

Abstract
We identified previously a patient with recurrent bacterial infections who failed to respond to gram-negative LPS in vivo, and whose leukocytes were profoundly hyporesponsive to LPS and IL-1 in vitro. We now demonstrate that this patient also exhibits deficient responses in a skin blister model of aseptic inflammation. A lack of IL-18 responsiveness, coupled with diminished LPS and/or IL-1–induced nuclear factor–κB and activator protein-1 translocation, p38 phosphorylation, gene expression, and dysregulated IL-1R–associated kinase (IRAK)–1 activity in vitro support the hypothesis that the defect lies within the signaling pathway common to toll-like receptor 4, IL-1R, and IL-18R. This patient expresses a “compound heterozygous” genotype, with a point mutation (C877T in cDNA) and a two-nucleotide, AC deletion (620–621del in cDNA) encoded by distinct alleles of the IRAK-4 gene (GenBank/EMBL/DDBJ accession nos. AF445802 and AY186092). Both mutations encode proteins with an intact death domain, but a truncated kinase domain, thereby precluding expression of full-length IRAK-4 (i.e., a recessive phenotype). When overexpressed in HEK293T cells, neither truncated form augmented endogenous IRAK-1 kinase activity, and both inhibited endogenous IRAK-1 activity modestly. Thus, IRAK-4 is pivotal in the development of a normal inflammatory response initiated by bacterial or nonbacterial insults.
Keywords: immunodeficiency, TLR4, IL-1R, inflammation, signal transduction
Introduction
In 1997, Kuhns et al. (1) described a patient with a 15-yr history of recurrent bacterial infections who exhibited a profound defect in the capacity to respond to LPS in vivo or LPS and rIL-1 in vitro, whereas responses to rTNF-α or platelet-activating factor remained intact. The patient's phagocytes expressed CD14 normally and bound LPS in a specific manner, suggesting a defect early in the signal transduction pathway. That paper was published before identification of toll-like receptor (TLR) 4 as the primary LPS signal transducing molecule (2, 3), or the recognition that TLR4 and IL-1R share a common signaling cascade that leads to nuclear factor (NF)–κB translocation, cytokine gene expression, and increased expression of costimulatory molecules (4). In response to LPS or IL-1, an adaptor molecule, MyD88, is recruited to and interacts with the intra-cytoplasmic toll/IL-1R (TIR) domain of TLR4 or IL-1 through its own COOH-terminal TIR domain (for review see reference 5). MyD88 also possesses an NH2-terminal death domain (DD) that enables recruitment of a serine/threonine kinase, IL-1R–associated kinase (IRAK)–1, to the receptor complex through a homotypic interaction via its NH2-terminal DD (for review see reference 6). Burns et al. (7) reported recently that the region of MyD88 that lies between its DD and TIR domains is essential for MyD88 binding of a related kinase, IRAK-4, that, in turn, phosphorylates IRAK-1 (7, 8). Once phosphorylated by IRAK-4, IRAK-1 autophosphorylates a different site, dissociates from the receptor complex, and interacts through its COOH terminus with TNF receptor–associated factor 6 (TRAF6), leading ultimately to NF-κB activation (for review see reference 6). A variant of MyD88 that lacks the region between the DD and TIR domains fails to bind IRAK-4 and, thereby, inhibits IRAK-4–mediated phosphorylation of IRAK-1 and downstream signaling (7). The importance of IRAK-4 in LPS and IL-1 signaling is further supported by the finding that mice with a targeted mutation in IRAK-4 exhibit an LPS- and IL-1–hyporesponsive phenotype (9) similar to that of our patient. We have identified two distinct IRAK-4 mutations in this patient as follows: (a) a point mutation (C21175T in genomic DNA; C877T in cDNA) and (b) a two-nucleotide AC deletion (15978–15979del in genomic DNA; 620–621del in cDNA) that are expressed on distinct alleles inherited from phenotypically normal parents. Both mutations result in truncations within the kinase domain of IRAK-4 protein, resulting in a recessive phenotype. In contrast to overexpression of WT IRAK-4, which reproducibly enhanced endogenous IRAK-1 kinase activity in HEK293T cells, both mutated forms not only failed to augment endogenous IRAK-1 kinase activity but also were modestly inhibitory. Thus, failure to express a WT IRAK-4 precludes recruitment and activation of key downstream signaling molecules and underlies the profoundly deficient inflammatory response seen in this patient.
Materials and Methods
Case Report.
The patient is a 21-yr-old woman who was the subject of an earlier work (1). At the time of the previous paper, she had 13 life-threatening infections in the first 15 yr of life characterized by minimal or no febrile responses. These included Streptococcus pneumoniae meningitis at 9 and 20 mo, endophthalmitis with Neisseria meningitidis at 13 mo, chronic serous otitis media, an abdominal abscess with gram-positive diplococci (culture-negative) at 3 yr, and episodes of Staphylococcus aureus cellulitis at 3, 6, and 12 yr. During treatment of an intraabdominal abscess at 3 yr, she developed neutropenia with an absolute neutrophil count of 1,000–2,000 cells/mm3 that has persisted to present. Other infections included Clostridium septicum infection with gangrene of the left leg at 9 yr, requiring total left leg amputation with hip disarticulation, associated with septic shock and acute respiratory distress syndrome. At 13 yr, she had nasal polyps culture positive for Streptococcus intermedicus and Gemella morbillorum. Since the original report, she has had five hospitalizations, but none with culture-positive infection. At 17 yr, she was hospitalized for a ruptured kidney cyst that required no antibiotics. She had two hospitalizations at 18 yr for vague abdominal complaints without diagnosis after a course of antibiotics for culture-negative cutaneous abscesses, one admission at 19 yr for ulcers of the left elbow, arm, and axilla (probably from poorly fitting crutches) and left eye inflammation of an unknown infection that was responsive to antibiotics, and one admission at 20 yr for chronic right knee pain with progressive symptoms of erythema and edema, and a diagnosis of degenerative arthritis, but without evidence of infection. Growth and development have been normal and she has gone through normal puberty with menarche, although her menstrual cycles have been irregular. She has done better clinically since she has passed through adolescence, as evidenced by lack of culture-positive severe infections. She has never had significant problems with viral or fungal infections. Her neutropenia persists and her neutrophil count rises to ∼4,000 cells/mm3 at times of acute inflammation. The patient, her mother, maternal grandmother, great maternal aunt, and great maternal grandmother have polycystic kidney disease. There is no family history of recurrent infections.
Reagents.
Protein-free Escherichia coli K235 LPS was prepared by the method of McIntire et al. (10). Human rIL-1β, rTNF-α, and rIL-18 were obtained from R&D Systems. PMA and ionomycin were purchased from Sigma-Aldrich. Abs to human IRAK-1 (Upstate Biotechnology), phosphorylated p38 (Promega), total ERK 1,2 (New England Biolabs, Inc.), and Flag (M2; Sigma-Aldrich) were used for Western blot analysis. All kits were used according to the manufacturers' instructions.
Isolation of PBMCs and Neutrophils.
Because the patient had a history of bacterial infection and was persistently neutropenic, leukopheresis and elutriation were not viable options and limited the yield of leukocytes collected per visit. Peripheral blood from normal volunteers was obtained through the Frederick Research Donor Program (Protocol OH00-C-N046) or the Warren Magnuson Clinical Center, National Institutes of Health (NIH; Protocol 99-CC-0168) and from the patient through the Warren Magnuson Clinical Center, NIH (Protocol 93-I-0119). Normal and patient blood were anticoagulated with citrate dextrose solution USP formula A (Baxter Healthcare). Whole blood, diluted with HBSS (BioWhittaker, Inc.) without divalent cations, was centrifuged over a discontinuous gradient cushion (LSM®; ICN Biomedicals) for 30 min at 400 g at room temperature. PBMCs were harvested from the top of the gradient and washed two times with HBSS. Both freshly prepared and cryopreserved cells were used in this study.
The neutrophil/erythrocyte pellet was suspended in an equal volume of HBSS. The cell suspension was diluted with dextran (1.5%final, mol wt = 200,000–500,000 g/mol; Amersham Biosciences) and allowed to sediment for 20 min before centrifugation at 200 g for 10 min. Contaminating erythrocytes were removed two times by hypotonic lysis using 0.2% PBS for 30 s, followed by an equal volume of 1.6% PBS to restore isotonicity. Neutrophils were counted on an automated cell counter (model T540; Coulter Corp.).
Induction of Blisters and Analysis of Inflammatory Response.
Blisters were raised on forearms of healthy, normal volunteers and the patient as described previously (11) after informed consent (NIH Clinical Center; Protocol No. 90-I-120). The roof of the blister was removed aseptically and a sterile, 8-well chamber device placed over the exposed dermal floor. Autologous, heat-inactivated serum (0.8–1.0 ml diluted to 70% with HBSS with divalent cations) was added to each well. At the indicated times, serum was collected from the wells and the exudate cells harvested by centrifugation. IL-6 and IL-8 levels in sera were determined by ELISA (R&D Systems; lower detection limits were 3.5 and 18.1 pg/ml, respectively). C5a levels were determined by radioimmunoassay (lower detection limit, 10 ng/ml; Amersham Biosciences). Exudate cells were resuspended in HBSS (without divalent cations), counted by hemocytometer, and identified by differential staining (Diff-Quik®, Baxter Scientific Products).
In Vitro Functional Assays.
To assess IL-18 responsiveness, T cells were purified from PBMCs by negative selection using T cell enrichment columns (R&D Systems), and cultured at 106 cells/ml in RPMI 1640 (BioWhittaker, Inc.) with 10% FCS (Hyclone), 50 μM 2−ME (Sigma-Aldrich), 10 μg/ml PHA (Sigma-Aldrich), and 1 ng/ml rIL-12 (R&D Systems). After 3 d, T cells were harvested, washed, and resuspended at 106 cells/ml RPMI 1640 with 10% FCS and cultured for 48 h in the presence of 20 ng/ml rIL-18 or PMA/1 μM ionomycin. IFN-γ was measured in culture supernatants by ELISA (R&D Systems).
For all other in vitro assays, except the IRAK-1 kinase assay, PBMCs were cultured in RPMI 1640 supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM Hepes, 0.3% sodium bicarbonate, and 10% human AB serum. For detection of NF-κB and activator protein-1 (AP-1) translocation, electrophoretic mobility shift assays (EMSAs) were performed as described previously (12). Western blot analysis of whole cell lysates was used to detect phosphorylated p38 kinase or total ERK 1,2 as described previously (13). To detect Flag-tagged IRAK4 proteins, HEK293T cells were cultured overnight in 100-mm Petri dishes (2.5 × 106 cells/dish) and cotransfected for 3 h with normal or mutated pRK7-IRAK4 expression vectors and control empty vector (pCDNA3; total amount of plasmid DNA was adjusted to 10 μg/dish by complementing with pCDNA3) using SuperFect Transfection Reagent (QIAGEN). After 48 h, cellular extracts were prepared (13). Proteins were resolved on 4–20% SDS-PAGE gels (Invitrogen), transferred to Immobilon P membranes, and analyzed by immunoblotting with 1 μg/ml anti-Flag mAb and secondary horseradish peroxidase–conjugated anti–mouse IgG (Amersham Biosciences; 1:12,500 dilution). Bands were detected with enhanced chemiluminescence plus reagents (Amersham Biosciences).
Steady-state levels of mRNA for LPS-inducible genes (e.g., TNF-α, GM-CSF, and cyclooxygenase-2 [Cox-2]), and for genes required for TLR4 and IL-1β signaling, were analyzed by reverse transcription (RT)-PCR with Southern blot analysis as described previously (14). A list of primer sets and probes used to detect specific mRNA species will be provided upon request.
Because the number of PBMCs was limited for any one experiment, the IRAK-1 kinase assay was performed on purified neutrophils exactly as described for elutriated monocytes (14). Fifty million neutrophils per time point were treated, and cell lysates were immunoprecipitated with anti–IRAK-1 Ab. Immunoprecipitates were assayed for the ability to phosphorylate myelin basic protein (MBP). The radioactive signal was detected using a PhosphorImager (Molecular Dynamics).
Cloning and Sequencing of IRAK-1 and IRAK-4 Genes.
Total RNA was isolated from PBMCs obtained from five healthy volunteers and the patient using RNeasy kit (QIAGEN), treated with RNase-free DNase I (QIAGEN) to eliminate genomic DNA, repurified, and quantified spectrophotometrically. cDNA was obtained by RT with random hexamers and avian myeloblastosis virus reverse transcriptase using Reverse Transcription System (Promega). For IRAK-1 sequencing, different portions of the coding sequence (CDS) were amplified by PCR using Pfu Turbo DNA polymerase (Stratagene) with the highest fidelity (15), Gen-N-hancer (Geno Technology, Inc.), and primers shown in Table SI (available at http://www.jem.org/cgi/content/full/jem.20030701/DC1). Sequences from multiple reactions were compared with each other and to GenBank/EMBL/DDBJ accession nos. L76191, NM_001569, dbSNP: 1059702, and P51617 and AAC41949 for IRAK-1 cDNA and protein sequences using Blast search programs (http://www.ncbi.nlm.nih.gov/BLAST; National Center for Biotechnology Information [NCBI], NIH).
The IRAK-4 mRNA CDS was amplified in PCR reactions using primer pairs shown in Table SII (available at http://www.jem.org/cgi/content/full/jem.20030701/DC1). PCR products were cloned for sequencing into the pCR4BluntTOPO vector using Zero Blunt TOPO PCR cloning kit (Invitrogen) followed by one shot chemical transformation into Top10 chemically competent cells. For every analysis, 10 colonies containing cloned PCR products from normal volunteers and 10 from the patient were picked and plasmid DNA isolated with QiaPrep Spin Miniprep kit (QIAGEN). After verification of the presence of the PCR products in the vector by restriction analysis with EcoRI, ≥3 samples of plasmid DNA from each group were sequenced with both M13 universal sequencing and gene-specific primers. The sequences obtained from normal volunteers and the patient were compared with each other as well as against the IRAK-4 GenBank/EMBL/DDBJ accession nos. AY186092 and AF445802 for IRAK-4 genomic DNA and cDNA, respectively, and Blast search programs. Sequencing was also performed on PCR products amplified from genomic DNA with primers encompassing the sequence of genomic DNA that corresponds to that of cDNA where mutations were identified. Genomic DNA was isolated with a Blood and Cell Culture DNA minikit (QIAGEN) and amplified by PCR using Pfu Turbo DNA polymerase and the following primers: 5′-ACAACTGGGTTGCCATTTTATTAGGTG-3′ (forward, start 20846) and 5′-CCAGATGCTCTAGGTTAAGACCTGCAT-3′ (reverse, start 21342), annealing temperature 60°C; 5′-GATCACCAAACAGAGTTCTCAAGAAAAA-3′ (forward, start 15662) and 5′-GACATAACCACATTTTTCTAGGGGTAGG-3′ (reverse, start 16208), annealing temperature 59°C; and 5′-TGGGTAGGTAGACTAGACCCACTCAAA-3′ (forward, start 15588) and 5′-GGCACACTATGAAGCAACTCTTTCTTT-3′ (reverse, start 16059), annealing temperature 59°C. PCR products and plasmids obtained by cloning PCR products into pCR4BluntTOPO vector were sequenced using Big Dye Terminator Cycle Sequencing Kit, v. 3.0 (Applied Biosystems). The entire IRAK-4 CDS PCR cloning and sequencing was repeated three times, whereas different portions of the IRAK-4 CDS were PCR-cloned and sequenced twice.
Sequencing was performed at the Biomedical Instrumentation Center (Uniformed Services University of the Health Sciences [USUHS]) and the Biopolymer Facility (University of Maryland, Baltimore). IRAK-4 mutations were confirmed in a Clinical Laboratory Improvement Amendments-certified laboratory (GeneDx, Inc.).
Site-directed Mutagenesis.
Site-directed mutagenesis of WT human IRAK-1 and IRAK-4 expression vectors (pRK5-IRAK-1 and pRK7-IRAK-4; Tularik, Inc.) was performed using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). The primers 5′-GAGCCCGCCCCTTTCCGTTTTGCTG-3′ (forward) and 5′-CAGCAAAACGGAAAGGGGCGGGCTC-3′ (reverse) were used to introduce T to C substitution at nucleotide 666 (i.e., 665–667, TTT into TCT) into pRK5-IRAK-1 expression vector, resulting in a F196S amino acid polymorphism that has been reported in the NCBI protein database (accession nos. P51617, F196 and AAC41949, S196). We designated the vector encoding F196, phenylalanine, as “pRK5-IRAK-1 (N)” and the vector encoding S196, serine, as “pRK5-IRAK-1 (M).”
The two IRAK-4 mutations found in the patient were introduced into the WT pRK7-IRAK4 expression vector (N) by site-directed mutagenesis and verified by sequencing. The primers 5′-TGCAAGATTGCTTAGGGTGCAGCTAATG-3′ (forward) and 5′-CATTAGCTGCACCCTAAGCAATCTTGCA-3′ (reverse) were used to introduce the C to T substitution at nucleotide 877 (877–879: CAG, normal; TAG, patient) into the WT vector [pRK7-IRAK4 (N)], which creates a stop codon at amino acid 293 [pRK7-IRAK4 (M #1)]. The primers 5′-ACGTAAATAACAACTGTGGCAGTGAAGAAG-3′ (forward) and 5′-CTTCTTCACTGCCACAGTTGTTATTTACGT-3′ (reverse) were used to introduce the AC deletion into pRK7-IRAK4 at nucleotides 620–621 [pRK7-IRAK4 (M #2)].
Reporter Assays.
As described previously (16), HEK293T cells (provided by Dr. N. Rice, NIH, Bethesda, MD) were seeded into 12-well plates (Costar) at 2 × 105 cells/well. After overnight incubation, cells were cotransfected for 3 h with pCDNA3/huCD14, pCDNA3/huTLR4, pEFBOS/Flag-huMD-2 plasmids (0.1 μg/well each), the NF-κB reporter (0.5 μg/well) and pCMV1-βGal reporter (0.5 μg/well), and varying amounts of pRK7-IRAK-4 (N) or pRK7IRAK4 (M #1) expression vectors using SuperFect transfection reagent. To study the response to IL-1β, the pCDNA3/huCD14, pCDNA3/huTLR4, and pEFBOS/Flag-huMD-2 plasmids were omitted. After transfection, cells were incubated for 20 h, washed, and restimulated with medium, LPS, or rIL-1β for the indicated times. Cells were lysed and luciferase activity was measured (16). Similar experiments were performed to determine the effect of pRK7-IRAK-4 (N), pRK7IRAK4 (M #1), and pRK7IRAK4 (M #2) expression on IL-1β–stimulated IRAK-1 kinase activity as described above in the In Vitro Functional Assays section for neutrophils.
Online Supplemental Material.
To compare the capacity of pRK5-IRAK-1 (N) and pRK5-IRAK-1 (M) vectors to reconstitute IRAK-1 signaling, transient transfection of the IRAK-1–deficient I1A clone of HEK293 cells (provided by Dr. X. Li, Cleveland Clinic Foundation, Cleveland, OH; references 17, 18) was performed as described above in the Reporter Assays section for HEK293T cells that express IRAK-1 (16).
Results
Patient Response to Skin Blister Induction Is Abnormal.
This patient was originally found to be hyporesponsive to i.v. challenge with LPS as evidenced by a subnormal febrile response, failure to mobilize a normal leukocytosis, and subnormal elevations in plasma levels TNF-α, IL-6, IL-8, lactoferrin, and G-CSF (1). Fig. 1 shows that the patient also had an abnormal inflammatory response to blister formation. Despite a normal early accumulation of exudate cells into the blisters, the later phase (>8 h) of neutrophil migration was markedly depressed (∼1.5 logs). The patient's blister fluid had normal levels of C5a, but failed to accumulate normal levels of IL-8, IL-6 (Fig. 1), and IL-1β (unpublished data). Thus, the patient's impaired inflammatory response can be observed in the absence of a bacterial stimulus.
Figure 1.

Patient exhibits impaired inflammatory response to blister formation in vivo. The panels show the accumulation of inflammatory cells and mediators in the medium bathing the exposed dermal surface versus time. In each panel, the value at t = 0 represents the background levels found in the 70% autologous serum. The shaded area represents the 95% confidence limits of the arithmetic mean (C5a; linear scale data) and geometric means (exudative cells, IL-8, and IL-6; log scale data) of the responses of normal subjects (n ≥ 7). Patient data (solid line) represents the mean of two experiments.
In Vitro Hyporesponsiveness to rIL-18.
The patient's leukocytes were shown previously to be LPS- and IL-1 hyporesponsive, whereas responses to other inflammatory stimuli were normal. In the same year as the identification of TLR4 as the LPS signal transducer, Thomassen et al. (19) found that, like IL-1, the IL-18 signaling pathway used IRAK-1 and TRAF6. Fig. 2 shows that normal T cells respond to rIL-18 with a dose-dependent induction of IFN-γ. In contrast, the patient's cells failed to respond to rIL-18, although the response to PMA plus ionomycin was normal.
Figure 2.

Patient exhibits impaired IL-18 responsiveness in vitro. T cells from normal (NL; open symbols) controls or the patient (PT; closed circles) were stimulated with rIL-18 and levels of IFN-γ measured in culture supernatants. PMA and ionomycin (ION) treatment was used as a positive control for nonreceptor-mediated stimulation of IFN-γ.
In 1998, positional cloning of murine tlr4 led to the identification of the elusive “Lps” gene (2, 3); however, the point mutation in the intracytoplasmic domain of TLR4 that renders C3H/HeJ mice LPS hyporesponsive did not affect sensitivity of C3H/HeJ macrophages to rIL-1β (20). Because our patient failed to respond to LPS and IL-1, as well as IL-18, we hypothesized that her hyporesponsiveness was not related to TLR4 per se, but rather, to a signaling component shared by the three receptor systems.
Downstream Effectors of TLR4 or IL-1 Signaling Are Poorly Activated in Patient's PBMCs and Result in Diminished LPS-induced Gene Expression.
Nuclear translocation of NF-κB and AP-1, as well as activation of mitogen-activated protein kinases, are consequences of both TLR and IL-1R signaling. Fig. 3 A compares the response of a control (normal) versus the patient's PBMCs to stimulation with LPS or rTNF-α for activation of NF-κB by EMSA. Control PBMCs responded robustly to LPS and TNF-α, whereas the patient responded to TNF-α only. Fig. 3 B shows that AP-1 translocation was strongly up-regulated by LPS in normal PBMCs, whereas the patient's PBMCs responded only slightly above the medium-treated background.
Figure 3.
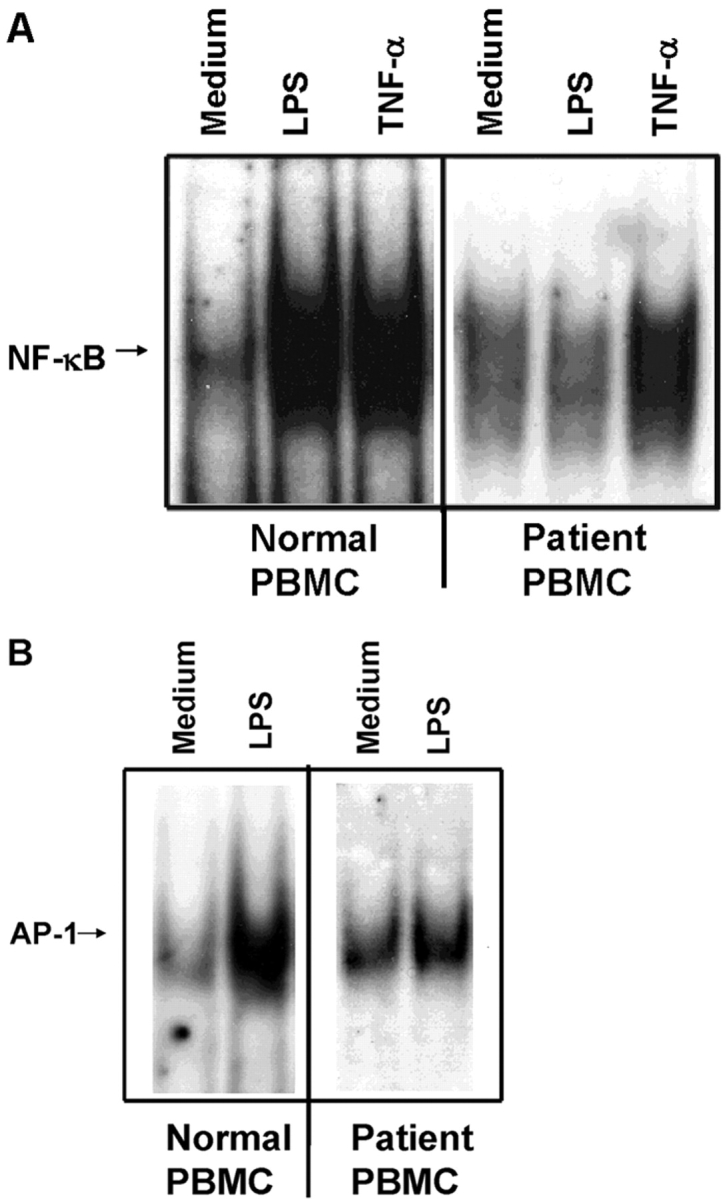
Patient's PBMCs exhibit impaired NF-κB (A) and AP-1 (B) translocation in response to LPS. PBMCs derived from a control or the patient were stimulated with 1 μg/ml LPS or 100 ng/ml rTNF-α for 60 min, and NF-κB and AP-1 translocation assessed by EMSA.
Fig. 4 illustrates that PBMCs from the patient show reduced phosphorylation of p38 mitogen-activated protein kinase in response to LPS or IL-1β. Although not completely eliminated, the patient's response was consistently lower when compared with an unrelated control (Fig. 4 A) or to her sister (Fig. 4 B). Fig. 4 B also confirms that the patient's responsiveness to rTNF-α to induce p38 phosphorylation was normal.
Figure 4.

Patient's PBMCs exhibit impaired p38 phosphorylation in response to LPS or IL-1. PBMCs derived from an unrelated control or the patient (A) or the patient's sister and the patient (B) were stimulated with 1 μg/ml LPS, 100 ng/ml rIL-1β, or 100 ng/ml rTNF-α for 15 min. Whole cell lysates were analyzed by Western blot analysis for phosphorylated p38 or total ERK-1,2.
TLR4- and IL-1–induced gene expression is mediated by the action of transacting factors such as NF-κB and AP-1, and of kinases such as p38 (for review see reference 5). Kuhns et al. (1) showed that this patient's PBMCs failed to secrete a number of proinflammatory cytokines when stimulated by LPS. Fig. 5 extends these findings at the level of gene expression and illustrates that her immediate family exhibits normal LPS responsiveness. Fig. 5 A shows that both parents were comparably LPS responsive, as indicated by the capacity of their PMBCs to respond to LPS to increase steady-state levels of TNF-α and Cox-2 mRNA, whereas steady-state mRNA levels for TLR4 and for the housekeeping gene, hypoxanthine-guanine phosphoribosyltransferase (HPRT), were not modulated, as we reported previously in elutriated monocytes (14). Fig. 5 B shows that PBMCs from the father, sister, and an unrelated donor all responded strongly and comparably to LPS to increase TNF-α, GM-CSF, and Cox-2 gene expression, indicating normal LPS responsiveness. In contrast, the patient's PBMCs responded poorly to LPS with minimal up-regulation of these genes. Steady-state mRNA levels for a number of signaling intermediates common to the TLR4 and IL-1R pathways (e.g., MyD88, Tollip, IRAK-1, and TRAF-6) were also measured. The patient's PBMCs expressed normal basal levels of these mRNA species, which were not modulated by LPS in either patient or control cells. Housekeeping genes (e.g., β-actin and HPRT) were expressed comparably in all samples. Collectively, these data strongly indicate that the parents and sibling of this LPS-hyporesponsive patient exhibit normal LPS responsiveness.
Figure 5.

Patient's PBMCs exhibit impaired LPS-induced gene expression; LPS-sensitivity of patient's family members is normal. PBMCs were stimulated with 100 ng/ml LPS for 3 h, and RNA was extracted for analysis of gene expression by RT-PCR with Southern blot analysis. (A) Comparison of responses of the patient's mother and father; (B) Comparison of father, sister, unrelated control, and patient responses.
LPS-induced IRAK-1 Kinase Activity Is Dysregulated in the Patient's Neutrophils.
After MyD88 recruitment to TLR4 or the IL-1R, there is a transient phosphorylation of IRAK-1 before it associates with TRAF6 and is ultimately targeted to the proteosome (21). Fig. 6 illustrates that the response of the patient's neutrophils to LPS, as measured by the capacity of immunoprecipitated IRAK-1 to phosphorylate the exogenous substrate, MBP, is dysregulated. In normal neutrophils, LPS induces a rapid and transient induction of IRAK-1 kinase activity that typically peaks 3–4 min after stimulation. This pattern of normal LPS-inducible IRAK-1–associated kinase activity was observed in >10 donors (unpublished data). In contrast, the patient's response to LPS was abnormal. In this experiment, the patient's baseline level of IRAK-1 kinase activity was initially higher than in the control, but kinase activity was rapidly lost upon LPS stimulation. By 5 min after stimulation, just as the control's kinase activity is decreasing to baseline levels, the patient's response is increasing to her baseline. Western blot analysis for total levels of ERK-1,2 in the cellular lysates used for anti–IRAK-1 immunoprecipitation show that total protein did not differ significantly from sample to sample in the normal versus patient cell lysates. Although an anti–IRAK-1 Ab was used to immunoprecipitate lysates for the kinase assay, recent papers have shown that IRAK-4 is necessary for activation of IRAK-1 kinase activity (8, 9) and that MyD88, IRAK-4, and IRAK-1 form a complex (7). Therefore, the observed phosphorylation of MBP may be attributable to the combined kinase activities of IRAK-1 and/or IRAK-4.
Figure 6.

Patient's neutrophils exhibit dysregulated IRAK-1 kinase activity. Control and patient neutrophils were stimulated with 100 ng/ml LPS for the indicated times. Cell lysates were immunoprecipitated with anti–IRAK-1 Ab, and immunoprecipitates were assayed for the capacity to phosphorylate MBP. Western blot analysis for total ERK-1,2 was performed on lysates before immunoprecipitation to ensure equal protein concentrations (n = 2).
Cloning and Sequencing of Genes Common to TLR4 and IL-1 Signaling Pathways.
When these studies were initiated, little was known about the signaling intermediates required for TLR4 and IL-1R–mediated signaling. Nonetheless, we cloned and sequenced candidate genes from controls and the patient as GenBank/EMBL/DDBJ sequences became available. Specifically, the transmembrane and intracytoplasmic CDS for both TLR4 and TLR2 were cloned from cDNA (i.e., we hypothesized that a mutation in one of these regions could sequester shared signaling molecules, thus precluding their interaction in response to LPS or IL-1β) and found the patient to be normal. We sequenced MyD88 from genomic DNA and cDNA and found no mutations, and when anti-MyD88 Ab became available, we confirmed that the patient's MyD88 migrated normally. We also cloned and sequenced Tollip, IRAK-2, IRAK-M, ECSIT, and TRAF6, and no polymorphisms were identified (unpublished data). However, when we sequenced IRAK-1 and IRAK-4 cDNA, we identified three mutations that will now be discussed in detail.
Previous papers in mice with a targeted mutation in IRAK-1 demonstrated that this protein is important in TLR4-, IL-1–, and IL-18–mediated signaling (22, 23). The IRAK-1 polymorphism identified in our patient was a single nucleotide substitution of T to a C at cDNA nucleotide 666 [Control: nucleotides 665–667 = TTT, amino acid = phenylalanine (F); Patient: nucleotides 665–667 = TCT, amino acid = serine (S)]. Based on sequence analysis of multiple clones, the patient was heterozygous for this polymorphism. Although this polymorphism had been reported in GenBank/EMBL/DDBJ (accession no. AAC41949), we did not know if its expression interfered with LPS- or IL-1–induced signaling. Transient transfection of the IRAK-1–deficient HEK293 cell line, IA1 (17), with either a “WT” IRAK-1 expression vector (pRK5-IRAK-1 [N]) or a vector generated by site-directed mutagenesis that encodes the alternate form expressed by the patient (pRK5-IRAK-1 [M]), or equal amounts of both vectors, led to full and comparable restoration of IL-1–induced NF-κB reporter gene expression (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20030701/DC1). Therefore, this IRAK-1 (T666C) polymorphism was not functionally meaningful.
When we cloned and sequenced the patient's IRAK-4 cDNA, two distinct mutations were identified. The first was a C to T substitution at cDNA nucleotide 877 (which corresponds to genomic DNA nucleotide 21175). This substitution creates an in-frame, stop codon (Fig. 7 A; nucleotides 877–879 are changed from CAG in the WT to TAG in the patient's gene). Five independent cloning experiments (including normal donors in each case) were performed in which random selection and sequencing of the patient's individual clones showed heterozygous expression of WT and mutated forms. In each experiment, an equivalent number of clones from normal individuals were selected and sequenced and in no case did we see expression of the mutated form in any of these clones. The presence of this mutation was confirmed by direct sequencing of PCR products derived from the patient's genomic DNA (i.e., there was a C/T overlap at nucleotide 21175). Also, direct cloning of these PCR products, followed by sequencing, revealed that approximately half the clones expressed C and the other half expressed T, confirming heterozygosity of this mutation (unpublished data).
Figure 7.


Identification and functionality of a point mutation in patient's IRAK-4. (A) Illustration of C877T substitution mutation (M #1), resulting in a truncated form of IRAK-4. Arrow indicates approximate location of truncation in IRAK-4 protein. (B) Vectors encoding the WT (N) or C877T mutation (M #1) forms of IRAK-4 were expressed in HEK293T cells (5 μg vector/transfection) and cell lysates subjected to Western blot analysis using anti-Flag Ab. (C) Overexpression of WT (N) or C877T (M #1) forms of IRAK-4 in HEK293T cells failed to induce NF-κB–induced reporter activity. Cells were transiently transfected with pELAM-Luc, pCMV-βGal, and the indicated amounts of expression vectors encoding either IRAK-1 or normal (N) and mutated (M) forms of IRAK-4 (total amount of plasmid DNA was kept constant at 1.5 μg per transfection). After recovery for 48 h, NF-κB reporter activity was measured. The data represent the mean ± SEM of a representative experiment (n = 3).
Functional Analysis of IRAK-4 Point Mutation.
The stop codon created by C877T is predicted to encode a truncated form of IRAK-4, resulting in a protein of 292 amino acids versus the WT form that is 460 amino acids, with estimated molecular weights of ∼30 kD and ∼50 kD, respectively. Attempts to detect endogenous full-length or truncated forms of IRAK-4 by Western blot analysis with an anti–IRAK-4 Ab (Tularik, Inc.) in the same immunoprecipitates used for the neutrophil IRAK-1 kinase assays were unsuccessful, probably due to low endogenous protein levels. Therefore, we used site-directed mutagenesis to introduce the C to T point mutation into the pFlagRK7-IRAK4 (N) WT expression vector. After verifying the sequences of these vectors, we expressed the normal or mutated (pFlagRK7-IRAK4 [M #1]) vectors in IRAK-1–competent HEK293T cells. Fig. 7 B shows that this single nucleotide substitution resulted in expression of a protein that was significantly smaller than WT IRAK-4, but slightly larger than predicted from the amino acid sequence only, suggesting additional posttranscriptional modification. Based on analysis of the IRAK-4 sequence using the Expasy Translate Tool and EMBL-EBI EMBOSS-Transseq programs (http://us.expasy.org/cgi-bin/dna_sequences and http://www.ebi.ac.uk/servicestmp, respectively), the DD encompasses amino acids 20–104, whereas the kinase domain spans amino acids 186–456. Therefore, this point mutation truncates the IRAK-4 molecule within the kinase domain.
S. Li et al. (8) reported that overexpression of pFlagRK7-IRAK4 (N) increased NF-κB reporter activity ∼10-fold in a subclone of HEK293 cells. They also showed that the response of another HEK293 clone to IL-1 could be inhibited in a dose-dependent manner by overexpression of a kinase-defective mutant (KK213AA) or a truncated form consisting of the NH2-terminal 191 amino acids. Although we were able to drive NF-κB reporter activity by IRAK-1 overexpression in our HEK293T cells, neither WT (N) nor mutant IRAK-4 (M #1) vectors activated NF-κB reporter activity (Fig. 7 C). Moreover, when we cotransfected the HEK293T cells with constructs that would enable LPS responsiveness (i.e., CD14, TLR4, and MD-2), expression of the WT IRAK-4 down-regulated LPS-induced NF-κB reporter activity, whereas expression of the mutated form (M #1) failed to inhibit (Fig. 8 A, left). This was also the case for IL-1–stimulated NF-κB reporter activity (Fig. 8 A, right). These findings were confirmed in another HEK293 cell line stably transfected with TLR4 and MD-2 (unpublished data), and suggests that the mutant lacks a domain that is critical for sequestration of signaling molecules under conditions of overexpression in our HEK293 cell lines.
Figure 8.


(A) Overexpression of WT (N), but not C877T mutant (M), IRAK-4 expression vector inhibits LPS- and IL-1–mediated signaling in HEK293T cells. Cells were transiently transfected as described in Fig. 7, permitted to recover for 48 h, and stimulated with medium, LPS, or rIL-1β for 5 h before measurement of NF-κB reporter activity. (B) Overexpression of WT (N), but not C877T mutant (M), IRAK-4 expression vectors differentially modulate IRAK-1 kinase activity. Cells were transiently transfected with control pCDNA3.1, pRK7-IRAK4 (N), or pRK7-IRAK4 (M; 5 μg per transfection). After 40 h recovery, cells were stimulated with rIL-1β for the indicated times, followed by lysis of cells and the IRAK-1 kinase assay (n = 3).
To test directly the capacity of the truncated IRAK-4 to activate IRAK-1 kinase activity, HEK293T cells were transfected with a control vector, pCDNA 3.1, the WT IRAK-4 vector, or the mutant (M #1) IRAK-4 vector. Transfectants were stimulated with rIL-1, and IRAK-1 was immunoprecipitated from cell lysates and assayed for the capacity to phosphorylate MBP. Fig. 8 B illustrates that, in contrast to the WT IRAK-4 construct that enhanced IL-1–induced IRAK-1 kinase activity in the HEK293T cells, overexpression of the mutant not only failed to enhance endogenous kinase activity, but also inhibited it.
Identification of a Second IRAK-4 Mutation.
Both the patient's parents responded vigorously to LPS (Fig. 5 A) and neither exhibited a history of recurrent infection. Therefore, we hypothesized that a second mutation on the patient's alternate IRAK-4 allele must be present to account for such a profound phenotype. A two-nucleotide deletion was identified in the cDNA at nucleotides 620–621, which corresponds to genomic DNA nucleotides 15978–15979. This second mutation was not seen in any clones that carried the point mutation, supporting the hypothesis that it was derived from the alternate allele. The predicted effect of this frameshift mutation would be to create a stop codon 36–39 nucleotides downstream in the IRAK-4 CDS, resulting in a second truncated form with a predicted size of 218 amino acids or ∼22 kD (Fig. 9 A) and would be predicted to express the entire DD, but a smaller region of the kinase domain than M #1. As in the case of the point mutation, the presence of the mutation was confirmed by sequencing PCR products amplified from genomic DNA that showed a C/A double peak at nucleotide 623 with a scrambled sequence thereafter (unpublished data). In addition, sequencing of cloned PCR products confirmed that approximately half expressed the deletion. Using site-directed mutagenesis, we introduced the deletion into the pRK7-FlagIRAK4 (N) expression vector to obtain the mutant vector, pRK7-IRAK4 (M #2) and overexpressed both in HEK293T cells to compare them functionally. Fig. 9 B shows that pRK7-IRAK4 (M #2) led to expression of two truncated Flag-tagged proteins with molecular weights of ∼26 and 30 kD, suggesting posttranscriptional modification, and/or possibly “read through” of the first stop codon. Fig. 9 C demonstrates that, unlike WT IRAK-4, this more-truncated form of IRAK-4 also failed to augment endogenous IRAK-1 kinase activity and inhibited endogenous basal and IL-1–induced IRAK-1 activity, although less than observed upon overexpression of pRK7-IRAK4 (M #1).
Figure 9.

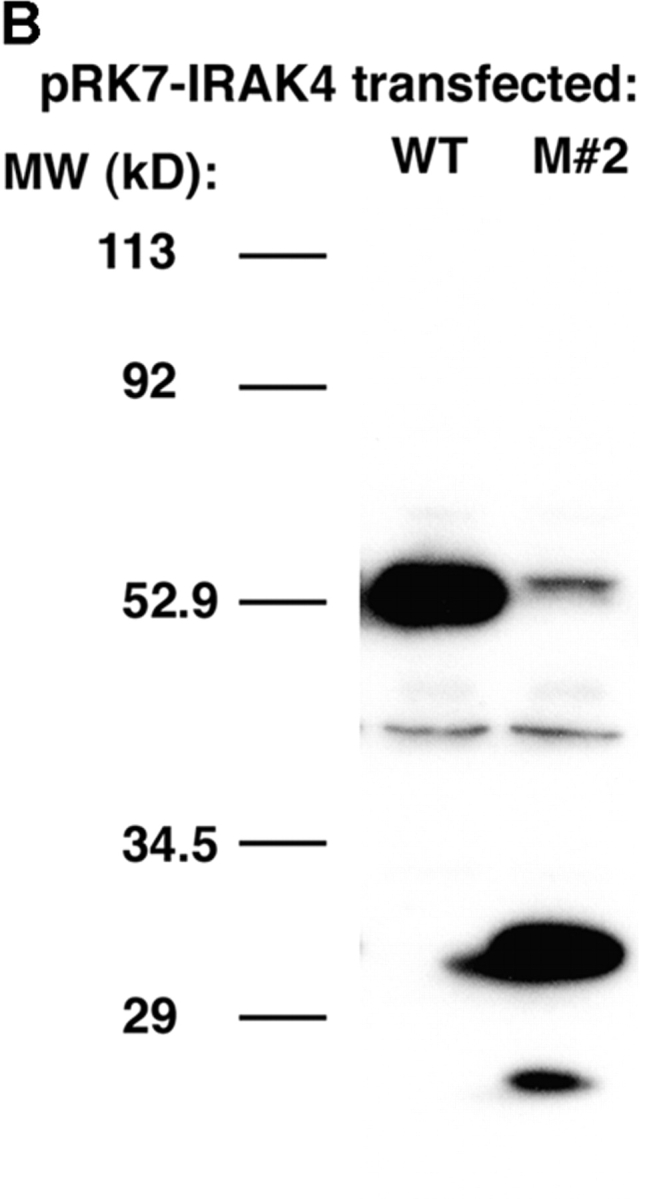

Identification and functionality of deletion mutation in patient's IRAK-4. (A) Illustration of effect of AC deletion at nucleotides 620-621 in the patient (620-621del), resulting in a truncated form of IRAK-4 (M #2). Arrow indicates approximate location of truncation in IRAK-4 protein. (B) Vectors encoding the WT (N) or mutated (M #2) forms of IRAK-4 were expressed in HEK293T cells (10 μg vector/transfection), and cell lysates subjected to Western blot analysis using anti-Flag Ab as described in Fig. 8. (C) Overexpression of WT (N), but not the 620-621 deleted mutant (M #2), IRAK-4 expression vectors (10 μg/transfection) differentially modulate IRAK-1 kinase activity (see Fig. 8 B; n = 3).
Discussion
We described previously our patient as having a long history of severe, recurrent bacterial infections and greatly diminished responses to LPS in vivo or to LPS and IL-1 in vitro (1), and speculated that this phenotype was due to a defect early in the LPS signal transduction pathway. In addition to abnormal LPS and IL-1 signaling, we now show that this patient's T cells failed to respond to rIL-18 to secrete IFN-γ (Fig. 2). Our findings that LPS- or IL-1–induced NF-κB and AP-1 translocation, p38 phosphorylation, or LPS-induced gene expression were all impaired in this patient's PBMCs (Figs. 3–5) provided strong evidence for an upstream signaling defect shared by all three receptor systems. This was further strengthened by the observation that this patient exhibited dysregulated neutrophil IRAK-1 kinase activity in response to LPS. Rather than being increased by LPS, IRAK-1 kinase activity was transiently down-regulated, suggesting an inhibitory effect of this mutation on IRAK-1 kinase activity. Although sequence analysis of this patient's IRAK-1 gene revealed a heterozygotic polymorphism (F196S) located in a critical region of the protein (i.e., just upstream of the region that encodes the kinase domain; reference 19), it fully reconstituted signaling in IRAK-1–deficient cells (Fig. S1).
However, this patient also possesses a “compound heterozygous” genotype in IRAK-4. The expression of distinct mutations on each of the patient's IRAK-4 alleles results in the failure to express a normal gene product (i.e., a “compound heterozygous genotype” that results in a recessive phenotype). Generally, inheritance of a compound heterozygous genotype leading to a recessive phenotype is more common than expression of homozygous recessive mutations, particularly in diseases where many different mutations have been reported (e.g., β-globin; references 24, 25). The two distinct mutations in this patient encode proteins that, when expressed, are truncated within the IRAK-4 kinase domain, yet retain the entire DD. Although overexpression of the WT IRAK-4 vector in HEK293T cells augmented IRAK-1 kinase activity induced by IL-1, overexpression of the mutant forms inhibited endogenous IRAK-1 kinase activity modestly, indicating that these molecules do not function normally and can interfere with WT IRAK-4 (i.e., exhibit dominant negative activity) when coexpressed with a normal IRAK-4 gene. These data are consistent with those of S. Li et al. (8) in which a 193 amino acid form of IRAK-4, or one that expresses mutations within the kinase domain, were found to inhibit IL-1–mediated signaling in a subline of HEK293 cells. Sequencing analysis revealed that the patient's father was heterozygous for the point mutation (i.e., +/C877T genotype), whereas her mother and sister were heterozygous for the deletion mutation (i.e., +/620–621del genotype), yet both parents and her sibling are fully LPS responsive, based on a history of normal febrile response to bacterial infections, no history of repeated infections, and normal LPS-inducible gene expression in vitro. One can only assume that compared with overexpression systems in vitro, the levels of expression of each of the mutated forms by the parents is insufficient to block activity of the their WT IRAK-4 such that LPS responsiveness is not measurably impaired. Therefore, gene therapy may be a viable approach for this patient.
The central role of IRAK-4 in LPS- and IL-1 signaling has only recently emerged. Work by S. Li et al. (8), published within the past year, indicates that IRAK-4 activation is required for phosphorylation of IRAK-1. This is consistent with earlier work of X. Li et al. (18), who showed that the IRAK-1 kinase domain is unnecessary for reconstitution of phosphorylation of IRAK-1 and signaling in IRAK-1–deficient cells. These authors postulated the existence of an upstream kinase that elicits the initial phosphorylation of IRAK-1 in response to IL-1. Although other IRAK family members (e.g., IRAK-2 and IRAK-M) can substitute for IRAK-1 (for review see reference 6), the necessity of IRAK-4 is further supported by the strong LPS- and IL-1–resistant phenotypes in mice with a targeted mutation in IRAK-4 (9). Burns et al. (7) demonstrated an interaction of MyD88, IRAK-4, and IRAK-1; IRAK-1 interacts with MyD88 through homotypic DD interactions, whereas the region of MyD88 between the TIR and DD is necessary for recruitment of IRAK-4, although the region on IRAK-4 that engages MyD88 was not reported. In coimmunoprecipitation experiments, we have found that overexpression of either truncated form (M #1 or M #2) in HEK293T cells results in a stronger association with MyD88 than WT IRAK-4 (unpublished data). This suggests that the DD of IRAK-4 enables it to bind to the region of MyD88 between its TIR and DD (7), and suggests the possibility that dominant negative inhibition may be facilitated through preferential or sustained binding of truncated forms. When coupled with the failure of the truncated form to compete for signaling components leading to inhibition of NF-κB reporter activity (Fig. 8 A; WT IRAK-4), their failure to augment IRAK-1 kinase activity, and their capacity to inhibit endogenous IRAK-1 kinase activity when overexpressed (Figs. 8 B and 9 C), a model of interaction between MyD88 and truncated IRAK-4, but not between truncated IRAK-4 and downstream signaling molecules, is suggested.
Quite recently, three unrelated patients with homozygous recessive IRAK-4 deficiencies were identified (26). Like our patient, these patients had recurrent pyogenic infections. Our patient expresses a unique pattern of IRAK-4 insufficiency as a consequence of her compound heterozygosity, further supporting the essentiality of IRAK-4 in the response to bacterial infection. Unquestionably, the most fascinating aspect of these two independent papers is that the substitution of T for C at nucleotide 877 of the IRAK-4 cDNA (C877T) is common to our patient and two of the patients described in the other paper. Because these patients are, to the best of our knowledge, unrelated, the data suggest that this site may represent a hypermutable site or “hot spot” in this gene. Coupled with the fact that our patient expresses a compound heterozygous genotype, these data suggest that there are likely to be additional patients that fail to express functional IRAK-4 and present with recurrent bacterial infections.
The data in this report also provide compelling new evidence for an important role for IRAK-4 in mobilizing the acute inflammatory response. The observation that our patient also fails to mount a normal inflammatory response in response to blister formation suggests that the role for IRAK-4 in inflammation is not restricted to pyogenic stimuli, as suggested by Picard et al. (26). Specifically, in response to skin blister induction, our patient had normal acute (3–5 h) cellular inflammation with normal levels of C5a, but impaired delayed (8–24 h) cellular inflammation with depressed levels of IL-8 and other proinflammatory cytokines. Normal levels of C5a, and presumably of other chemoattractants not measured (i.e., leukotriene B4), that do not require the TLR pathway for synthesis, likely account for the normal, early phase of inflammation. Our patient's diminished capacity to produce IL-8 and other proinflammatory cytokines likely explains the defective later phase of acute inflammation in the blister model. The defective production of inflammatory mediators identified in our patient as a result of mutations in IRAK-4 provides strong evidence for the importance of IRAK-4 and products of the TLR pathway in mobilizing the inflammatory response in vivo. It is also possible that the release of endogenous, cell-derived TLR agonists released as a consequence of the inflammatory response to blister formation provides the necessary stimulus for this later inflammatory response. In this regard, Kuhns et al. (27) and Smiley et al. (28) reported that fibrinogen is a potent TLR4 agonist, leading to the induction of chemokine expression; our patient also fails to respond to fibrinogen in vitro (unpublished data). Finally, the absence of increased susceptibility to viral and fungal infections in our patient further suggests that IRAK-4–independent pathways must also be triggered in host defense against these infections.
Acknowledgments
We give very special thanks to the patient and family for their cooperation and interest in identifying the molecular basis for this immune deficiency, and for their extreme patience during this work. We thank V. Anderson, R.N. and S.M. Holland, M.D. for their assistance in the care of this patient. We are indebted to those who provided important reagents (Tularik, Inc. and Dr. X. Li) and technical guidance, especially, Mr. M. Flora (USUHS). We appreciate the input of Drs. M. Blitzer and E. Stroval, and, lastly, we thank our many scientific colleagues for their helpful discussions over the past five years.
This work was supported in part by the National Cancer Institute, National Institutes of Health (NIH), contract no. N01-CO-56000 and by extramural NIH grants AI-18797 (to S.N. Vogel) and HL-66436 (to M. Arditi).
Abbreviations used in this paper: AP-1, activator protein-1; CDS, coding sequence; Cox-2, cyclooxygenase-2; DD, death domain; EMSA, electrophoretic mobility shift assay; IRAK, IL-1R–associated kinase; MBP, myelin basic protein; NF, nuclear factor; TIR, toll/IL-1R; TLR, toll-like receptor; TRAF6, TNF receptor–associated factor 6.
The online version of this article includes supplemental material.
References
- 1.Kuhns, D.B., D.A. Long-Priel, and J.I. Gallin. 1997. Endotoxin and IL-1 hyporesponsiveness in a patient with recurrent bacterial infections. J. Immunol. 158:3959–3964. [PubMed] [Google Scholar]
- 2.Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 3.Qureshi, S.T., L. Lariviere, G. Leveque, S. Clermont, K.J. Moore, P. Gros, and D. Malo. 1999. Endotoxin-tolerant mice have mutations in toll-like receptor 4 (Tlr4). J. Exp. Med. 189:615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Medzhitov, R., P. Preston-Hurlburt, E. Kopp, A. Stadleln, C. Chen, S. Ghosh, and C.A. Janeway, Jr. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell. 2:253–258. [DOI] [PubMed] [Google Scholar]
- 5.Medzhitov, R.P., and C.A. Janeway, Jr. 2000. Innate immunity. N. Engl. J. Med. 343:338–344. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki, N., S. Suzuki, and W.C. Yeh. 2002. IRAK-4 as the central TIR signaling mediator in innate immunity. Trends Immunol. 23:503–506. [DOI] [PubMed] [Google Scholar]
- 7.Burns, K., S. Janssens, B. Brissoni, N. Olivos, R. Bayaert, and J. Tschopp. 2003. Inhibition of interleukin 1 receptor/toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med. 197:263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li, S., A. Strelow, E.J. Fontana, and H. Wesche. 2002. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. USA. 99:5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki, N., S. Suzuki, G.S. Duncan, D.G. Millar, T. Wada, C. Mirtsos, H. Takada, A. Wakeham, A. Itie, S. Li, et al. 2002. Severe impairment of interleukin-1 and toll-like receptor signaling in mice lacking IRAK-4. Nature. 416:750–756. [DOI] [PubMed] [Google Scholar]
- 10.McIntire, F.C., G.H. Barlow, H.W. Sievert, R.A. Finley, and A.L. Yoo. 1967. Chemical, physical, and biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry. 6:2363–2372. [DOI] [PubMed] [Google Scholar]
- 11.Kuhns, D.B., E. DeCarlo, D.M. Hawk, and J.I. Gallin. 1992. Dynamics of the cellular and humoral components of the inflammatory response elicited in skin blisters in humans. J. Clin. Invest. 89:1734–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medvedev, A.E., J.C.G. Blanco, N. Qureshi, and S.N. Vogel. 1999. Limited role of ceramide in lipopolysaccharide-mediated mitogen-activated protein kinase activation, transcription factor induction, and cytokine release. J. Biol. Chem. 274:9342–9350. [DOI] [PubMed] [Google Scholar]
- 13.Medvedev, A.E., K.M. Kopydlowski, and S.N. Vogel. 2000. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and Toll-like receptor 2 and 4 gene expression. J. Immunol. 164:5564–5574. [DOI] [PubMed] [Google Scholar]
- 14.Medvedev, A.E., A. Lentschat, L.M. Wahl, D.T. Golenbock, and S.N. Vogel. 2002. Dysregulation of LPS-induced toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J. Immunol. 169:5209–5216. [DOI] [PubMed] [Google Scholar]
- 15.Cline, J., J.C. Braman, and H.H. Hogrefe. 1996. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 24:3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medvedev, A.E., and S.N. Vogel. 2003. Overexpression of CD14, TLR4, and MD-2 in HEK 293T cells does not prevent induction of in vitro endotoxin tolerance. J. Endotoxin Res. 9:60–64. [DOI] [PubMed] [Google Scholar]
- 17.Li, X., M. Commane, C. Burns, K. Vithalani, Z. Cao, and G.R. Stark. 1999. Mutant cells that do not respond to interleukin-1 (IL-1) reveal a novel role of IL-1 receptor-associated kinase. Mol. Cell. Biol. 19:4643–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, X., M. Commane, Z. Jiang, and G.R. Stark. 2001. IL-1-induced NFκB and c-Jun N-terminal kinase (JNK) activation diverge at IL-1 receptor-associated kinase (IRAK). Proc. Natl. Acad. Sci. USA. 98:4461–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomassen, E., T.A. Bird, B.R. Renshaw, J.M.K. Kennedy, and J.E. Sims. 1998. Binding of interleukin-18 to the interleukin 1 receptor homologous receptor IL-1Rrp1 leads to activation of signaling pathways similar to those used by interleukin-1. J. Interferon Cytokine Res. 18:1077–1088. [DOI] [PubMed] [Google Scholar]
- 20.Vogel, S.N., D. Johnson, P.-Y. Perera, A. Medvedev, L. Lariviere, S.T. Qureshi, and D. Malo. 1999. Cutting edge: functional characterization of the effect of the C3H/HeJ defect in mice that lack an Lpsn gene: in vivo evidence for a dominant negative mutation. J. Immunol. 162:5666–5670. [PubMed] [Google Scholar]
- 21.Yamin, T.-T., and D.K. Miller. 1997. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J. Biol. Chem. 272:21540–21547. [DOI] [PubMed] [Google Scholar]
- 22.Thomas, J.A., J.L. Allen, M. Tsen, T. Dubnicoff, J. Danao, X.C. Liao, Z. Cao, and S.A. Wasserman. 1999. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J. Immunol. 163:978–984. [PubMed] [Google Scholar]
- 23.Swantek, J.L., M.F. Tsen, M.H. Cobb, and J.A. Thomas. 2000. IL-1 receptor-associated kinase modulates host responsiveness to endotoxin. J. Immunol. 164:4301–4306. [DOI] [PubMed] [Google Scholar]
- 24.Nussbaum, R.L., R.R. McIness, and H.F. Willard. 2001. Thompson and Thompson Genetics in Medicine. 6th ed. W. B. Saunders Co., Philadelphia. 444 pp.
- 25.Jorde, L.B., J.C. Carey, M.J. Bamshad, and R.L. White. 2000. Medical Genetics. Wm. Schmitt, editor. Mosby, St. Louis. 372 pp.
- 26.Picard, C., P.A. Puel, M. Bonnet, C.L. Ku, J. Bustamante, K. Yang, C. Soudais, S. Dupuis, J. Feinberg, C. Fieschi, et al. 2003. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 299:2076–2079. [DOI] [PubMed] [Google Scholar]
- 27.Kuhns, D.B., E.L. Nelson, W.G. Alvord, and J.I. Gallin. 2001. Fibrinogen induces IL-8 synthesis in human neutrophils stimulated with formyl-methionyl-phenylalanine or leukotriene B4. J. Immunol. 167:2869–2878. [DOI] [PubMed] [Google Scholar]
- 28.Smiley, S.T., J.A. King, and W.W. Hancock. 2001. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 167:2887–2894. [DOI] [PubMed] [Google Scholar]