asthma has remained unclear. PGD2, its metabolite 15d-PGJ2, or both can inhibit production of IL-12, a cytokine involved in Th1 cell differentiation, by macrophages and dendritic cells (46, 47). Therefore, PGD2 as such or its metabolites may function to skew the Th2 cell differentiation of naive T cells (5, 6). However, we observed no effect of PGD2 on cytokine production by antigen-activated T cells obtained from the spleen of sensitized mice, indicating that PGD2 did not affect Th2 cytokine production by already-differentiated T cells. Hence, the Th1-inhibiting activity of PGD2, if any, did not contribute significantly to the PGD2-induced augmentation of Th2 type inflammation. In addition, because the numbers of mast cells in pulmonary tissue were not altered by PGD2 treatment, and PGD2 had no effect on Th2 cytokine production by BMMCs in vitro (unpublished data), this PGD2-promoted Th2 type inflammation apparently did not involve mast cells.
Vasodilation and increased vascular permeability are well-known effects of PGD2. In allergic situations, released PGD2 may facilitate transendothelial migration of inflammatory cells, such as eosinophils, mast cells, lymphocytes, and monocytes, possibly via DP receptor–mediated vasodilation-extravasation (48–51). On the other hand, recent evidence suggests that PGD2 directly attracts Th2 cells into the lung (37). In addition, a G protein–coupled, seven-transmembrane–type receptor, CRTH2, which is preferentially expressed in Th2 cells, eosinophils, and basophils (52), has also been shown to contribute to the PGD2-mediated chemotaxis of these cells (53). In this work, we found increased numbers of eosinophils, lymphocytes, and macrophages in BAL fluid from PGD2-pretreated, low-dose OVA-challenged mice in a manner dependent on PGD2 pretreatment. This Th2-skewed inflammation might result from a direct chemotactic effect of PGD2 on Th2 cells and eosinophils via DP receptor, CRTH2 receptor, or both. However, we found no obvious inflammatory changes or increases in Th2 cytokines after treatment with 10−5 M PGD2 alone, without antigen challenge. These results suggested that PGD2 by itself has no chemotactic activity, but induces chemoattractants for antigen-activated cells, an idea that clearly supports the indirect recruitment of Th2 cells by PGD2.
The CCR3 receptor ligand eotaxin is known to act on Th2 cells, eosinophils, and mast cells (54). In our analysis, eotaxin was detected in BAL fluid from PGD2-pretreated, OVA-challenged mice. Nonetheless, eotaxin, a chemokine produced in response to Th2 cytokines such as IL-4 and -13 (55), was most likely not induced by PGD2 but produced secondarily in response to massive infiltration of Th2 cells. This assumption is supported by the absence of eotaxin in BAL fluid from sensitized mice treated with PGD2 alone. We observed the appearance of OVA-specific T cells and increased numbers of T1/ST2+CD3+ T cells as Th2 cells in the spleen 12 d after the final intraperitoneal immunization with OVA (unpublished data). Interestingly, at this time point T1/ST2+CD3+ T cells were also observed in lung tissue, even though the mice had not received PGD2 and showed no evidence of inflammation in the lung. This phenomenon is consistent with the results of previous papers showing that after immunization memory T cells are retained in peripheral tissues, including lung (56–58). Although the number of intrapulmonary T1/ST2+CD3+ cells did not change, even after PGD2 pretreatment and antigen challenge, the number of T1/ST2+ CD3+ T cells in BAL fluid increased significantly after PGD2 pretreatment and antigen challenge. This increase in T1/ST2+CD3+ T cells in BAL fluid was strongly inhibited by anti-MDC antibody. These results suggest that antigen-activated Th2 cells, i.e., T1/ST2+ CD3+ T cells, which had circulated in pulmonary tissue after sensitization, migrated into the airway lumen in response to MDC induced locally by PGD2. Because PGD2 alone in naive mice caused neither tissue inflammatory changes nor Th2 cytokine production, antigen-driven activation of T cells or their distribution (or both) in pulmonary tissue is considered prerequisite for PGD2-promoted Th2 type pulmonary inflammation. The identification of MDC as a downstream mediator of PGD2 in this paper suggests that one of the roles of antigen-induced T cell activation may be to induce receptor(s) for chemokine(s).
We found that PGD2 directly induced MDC mRNA, but not TARC mRNA, in bronchial epithelial cells in vitro as well as in MDC proteins in vivo in an asthma model. The fact that MDC protein in epithelium was most conspicuous in the lung suggested that bronchial epithelial cell–derived MDC mediates the effect of PGD2 on the infiltration of Th2 type cells, leading to the development of Th2 type inflammation. In fact, neutralization of MDC in vivo with anti-MDC antibody treatment inhibited not only the PGD2-promoted transepithelial migration of eosinophils, Th2 cells, and macrophages but also the associated Th2 cytokine production and AHR. In line with our conclusion, the importance of MDC has been pointed out by Gonzalo et al., who reported that anti-MDC antibody potently inhibits the development of asthma in another murine model (59). MDC acts on its target cells through CCR4 (60). Thus, the finding that CCR4 deficiency does not protect against asthma (61) seems to conflict with our conclusion. However, CCR4 may not be the sole receptor for MDC (62). In fact, TARC, a chemokine that also binds to CCR4, may also serve as a ligand for CCR8 (63), although this remains controversial (64, 65). Perhaps MDC may also bind to CCR8 or to receptors other than CCR4. However, works using CCR8 mutant animals or antibodies against TCA-3, a murine high affinity CCR8 ligand, do not support a role of CCR8 ligands in models of asthma (66, 67), despite initial expectations (68). One can still argue that CCR4, its ligands, or both might compensate for CCR8 and vice versa, a prediction that can only be verified by studies in mice lacking both CCR4 and CCR8. Perhaps receptors other than CCR4 and CCR8 transduce MDC signals. Only further analysis can conclusively resolve these points of controversy. Apart from the specific receptor for MDC and its role in asthma, however, our results and those of Gonzalo et al. (59) clearly indicate a nonredundant function of MDC in Th2 type pulmonary inflammation and suggest that MDC may be a therapeutic target for the management of asthma in humans.
Importantly, our work revealed that MDC serves as a downstream effector molecule of PGD2 in Th2 type inflammation, a previously unknown finding. Identification of such a molecular link between PGD2 and Th2 recruitment unequivocally demonstrates that PGD2 acts indirectly. Furthermore, this link suggests that the infiltration of Th2 cells, at least during the early phase of inflammation, and that of other inflammatory cells occur through distinct mechanisms. Thus, Th2 cells actively transmigrate through endothelium in response to PGD2-induced epithelial MDC. The recruited Th2 cells induce other chemokines, such as eotaxin, in situ that attract other inflammatory cells. Vasodilatation and increased vascular permeability in response to PGD2 (69) would facilitate both of these mechanisms. Finally, our results showing that PGD2 has a role in the induction of MDC but not in that of its related chemokine TARC suggest that future work should examine how PGD2 differentially regulates the transcription of these chemokines. The results of such work may provide important clues suggesting how regulation of MDC transcription could be used for the treatment of asthma in humans.
Acknowledgments
We wish to thank Dr. T. Coyle for anti-T1/ST2 antibody.
This work was supported in part by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan.
Abbreviations used in this paper: AHR, airway hyperresponsiveness; BAL, bronchoalveolar lavage; BMMC, bone marrow mast cell; CCR, CC chemokine receptor; MCP, monocyte chemotactic protein; MDC, macrophage-derived chemokine; PGD2, prostaglandin D2; RANTES, regulated upon activation normal T cell expressed and secreted; RT, reverse transcriptase; TARC, thymus- and activation-regulated chemokine.
K. Honda and M. Arima contributed equally to this work.
References
- 1.Till, S., R. Dickason, D. Huston, M. Humbert, D. Robinson, M. Larche, S. Durham, A.B. Kay, and C. Corrigan. 1997. IL-5 secretion by allergen-stimulated CD4+ T cells in primary culture: relationship to expression of allergic disease. J. Allergy Clin. Immunol. 99:563–569. [DOI] [PubMed] [Google Scholar]
- 2.Corrigan, C.J., and A.B. Kay. 1990. CD4 T-lymphocyte activation in acute severe asthma. Relationship to disease severity and atopic status. Am. Rev. Respir. Dis. 141:970–977. [DOI] [PubMed] [Google Scholar]
- 3.Kuo, M.L., J.L. Huang, K.W. Yeh, P.S. Li, and K.H. Hsieh. 2001. Evaluation of Th1/Th2 ratio and cytokine production profile during acute exacerbation and convalescence in asthmatic children. Ann. Allergy Asthma Immunol. 86:272–276. [DOI] [PubMed] [Google Scholar]
- 4.Oshikawa, K., K. Kuroiwa, K. Tago, H. Iwahana, K. Yanagisawa, S. Ohno, S.I. Tominaga, and Y. Sugiyama. 2001. Elevated soluble ST2 protein levels in sera of patients with asthma with an acute exacerbation. Am. J. Respir. Crit. Care Med. 164:277–281. [DOI] [PubMed] [Google Scholar]
- 5.Abbas, A.K., K.M. Murphy, and A. Sher. 1996. Functional diversity of helper T lymphocytes. Nature. 383:787–793. [DOI] [PubMed] [Google Scholar]
- 6.Mosmann, T.R., and S. Sad. 1996. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today. 17:138–146. [DOI] [PubMed] [Google Scholar]
- 7.Townsend, M.J., P.G. Fallon, D.J. Matthews, H.E. Jolin, and A.N. McKenzle. 2000. T1/ST2 in developing primary T helper cell type 2 responses. J. Exp. Med. 191:1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda, K., T. Tanaka, W. Shi, M. Matsumoto, M. Minami, S. Kashiwamura, K. Nakanishi, N. Yoshida, T. Kishimoto, and S. Akira. 1996. Essential role of Stat6 in IL-4 signalling. Nature. 380:627–630. [DOI] [PubMed] [Google Scholar]
- 9.Bellini, A., E. Vittori, M. Marini, V. Ackerman, and S. Mattoli. 1993. Intraepithelial dendritic cells and selective activation of Th2-like lymphocytes in patients with atopic asthma. Chest. 103:997–1005. [DOI] [PubMed] [Google Scholar]
- 10.Del Prete, G.F., M. De Carli, M.M. D'Elios, P. Maestrelli, M. Ricci, L. Fabbri, and S. Romagnani. 1993. Allergen exposure induces the activation of allergen-specific Th2 cells in the airway mucosa of patients with allergic respiratory disorders. Eur. J. Immunol. 23:1445–1449. [DOI] [PubMed] [Google Scholar]
- 11.Hogg, J.C. 1997. The pathology of asthma. APMIS. 105:735–745. [DOI] [PubMed] [Google Scholar]
- 12.Humbert, M., S.R. Durham, S. Ying, P. Kimmitt, J. Barkans, B. Assoufi, R. Pfister, G. Menz, D.S. Robinson, A.B. Kay, and C.J. Corrigan. 1996. IL-4 and IL-5 mRNA and protein in bronchial biopsies from patients with atopic and nonatopic asthma: evidence against “intrinsic” asthma being a distinct immunopathologic entity. Am. J. Respir. Crit. Care Med. 154:1497–1504. [DOI] [PubMed] [Google Scholar]
- 13.Jaffar, Z.H., L. Stanciu, A. Pandit, J. Lordan, S.T. Holgate, and K. Roberts. 1999. Essential role for both CD80 and CD86 costimulation, but not CD40 interactions, in allergen-induced Th2 cytokine production from asthmatic bronchial tissue: role for alphabeta, but not gammadelta, T cells. J. Immunol. 163:6283–6291. [PubMed] [Google Scholar]
- 14.Huang, S.K., H.Q. Xiao, J. Kleine-Tebbe, G. Paciotti, D.G. Marsh, L.M. Lichtenstein, and M.C. Liu. 1995. IL-13 expression at the sites of allergen challenge in patients with asthma. J. Immunol. 155:2688–2694. [PubMed] [Google Scholar]
- 15.Prieto, J., C. Lensmar, A. Roquet, I. van der Ploeg, D. Gigliotti, A. Eklund, and J. Grunewald. 2000. Increased interleukin-13 mRNA expression in bronchoalveolar lavage cells of atopic patients with mild asthma after repeated low-dose allergen provocations. Respir. Med. 94:806–814. [DOI] [PubMed] [Google Scholar]
- 16.Robinson, D.S., S. Ying, A.M. Bentley, Q. Meng, J. North, S.R. Durham, A.B. Kay, and Q. Hamid. 1993. Relationships among numbers of bronchoalveolar lavage cells expressing messenger ribonucleic acid for cytokines, asthma symptoms, and airway methacholine responsiveness in atopic asthma. J. Allergy Clin. Immunol. 92:397–403. [DOI] [PubMed] [Google Scholar]
- 17.Robinson, D.S., Q. Hamid, S. Ying, A. Tsicopoulos, J. Barkans, A.M. Bentley, C. Corrigan, S.R. Durham, and A.B. Kay. 1992. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N. Engl. J. Med. 326:298–304. [DOI] [PubMed] [Google Scholar]
- 18.Robinson, D.S., S. Ying, A.M. Bentley, Q. Meng, J. North, S.R. Durham, A.B. Kay, and Q. Hamid. 1993. Relationships among numbers of bronchoalveolar lavage cells expressing messenger ribonucleic acid for cytokines, asthma symptoms, and airway methacholine responsiveness in atopic asthma. J. Allergy Clin. Immunol. 92:397–403. [DOI] [PubMed] [Google Scholar]
- 19.Walker, C., W. Bauer, R.K. Braun, G. Menz, P. Braun, F. Schwarz, T.T. Hansel, and B. Villiger. 1994. Activated T cells and cytokines in bronchoalveolar lavages from patients with various lung diseases associated with eosinophilia. Am. J. Respir. Crit. Care Med. 150:1038–1048. [DOI] [PubMed] [Google Scholar]
- 20.Ying, S., S.R. Durham, C.J. Corrigan, Q. Hamid, and A.B. Kay. 1995. Phenotype of cells expressing mRNA for TH2-type (interleukin 4 and interleukin 5) and TH1-type (interleukin 2 and interferon gamma) cytokines in bronchoalveolar lavage and bronchial biopsies from atopic asthmatic and normal control subjects. Am. J. Respir. Cell Mol. Biol. 12:477–487. [DOI] [PubMed] [Google Scholar]
- 21.Panina-Bordignon, P., A. Papi, M. Mariani, P. Di Lucia, G. Casoni, C. Bellettato, C. Buonsanti, D. Miotto, C. Mapp, A. Villa, et al. 2001. The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J. Clin. Invest. 107:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berin, M.C., L. Eckmann, D.H. Broide, and M.F. Kagnoff. 2001. Regulated production of the T helper 2-type T-cell chemoattractant TARC by human bronchial epithelial cells in vitro and in human lung xenografts. Am. J. Respir. Cell Mol. Biol. 24:382–389. [DOI] [PubMed] [Google Scholar]
- 23.Sekiya, T., M. Miyamasu, M. Imanishi, H. Yamada, T. Nakajima, M. Yamaguchi, T. Fujisawa, R. Pawankar, Y. Sano, K. Ohta, et al. 2000. Inducible expression of a Th2-type CC chemokine thymus- and activation-regulated chemokine by human bronchial epithelial cells. J. Immunol. 165:2205–2213. [DOI] [PubMed] [Google Scholar]
- 24.Bonecchi, R., G. Bianchi, P.P. Borignon, D. D'Ambrosio, R. Lang, A. Borsatti, S. Sozzani, P. Allavena, P.A. Gray, A. Mantovani, and F. Sinigaglia. 1998. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 187:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang, M.S., J. McNinch, C. Elias, C.L. Manthey, D. Grosshans, T. Meng, T. Boone, and D.P. Andrew. 1997. Molecular cloning and functional characterization of a novel CC chemokine, stimulated T cell chemotactic protein (STCP-1) that specifically acts on activated T lymphocytes. J. Biol. Chem. 272:25229–25237. [DOI] [PubMed] [Google Scholar]
- 26.Andrew, D.P., M.S. Chang, J. McNinch, S.T. Wathen, M. Rihanek, J. Tseng, J.P. Spellberg, and C.G. Elias III. 1998. STCP-1 (MDC) CC chemokine acts specifically on chronically activated Th2 lymphocytes and is produced by monocytes on stimulation with Th2 cytekines IL-4 and IL-13. J. Immunol. 161:5027–5038. [PubMed] [Google Scholar]
- 27.Imai, T., M. Nagira, S. Takagi, M. Kakizaki, M. Nishimura, J. Wang, P.W. Gray, K. Matsushima, and O. Yoshie. 1999. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int. Immunol. 11:81–88. [DOI] [PubMed] [Google Scholar]
- 28.Sekiya, T., H. Yamada, M. Yamaguchi, K. Yamamoto, A. Ishii, O. Yoshie, Y. Sano, A. Morita, K. Matsushima, and K. Hirai. 2002. Levels of a TH2-type CC chemokine thymus and activation-regulated chemokine (TARC) in serum and induced sputum of asthmatics. Allergy. 57:173–177. [DOI] [PubMed] [Google Scholar]
- 29.Romagnani, S. 2002. Cytokines and chemoattractants in allergic inflammation. Mol. Immunol. 38:881–885. [DOI] [PubMed] [Google Scholar]
- 30.Kurashima, K., M. Fujimura, S. Myou, K. Kasahara, H. Tachibana, N. Amemiya, Y. Ishiura, N. Onai, K. Matsushima, and S. Nakao. 2001. Effects of oral steroids on blood CXCR3+ and CCR4+ T cells in patients with bronchial asthma. Am. J. Respir. Crit. Care Med. 164:754–758. [DOI] [PubMed] [Google Scholar]
- 31.Cheng, G., M. Arima, K. Honda, H. Hirata, F. Eda, N. Yoshida, F. Fukushima, Y. Ishii, and T. Fukuda. 2002. Anti-interleukin-9 antibody treatment inhibits airway inflammation and hyperreactivity in mouse asthma model. Am. J. Respir. Crit. Care Med. 166:409–416. [DOI] [PubMed] [Google Scholar]
- 32.Lewis, R.A., N.A. Soter, P.T. Diamond, K.F. Austen, J.A. Oates, and L.J. Roberts. 1982. Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J. Immunol. 129:1627–1631. [PubMed] [Google Scholar]
- 33.Holgate, S.T., G.B. Burns, C. Robinson, and M.K. Church. 1984. Anaphylactic- and calcium-dependent generation of prostaglandin D2 (PGD2), thromboxane B2, and other cyclooxygenase products of arachidonic acid by dispersed human lung cells and relationship to histamine release. J. Immunol. 133:2138–2144. [PubMed] [Google Scholar]
- 34.Gundel, R.H., P. Kinkade, C.A. Torcellini, C.C. Clarke, J. Watrous, S. Desai, C.A. Homon, P.R. Farina, and C.D. Wegner. 1991. Antigen-induced mediator release in primates. Am. Rev. Respir. Dis. 144:76–82. [DOI] [PubMed] [Google Scholar]
- 35.Miadonna, A., A. Tedeschi, C. Brasca, G. Folco, A. Sala, and R.C. Murphy. 1990. Mediator release after endobronchial antigen challenge in patients with respiratory allergy. Allergy Clin. Immunol. 85:906–913. [DOI] [PubMed] [Google Scholar]
- 36.Turner, N.C., R.W. Fuller, and D.M. Jackson. 1995. Eicosanoid release in allergen-induced bronchoconstriction in dogs. Its relationship to airways hyperreactivity and pulmonary inflammation. J. Lipid Mediat. Cell Signal. 11:93–102. [DOI] [PubMed] [Google Scholar]
- 37.Matsuoka, T., M. Hirata, H. Tanaka, Y. Takahashi, T. Murata, K. Kabashima, Y. Sugimoto, T. Kobayashi, F. Usikubi, Y. Aze, et al. 2000. Prostaglandin D2 as a mediator of allergic asthma. Science. 287:2013–2017. [DOI] [PubMed] [Google Scholar]
- 38.Fujitani, Y., T. Kanaoka, K. Aritake, N. Uodome, K. Okazaki-Hatake, and Y. Urade. 2002. Pronounced eosinophilic lung inflammation and Th2 cytokine release in human lipcalin-type prostaglandin D synthase transgenic mice. J. Immunol. 168:443–449. [DOI] [PubMed] [Google Scholar]
- 39.Ohkawara, Y., X.F. Lei, M.R. Stampfli, J.S. Marshall, Z. Xing, and M. Jordana. 1997. Cytokine and eosinophil responses in the lung, peripheral blood, and bone marrow compartments in a murine model of allergen-induced airway inflammation. Am. J. Respir. Cell Mol. Biol. 16:510–520. [DOI] [PubMed] [Google Scholar]
- 40.Fukuda, T., T. Yoshida, S. Okada, M. Hatano, T. Miki, K. Ishibashi, S. Okabe, H. Koseki, S. Hirosawa, M. Taniguchi, et al. 1997. Disruption of the Bcl6 gene results in an impaired germinal center formation. Oncogene. 186:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stassen, M., M. Arnold, L. Hultner, C. Muller, C. Neudorfl, T. Reineke, and E. Schmitt. 2000. Murine bone marrow-derived mast cells as potent producers of IL-9: costimulatory function of IL-10 and kit ligand in the presence of IL-1. J. Immunol. 164:5549–5555. [DOI] [PubMed] [Google Scholar]
- 42.Arima, M., J. Plitt, C. Stellato, C. Bickel, S. Motojima, S. Makino, T. Fukuda, and R.P. Schleimer. 1999. Expression of interleukin-16 by human epithelial cells. Inhibition by dexamethasone. Am. J. Respir. Cell Mol. Biol. 21:684–692. [DOI] [PubMed] [Google Scholar]
- 43.Jacky, J.P. 1978. A plethysmograph for long-term measurement of ventilation in unrestrained animals. J. Appl. Physiol. 45:644–647. [DOI] [PubMed] [Google Scholar]
- 44.Hamelmann, E., J. Schwartz, K. Takeda, A. Oshiba, G.L. Larsen, C.G. Irvin, and E.W. Gelfand. 1997. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care Med. 156:766–775. [DOI] [PubMed] [Google Scholar]
- 45.Lee, J.J., M.P. McGarry, S.C. Farmer, K.L. Denzler, K.A. Larson, P.E. Carrigan, I.E. Brennesise, M.A. Horton, A. Haczku, E.W. Gelfand, and N.A. Lee. 1997. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathogenic of asthma. J. Exp. Med. 185:2143–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Azuma, Y., M. Shinohara, P.L. Wang, and K. Ohura. 2001. 15-Deoxy-delta (12,14)-prostaglandin J (2) inhibits IL-10 and IL-12 production by macrophages. Biochem. Biophys. Res. Commun. 283:344–346. [DOI] [PubMed] [Google Scholar]
- 47.Fougeray, V.A., J. Fontaine, G. Chinetti, P. Gosset, P. Delerive, C. Maliszewski, M. Capron, B. Staels, M. Moser, and F. Trottein. 2000. Peroxisome proliferator-activated receptor γ activators inhibit interleukin-12 production in murine dendritic cells. FEBS Lett. 486:261–266. [DOI] [PubMed] [Google Scholar]
- 48.Beasley, C.R., C. Robinson, R.L. Featherstone, J.G. Varley, C.C. Hardy, M.K. Church, and S.T. Holgate. 1987. 9α, 11βPprostaglandin F2, a novel metabolite of prostaglandin D2, is a potent contractile agonist of human and guinea pig airways. J. Clin. Invest. 79:978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emery, D.L., T.D. Djokic, D.F. Graf, and J.A. Nadel. 1989. Prostaglandin D2 causes accumulation of eosinophils in the lumen of the dog trachea. J. Appl. Physiol. 67:959–962. [DOI] [PubMed] [Google Scholar]
- 50.Pons, F., T.J. Williams, S.A. Kirk, F. McDonald, and A.G. Rossi. 1994. Pro-inflammatory and anti-inflammatory effects of the stable prostaglandin D2 analogue, ZK 118.182. Eur. J. Pharmacol. 261:237–247. [DOI] [PubMed] [Google Scholar]
- 51.Sampson, S.E., A.P. Sampson, and J.F. Costello. 1997. Effect of inhaled prostaglandin D2 in normal and atopic subjects, and of pretreatment with leukotriene D4. Thorax. 52:513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagata, K., H. Hirai, K. Tanaka, K. Ogawa, T. Aso, K. Suganuma, M. Nakamura, and S. Takano. 1999. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Lett. 459:195–199. [DOI] [PubMed] [Google Scholar]
- 53.Hirai, H., K. Tanaka, O. Yoshie, K. Ogawa, K. Kenmotsu, Y. Takamori, M. Ichimasa, K. Sugamura, M. Nakamura, S. Takano, and K. Nagata. 2001. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 193:255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romagnani, S. 2002. Cytokines and chemoattractants in allergic inflammation. Mol. Immunol. 38:881–885. [DOI] [PubMed] [Google Scholar]
- 55.Matsukura, S., C. Stellato, J.R. Plitt, C. Bickel, K. Miura, S.N. Georas, V. Casolaro, and R.P. Schleimer. 1999. Activation of eotaxin gene transcription by NF-kappa B and STAT6 in human airway epithelial cells. J. Immunol. 163:6876–6883. [PubMed] [Google Scholar]
- 56.Masopust, D., V. Vezys, A.L. Marzo, and L. Lefrançois. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 5512:2413–2417. [DOI] [PubMed] [Google Scholar]
- 57.Leereinhardt, R., A. Khoruts, R. Merica, T. Zell, and M. Jenkins. 2001. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 410:101–105. [DOI] [PubMed] [Google Scholar]
- 58.Usherwood, E.J. 2002. A new approach to epitope confirmation by sampling effector/memory T cells migrating to the lung. J. Immunol. Methods. 266:135–142. [DOI] [PubMed] [Google Scholar]
- 59.Gonzalo, J.A., Y. Pan, C.M. Lloyd, G.Q. Jia, G. Yu, B. Dussault, C.A. Powers, A.E.I. Proudfoot, A.J. Coyle, D. Gearing, and J.C. Gutierrez-Ramos. 1999. Mouse monocyte-derived chemokine is involved in airway hyperreactivity and lung inflammation. J. Immunol. 163:403–411. [PubMed] [Google Scholar]
- 60.Imai, T., D. Chantry, C.J. Raport, C.L. Wood, M. Nishimura, R. Godiska, O. Yoshie, and P.W. Gray. 1998. Macrophage-derived chemokine is a functional ligand for the CC chemokine receptor 4. J. Biol. Chem. 273:1764–1768. [DOI] [PubMed] [Google Scholar]
- 61.Chvatchko, Y., A.J. Hoogewerf, A. Meyer, S. Alouani, P. Juillard, R. Buser, F. Conquet, A.E. Proudfoot, T.N. Wells, and C.A. Power. 2000. A key role for CC chemokine receptor 4 in lipopolysaccharide-induced endotoxic shock. J. Exp. Med. 191:1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bochner, B.S., C.A. Bickel, M.L. Taylor, D.W. MacGlashan, Jr., P.W. Gray, C.J. Raport, and R. Godiska. 1999. Macrophage-derived chemokine induces human eosinophil chemotaxis in a CC chemokine receptor 3- and CC chemokine receptor 4-independent manner. J. Allergy Clin. Immunol. 103:527–532. [DOI] [PubMed] [Google Scholar]
- 63.Bernardini, G., J. Hedrick, S. Sozzani, W. Luini, G. Spinetti, M. Weiss, S. Menon, A. Zlotnik, A. Mantovani, A. Santoni, and M. Napolitano. 1998. Identification of the CC chemokines TARC and macrophage inflammatory protein-1 beta as novel functional ligands for the CCR8 receptor. Eur. J. Immunol. 28:582–588. [DOI] [PubMed] [Google Scholar]
- 64.Dairaghi, D.J., R.A. Fan, B.E. McMaster, M.R. Hanley, and T.J. Schall. 1999. HHV8-encoded vMIP-I selectively engages chemokine receptor CCR8. Agonist and antagonist profiles of viral chemokines. J. Biol. Chem. 274:21569–21574. [DOI] [PubMed] [Google Scholar]
- 65.Murphy, P.M., M. Baggiolini, I.F. Charo, C.A. Hebert, R. Horuk, K. Matsushima, L.H. Miller, J.J. Oppenheim, and C.A. Power. 2000. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 52:145–176. [PubMed] [Google Scholar]
- 66.Chung, C.D., F. Kuo, J. Kumer, A.S. Motani, C.E. Lawrence, W.R. Henderson, Jr., and C. Venkataraman. 2003. CCR8 is not essential for the development of inflammation in a mouse model of allergic airway disease. J. Immunol. 170:581–587. [DOI] [PubMed] [Google Scholar]
- 67.Goya, I., R. Villares, A. Zaballos, J. Gutierrez, L. Kremer, J.A. Gonzalo, R. Varona, L. Carramolino, A. Serrano, P. Pallares, L.M. Criado, R. Kolbeck, M. Torres, A.J. Coyle, J.C. Gutierrez-Ramos, C. Martinez-A, and G.J. Marquez. 2003. Absence of CCR8 does not impair the response to ovalbumin-induced allergic airway disease. J. Immunol. 170:2138–2146. [DOI] [PubMed] [Google Scholar]
- 68.Chensue, S.W., N.W. Lukacs, T.Y. Yang, X. Shang, K.A. Frait, S.L. Kunkel, T. Kung, M.T. Wiekowski, J.A. Hedrick, D.N. Cook, et al. 2001. Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in CC chemokine receptor 8 knockout mice. J. Exp. Med. 193:573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Flower, R.J., E.A. Harvey, and W.P. Kingston. 1976. Inflammatory effects of prostaglandin D2 in rat and human skin. Br. J. Pharmacol. 56:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]