

Abstract
Many tumor-associated antigens are derived from nonmutated “self” proteins. T cells infiltrating tumor deposits recognize self-antigens presented by tumor cells and can be expanded in vivo with vaccination. These T cells exist in a functionally tolerant state, as they rarely result in tumor eradication. We found that tumor growth and lethality were unchanged in mice even after adoptive transfer of large numbers of T cells specific for an MHC class I–restricted epitope of the self/tumor antigen gp100. We sought to develop new strategies that would reverse the functionally tolerant state of self/tumor antigen-reactive T cells and enable the destruction of large (with products of perpendicular diameters of >50 mm2), subcutaneous, unmanipulated, poorly immunogenic B16 tumors that were established for up to 14 d before the start of treatment. We have defined three elements that are all strictly necessary to induce tumor regression in this model: (a) adoptive transfer of tumor-specific T cells; (b) T cell stimulation through antigen-specific vaccination with an altered peptide ligand, rather than the native self-peptide; and (c) coadministration of a T cell growth and activation factor. Cells, vaccination, or cyto-kine given alone or any two in combination were insufficient to induce tumor destruction. Autoimmune vitiligo was observed in mice cured of their disease. These findings illustrate that adoptive transfer of T cells and IL-2 can augment the function of a cancer vaccine. Furthermore, these data represent the first demonstration of complete cures of large, established, poorly immunogenic, unmanipulated solid tumors using T cells specific for a true self/tumor antigen and form the basis for a new approach to the treatment of patients with cancer.
Keywords: adoptive cell transfer, immunotherapy, IL-2, recombinant poxvirus, T cell epitope
Introduction
The observation that many tumor-associated antigens are nonmutated “self-antigens” has raised the question of how to induce large numbers of highly active, self/tumor antigen-specific T cells capable of destroying large, established tumors. Nonmutated self-antigens are particularly attractive targets for immunotherapy, because they are shared among patients, obviating the need for personalized vaccine development (1–6). We and others have attempted numerous vaccination approaches targeting self/tumor antigens. However, despite the increased numbers of self/tumor antigen-specific T cells, the reproducible induction of significant destruction of large, established cancers in mouse or in man using any self-antigen vaccine-based approach remains an elusive goal (7–9). A state of functional tolerance apparently exists in the tumor-bearing host.
This state of functional tolerance, defined here as the coexistence of tumor-specific T cells and growing tumor cells, may be incomplete. There is evidence indicating that antigen expression by tumors leads to limited T cell activation without tumor regression (10–12). Work in the spontaneous insulinoma model has indicated that effector “exhaustion,” rather than tumor escape is the underlying mechanism of functional tolerance (13). Work using other antigenic systems indicates that antigens expressed by tumors actively tolerize tumor-specific T cells (14). Antigenic “ignorance” to tumors has also been proposed as a mechanism for functional tolerance (15). Thus, the mechanisms underlying the observed functional tolerance of antitumor T cells remain unclear.
The modeling of self-reactivity is likely to be important in the development of new immunotherapies that target self/tumor antigens, but many of the currently available tumor models that model the complete destruction of established tumors target “foreign” antigens. The antigenic systems most commonly used until now by us and others include chicken egg ovalbumin, β-galactosidase from Escherichia coli, the Ld major histocompatibility antigen, and hemagglutinin or nucleoprotein derived from influenza A virus (14, 16–20). These models have shed valuable light on basic immunologic principles (21), but it is unclear to what extent the results obtained from these models reflect immune responses against true self/tumor antigens. For example, the high levels of expression of foreign antigens under the control of constitutively activated viral promoters may not mimic the expression of self-antigens that are the targets of tumor-specific T cells. Most importantly, each of these model antigens has been selected for study because they induce unusually strong T cell responses, perhaps not reflecting T cell responses characteristic of those observed against tumor antigens.
We sought to study the activation and proliferation of self-specific T cells that can infiltrate and destroy large, established tumors. Here, we investigate parameters that can be manipulated in vivo to transform a completely ineffective antitumor response into a productive one. To utilize a tumor model that is more physiologically relevant, we used the poorly immunogenic B16 melanoma, a highly aggressive tumor in C57BL/6 mice (22). A much-studied tumor, B16 expresses no MHC class II and very low levels of MHC class I, although both are inducible upon treatment with interferon-γ (23). The B16 melanoma expresses the mouse homologue (pmel-17) of human gp100, an enzyme involved in pigment synthesis that is expressed by the majority of malignant melanoma cells, as well as by normal melanocytes. gp100 is a member of a family of “self” (i.e., unmutated), melanoma/melanocyte differentiation antigens that are widely expressed by melanoma cells. These antigens are frequently the targets of T cells that infiltrate human tumors (24).
The generation of autoreactive T cells in vivo may be difficult because of the mechanisms of central and peripheral tolerance and the impact of antigen expression by tumor tissue on T cell function (14, 25–27). Altered peptide ligands have been used to improve the reactivity of T cells specific for self/tumor antigens in mouse and man (9, 28–30). For gp100, H-2Db-restricted CD8+ T cells capable of recognizing B16 melanoma and normal melanocytes could only be elicited when the altered peptide was used (31). We described previously the cloning of the unmutated mouse homologue of gp100 (mgp100) from B16 melanoma (32). We have also described the identification of a peptide derived from human (h)gp100, KVPRNQDWL (hgp10025–33), that represents an altered peptide ligand form of mgp10025–33, EGSRNQDWL (31). Upon establishing this model for vaccination against self/tumor antigen, our next goal was to identify immunotherapeutic strategies that would induce the regression of large established tumors using CD8+ T cells specific for a self-antigen.
Materials and Methods
Generation of Pmel-1 TCR Transgenic Mice.
To study self/tumor antigen-specific T cell responses to melanoma, we developed a transgenic mouse strain on a C57BL/6 background and named it pmel-1. RNA was isolated from clone 9, a gp10025–33-specific, H-2Db–restricted CD8+ T cell clone (31), and α and β TCR regions were amplified by 5′-Rapid Amplification of cDNA Ends (5′-RACE, Life Technologies) using constant region anti-sense primers α1 (5′-GGCTACTTTCAGCAGGAGGA-3′) and β1 (5′-AGGCCTCTGCACTGATGTTC-3′), respectively. 5′-RACE products were amplified with nested TCR α and β constant region primers α2 (5′-GGGAGTCAAAGTCGGTGAAC-3′) and β2 (5′-CCACGTGGTCAGGGAAGAAG-3′), and cloned into pCR4TOPO TA sequencing vectors (Invitrogen). TCR α and β transcripts were sequenced as Vα1/JαTA19/Cα and Vβ13S1/Dβ1/Jβ1S6/Cβ1. Genomic cloning PCR primers were designed based on the method described previously by Kouskoff et al. (33). The α and β genomic variable domains were PCR amplified (Perkin-Elmer) with primers gα1 (5′-TCTCCCGGGCTTCTCACTGCCTAGCCATGATGAAATCCTTGAGTGTTTC-3′) and gα2 (5′-GTAGCGGCCGCGTAAAATCTATCCTAGTGTTCCCCAGA-3′) or gβ1 (5′-GATCTCGAGAATCTGCCATGGGCACCAG-3′) and gβ2 (5′-GATACCGCGGTTCCTTTCCAAGACCAT-3′), respectively. The genomic variable domains were TA-cloned into pCR4TOPO (Invitrogen), validated by sequencing, subcloned into TCR cassette vectors provided by Dr. D. Mathis (Harvard Medical School, Boston, MA; reference 33), and co-injected into fertilized C57BL/6 embryos (Science Applications International Corporation) yielding three TCR transgenic founder lines. Unpublished transgenic mice expressing the α+/β+ transgenic TCR with specificity for a Kb-restricted epitope from β-galactosidase (34) were identically constructed in our laboratory and were used as controls in some experiments.
Mice and Tumor Cells.
C57BL/6 and pmel-1 TCR transgenic mice were bred and housed at the National Institutes of Health (NIH) and Netherlands Cancer Institute animal facilities. Rag-1−/− mice were obtained from Jackson ImmunoResearch Laboratories. B16 (H-2b) is a gp100+ spontaneous murine melanoma obtained from the National Cancer Institute tumor repository and maintained in culture media (CM) comprised of RPMI 1640 with 10% heat-inactivated fetal bovine serum (Biofluids), 0.03% l-glutamine, 100 μg/ml streptomycin, 100 μg/ml penicillin, and 50 μg/ml gentamicin sulfate (NIH Media Center).
Peptides and Recombinant Poxviruses.
All synthetic peptides were synthesized using regular F-MOC chemistry. The synthetic, H-2Db–restricted peptides hgp10025–33, KVPRNQDWL, mgp10025–33, EGSRNQDWL, and NP366–374, ASNENMETM, were synthesized by Peptide Technologies to a purity >99% by HPLC and amino acid analysis. All recombinant vaccinia viruses (rVVs) and recombinant fowlpox viruses (rFPVs) used in this paper have been described previously (31) and were either generated in our laboratory or provided by D. Panicali, L. Gritz, and A. Gomez-Yafal (Therion Biologics, Cambridge, MA). These viruses were constructed and purified as described by Earl et al. (35).
In Vitro Activation of Pmel-1 T Cells and Cytokine Release Assay.
Peripheral blood mononuclear cells or splenocytes from mice were depleted of erythrocytes by hypotonic lysis, cultured in CM with 30 IU/ml rhIL-2 in the presence of 1 μM hgp10025–33 peptide, and used on days 5–10 after start of the culture. For cytokine release assays, 105 T cells were cocultured in CM with 105 target cells or 1 μM of the indicated peptide. Supernatants were collected after 24 h and tested using an mIFN-γ ELISA kit (Endogen) according to the manufacturer's protocol.
Histology.
Skin and tumor samples were analyzed as described previously (36). In brief, 4-μm cryostat sections were air-dried overnight and fixed in acetone for 10 min at room temperature, preincubated in 5% (vol/vol) normal goat serum (Central Laboratory of the Netherlands Red Cross Blood Transfusion Service) and stained with 0.5 μg/ml anti–mouse vβ13 mAb-FITC or mIgG1-FITC control Ab (BD Biosciences) in PBS/1% BSA followed by rabbit anti-FITC (1:40,000; DakoCytomation), biotinylated goat anti–rabbit antibody (1:400; DakoCytomation) and streptavidin–biotin-conjugated alkaline phosphatase complex (1:100, ABC-protocol; DakoCytomation). Color was developed using Permanent red chromogen substrate (PRC kit; Cell Marque Corporation) and sections were counterstained with hematoxylin.
Flow Cytometry and Intracellular IFN-γ Assay.
To obtain appropriate lymphocyte samples, mice were either tail-bled on indicated days after vaccination or killed and splenectomized for fresh splenocytes. Where designated, splenocytes were cultured as described previously (31). Erythrocytes were removed by hypotonic lysis or ficoll gradient separation, and cells were stained with the indicated dilutions of mAbs against CD8a (1:20, clone 53–6.7), CD4 (1:10, clone H129.19), Vβ13 (1:10, clone MR12–3), CD25 (1:10, clone PC61), CD44 (1:10, clone IM7), CD62L (1:10, clone MEL-14), and CD69 (1:10, clone H1.2F3). All antibodies were purchased from BD Biosciences. H2-Db–mgp10025–33 tetramers were a gift from M. Toebes and T.N.M. Schumacher (Netherlands Cancer Institute, Amsterdam, Netherlands). Propidium iodide staining cells were excluded from analysis. Intracellular IFN-γ assay was performed using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's recommendations. Samples were analyzed using a FACScalibur™ flow cytometer and CELLquest™ software.
Adoptive Transfer and Tumor Treatment.
Mice were injected subcutaneously with 1–5 × 105 B16 melanoma cells and treated with intravenous adoptive transfer of freshly isolated 106–7 fresh splenocytes (∼2 × 106 CD8+ Vβ13+ T cells) or in vitro–activated pmel-1 splenocytes (106–7 CD8+ Vβ13+ T cells). Mice (n = 5 for all groups) were vaccinated by intravenous injection of 2 × 107 plaque-forming units of rVV or rFPV encoding mgp100, hgp100, or β-galactosidase (31) or by subcutaneous injection with 100 μl water/IFA emulsion containing 100 μg of mgp10025–33, hgp10025–33, or β-gal96–103 peptide followed by two daily intraperitoneal injections of 100 μg anti-CD40 mAb purified from FGK45 hybridoma culture supernatant. rhIL-2 (a gift from Chiron Corp.) was administered intraperitoneally directly after vaccination (100,000 Cetus Units or 600,000 IU rhIL-2 in PBS, twice daily for 3–5 d). Tumors were measured with calipers and the products of perpendicular diameters were recorded. Mice were killed once tumors reached 400 mm2. All experiments were performed in a blinded, randomized fashion (measuring investigator had no knowledge of the experimental group) and performed independently at least twice with similar results.
Results
Despite Large Numbers of gp100-specific T Cells, B16 Melanoma Grows Normally in Pmel-1 TCR Transgenic Mice.
To study self/tumor antigen-specific T cell responses to melanoma, we developed a transgenic mouse strain on a C57BL/6 background and named it pmel-1. Pmel-1 transgenic mice expressed the Vα1Vβ13 TCR from a cloned T cell (clone 9) described previously (31). Clone 9, like the pmel-1 TCR transgenic cells derived from it, recognized an H-2Db–restricted epitope corresponding to amino acids 25–33 of gp100. Virtually all (>95%) of the CD8+ T cells in pmel-1 TCR transgenic mice were Vβ13+, amounting to ∼20% of all splenocytes (Fig. 1 A). Pmel-1 T cells in blood and spleen generally expressed baseline levels of the activation/effector markers CD25, CD44, and CD69, indicating that most of the transgenic cells were in the naive state. The CD62L levels were as high if not higher than those found in normal splenocytes, which was also consistent with the finding that pmel-1 cells were largely naive.
Figure 1.

Despite large numbers of gp100-specific T cells, B16 melanoma grows normally in pmel-1 TCR transgenic mice. (A) Generation and characterization of pmel-1 TCR transgenic mice. Single cell suspensions of spleens from a 6-wk-old pmel-1 mouse and a nontransgenic C57BL/6 mouse littermate as well as pmel-1 splenocytes cultured with hgp10025–33 peptide were stained for CD8, Vβ13, and the activation markers CD25, CD44, CD62L, and CD69, and analyzed by FACS®. (B) Recognition of gp100 peptide by pmel-1 T cells. PBL from pmel-1 “D8” founder were cultured for 7 d with mgp10025–33 peptide, washed, and incubated with titrated doses of mgp10025–33 (native) or hgp10025–33 (altered) peptide. IFN-γ production was measured by ELISA on culture supernatants. (C) Pmel-1 T cells specifically recognize B16 melanoma. Cultured pmel-1 splenocytes (gray bars) or clone 9 (black bars) were coincubated with B16 melanoma, EL-4 thymoma, MCA207 sarcoma, MC38 colon carcinoma cells, or CM. Supernatants were assessed for IFN-γ production by ELISA. (D) B16 melanoma grows progressively in pmel-1 TCR transgenic mice. B16 cells were implanted in 6-wk-old pmel-1 T cell receptor transgenic mice and littermates not expressing the transgene. All experiments shown were performed independently at least two times with similar results.
Upon in vitro stimulation with 1 μM of the hgp10025–33 peptide, T cells from pmel-1 TCR transgenic mice proliferated extensively and developed an effector phenotype that included the up-regulation of CD25, CD44, and CD69 and the partial down-regulation of CD62L (Fig. 1 A). Functionally, pmel-1 T cells released IFN-γ after stimulation with mgp100, hgp100 peptide (Fig. 1 B), or B16 melanoma cells, which are naturally mgp100+ (Fig. 1 C). The transgenic T cells resembled the reactivity of the original clone 9, from which their α/β TCR was cloned (Fig. 1 C and not depicted). Thus, the pmel-1 transgenic mouse could be used as a source of naive T cells with specificity for the gp10025–33 epitope.
To determine the role of T cell precursor frequency in the ability of the immune system to reject tumors, we injected B16 tumor cells subcutaneously into pmel-1 TCR transgenic mice and nontransgenic littermates. Surprisingly, tumors grew at the same rate in pmel-1 mice despite the presence of overwhelming numbers of CD8+ gp100-specific T cells (Fig. 1 D). Likewise, the adoptive transfer of naive or in vitro–activated gp100-specific pmel-1 splenocytes into tumor-bearing mice alone or in combination with IL-2 had minimal effects on tumor growth or the survival of tumor-bearing mice. Thus, the mere presence of naive or activated tumor-specific T cells alone was insufficient to cause the regression of the subcutaneous tumor, thereby indicating that the pmel-1 cells were functionally tolerant to the tumor.
Immunization with Altered Peptide Ligand Results in Higher Numbers of Self/Tumor Antigen-Specific T Cells That Have Limited Antitumor Efficacy.
In an effort to break the functionally tolerant state of pmel-1 cells, we immunized tumor-bearing wild-type C57BL/6 mice with rVVs or rFPVs, encoding either the self-antigen, mgp100, or the altered ligand, hgp100 (31). To track the activation and antitumor activity of naive pmel-1 T cells in vivo, we adoptively transferred pmel-1 splenocytes into otherwise normal, unmanipulated mice bearing subcutaneous B16 tumors established for 3 d. Vaccination with rVVmgp100 did not cause any significant antitumor effect. However, vaccination with rVVhgp100 induced a modest delay in subcutaneous tumor growth in some experiments, one of which is shown (Fig. 2 A).
Figure 2.

Enhanced T cell and antitumor response by adoptively transferred T cells after vaccination with altered peptide ligand. (A) Enhanced antitumor efficacy upon vaccination with rVV encoding altered peptide ligand. Tumor growth was measured in C57BL/6 mice bearing 3-d B16 tumors that received pmel-1 splenocytes followed immediately by vaccination with rVVLacZ, rVVmgp100, rVVhgp100, or no treatment. (B) Increased numbers of pmel-1 T cells from mice immunized with rVV encoding altered peptide ligand. Lymphocytes from mice described in A were isolated from peripheral blood and stained for CD8 and Vβ13 before FACS® analysis. Numbers of CD8+Tm+ cells are depicted through time as percentage of total CD8+ cells. (C) Prolonged peptide/MHC dissociation time of altered peptide ligand. RMA-S cells were incubated with hgp10025–33 (closed symbols) or mgp10025–33 (open symbols, shown at corresponding concentrations) at 25°C overnight. The cells were washed three times, incubated at 37°C for the time designated, and added to 7-d cultured pmel-1 T cells for 24 h. IFN-γ in culture supernatants was quantified by ELISA. Experiments were performed independently at least twice with similar results.
FACS® analysis using mgp100-specific MHC class I tetramers showed that vaccination with rVVmgp100 induced a weak and sometimes undetectable T cell response, whereas use of rVVhgp100 markedly increased specific T cell numbers (Fig. 2 B). Similar results were obtained when we used a “minigene” rVV encoding the nine amino acid minimal peptide determinant preceded by an ER-insertion sequence (31; unpublished data), or when we used the synthetic 9-mer peptides emulsified in IFA and given with anti-CD40 antibody (37). Only immunization with the hgp10025–33 effectively induced gp100-specific T cells, and these T cell responses were cross-reactive with mgp10025–33. These findings confirmed earlier observations that gp100 sequences outside the minimal determinant do not contribute to the differential immunogenicity of rVVmgp100 and rVVhgp100 (31).
Both hgp10025–33 and mgp10025–33 peptides contain the optimal MHC anchor residues at the dominant anchor positions, 5 (N) and 9 (L). However, the difference in the three amino-terminal residues resulted in T cell recognition at >100-fold lower concentrations of the hgp10025–33 peptide when compared with mgp10025–33 (Fig. 1 B). We have assessed previously the binding efficiency of the peptides for the H-2 Db class I molecule, finding that the human peptide bound to H-2 Db with 100-fold greater efficiency than the mouse peptide (31). Here, we extended these observations by evaluating the dissociation time of peptide from MHC I molecules on RMA-S cells, again observing significantly decreased binding at time zero of mgp10025–33, compared with hgp10025–33, followed by an accelerated dissociation time for the mgp10025–33 peptide (Fig. 2 C).
Together, these results suggest that pmel-1 T cells can escape central and peripheral tolerance and are not completely refractory to stimulation by self-antigen because the immunogen encoding the altered peptide ligand activated and expanded these self-reactive T cells and conferred limited antitumor efficacy.
Stimulation of Adoptively Transferred T Cells through Antigen-specific Vaccination and IL-2 Causes Regression and Long-term Cures of Large Established Tumors.
Enhanced activation of adoptively transferred pmel-1 T cells by immunization with an altered peptide ligand resulted in inconsistent and limited antitumor effects, leading to growth retardation but not regression of small tumors. Therefore, we set out to determine other factors that might enhance self-specific T cell activation and antitumor effect. We have explored previously the uses of a variety of T cell costimulatory, growth, and activation factors and found that in mouse models, IL-2 can augment the function of recombinant virus–based vaccines (38). To determine if we could enhance the activation of antitumor T cells in a way that would enable them to treat large, established, vascularized tumors, we adoptively transferred pmel-1 splenocytes into mice bearing subcutaneous B16 tumors established for 7 (Fig. 3, A and B) or 14 d (Fig. 3, C and D). This adoptive transfer was followed by vaccination with rFPV encoding the hgp100-altered peptide ligand and administration of IL-2. This combined treatment reproducibly induced tumor regression and even long-term cures of mice bearing established tumors larger than 50 mm2 (Fig. 3 C).
Figure 3.

Adoptive transfer of tumor-specific T cells combined with vaccination and IL-2 causes regression and cure of large, established tumors. B16 tumor was implanted subcutaneously into C57BL/6 mice treated by adoptive transfer of fresh pmel-1 splenocytes ± vaccination with rFPVhgp100 either 7 (A and B) or 14 d (C and D) after tumor inoculation. IL-2 was administered twice daily for six doses. Fresh or cultured splenocytes were effective in the treatment of large, established tumors. Splenocytes derived from an identically constructed TCR transgenic mouse with specificity for β-galactosidase were used as a control in some experiments (C and D) and were not therapeutic. Statistically significant tumor regression was seen in mice treated with pmel-1 cells given in combination with rFPVhgp100 and IL-2 in >20 independently performed experiments. There were at least five mice/group in all experiments. Mouse survival consistently correlated with tumor growth reduction.
Invariably, as observed in >20 independently performed experiments, the combination of adoptive transfer of self/tumor-specific CD8+ T cells, antigenic stimulation with an altered peptide ligand, and administration of IL-2 was required to induce this strong antitumor effect. The regimen is ineffective against tumors that are gp100-negative (unpublished data). Administration of either pmel-1 cells alone, IL-2 alone, vaccine alone, or any combination of just two of the three of these immunotherapeutic components induced weak, if any, antitumor effects (Fig. 3, A and B). In an effort to use the model in a clinically relevant way, we have focused on the adoptive transfer of T cells. However, in mice transgenic for the pmel-1 TCR, IL-2 given in combination with vaccination using the altered form of gp100 was also therapeutically effective in the treatment of established B16 tumors (unpublished data).
T cells derived from fresh, naive splenocytes, or from cultured splenocytes activated ex vivo, could mediate the regression of large, established, subcutaneous tumors in vivo. All mice in Fig. 4 A that were treated with the combined regimen of adoptive transfer of pmel-1 cells (either with fresh or cultured T cells), IL-2, and vaccination with rFPVhgp100 were complete and long-term responders. These responses have been followed for >1 yr, and are often accompanied by the development of limited vitiligo, which generally begins at the site of tumor regression, and then spreads in an unpredictable pattern throughout the mouse. Vitiligo is not observed when mice are treated with control β-galactosidase–specific T cells (unpublished data). A representative cohort of complete responder mice observed >1 yr after treatment corresponds to the group treated with cultured T cells, IL-2, and vaccination with rFPVhgp100, shown in Fig. 4 B.
Figure 4.
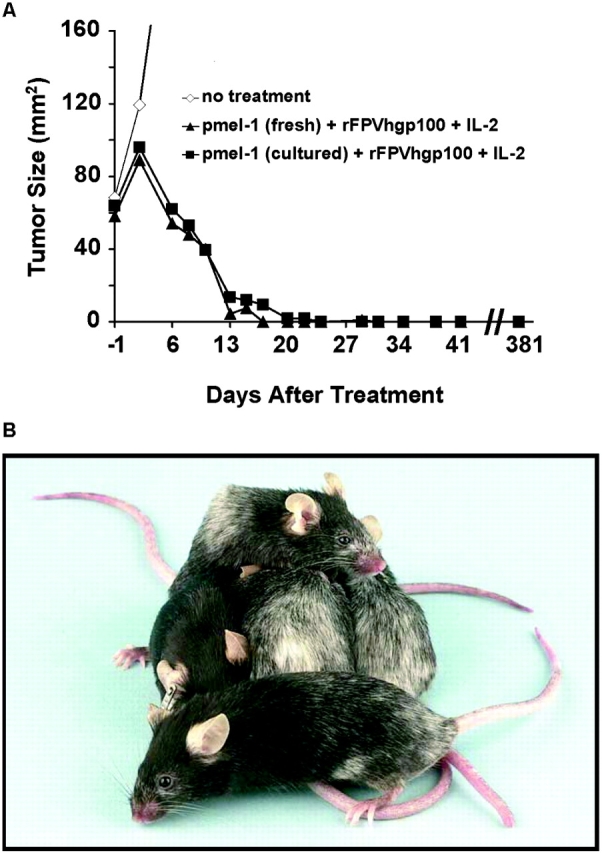
Long-term (>1 yr) survival of mice bearing large, established B16 tumors after treatment with adoptive transfer of tumor-specific T cells combined with vaccination and IL-2 is associated with the development of vitiligo. C57BL/6 mice were treated with adoptive transfer of fresh or cultured pmel-1 transgenic splenocytes 14 d after inoculation with B16 melanoma and vaccinated with rFPVhgp100. IL-2 was administered twice daily for six doses. Mice treated with fresh naive or cultured transgenic T cells were cured of B16, and vitiligo was observed, which started at the former tumor site. At >1-yr after therapy, these mice remain tumor-free with progressive vitiligo. To illustrate the autoimmune vitiligo, a photograph of the cohort of 5/5 surviving mice treated with cultured pmel-1 cells from A is shown in B.
It is important to note that complete cures of tumors are not always observed when using the triple combination of cells, vaccine, and cytokine, especially when doses of individual components are reduced (unpublished data). Thus, it may be useful to elucidate the minimal optimal treatment protocol required to reproducibly induce the regression of large B16 tumors established for 14 d. 105 Cetus units (6 × 105 IU) twice a day for 3 d was clearly sufficient when given in combination with 107 fresh or cultured pmel-1 cells and 2 × 107 plaque-forming units of rFPVhgp100 to reproducibly induce the regression of large B16 tumors established for 14 d. We used adoptively transferred cells in an effort to model a clinical possibility, but as mentioned earlier, vaccination plus IL-2 is effective in pmel-1 TCR transgenic mice bearing established tumors.
The administration of a combination of cultured pmel-1 T cells, vaccination, and IL-2 was required even in the absence of endogenous host B or T lymphocytes, as demonstrated by experiments in C57BL/6 Rag-1−/− knockout mice bearing subcutaneous B16 tumors established for 14 d (Fig. 5) . The cultured cells that were transferred were >98% CD8+ by FACS® (unpublished data). Tumors shown in Fig. 5 were treated with a suboptimal number (106) of pmel-1 cells, resulting in resurgent tumor growth. However, in experiments repeated more than five times, antitumor effects were at least as strong (and generally stronger) in Rag-1−/− mice as those seen in wild-type C57BL/6 mice (unpublished data).
Figure 5.

Endogenous host B or T lymphocytes are not required for the treatment of established B16 tumors. C57BL/6 Rag-1−/− knockout mice bearing subcutaneous B16 tumors established for 14 d were treated with 106 cultured pmel-1 cells given in combination with rFPVhgp100 and IL-2 as described previously. Similar results were obtained in five independently completed experiments.
IL-2 Increases the Number and Function of Self-specific, Tumoricidal T Cells.
To further understand the mechanisms of how the antitumor effector function of pmel-1 cells was enhanced by IL-2, we monitored the levels of pmel-1 T cells in the blood of mice using tetramer analysis to follow the transferred cells after treatment. Data shown in Fig. 6 (A–C) are obtained from the same, representative experiment. In this treatment model, using B16 tumors established for 3 d, cell transfer combined with immunization with the altered peptide ligand and IL-2 was required to observe the maximal therapeutic effect (Fig. 6 A). In other experiments, IL-2 could be replaced with either IL-7 or IL-15 (unpublished data). Cells plus rVV encoding the altered peptide ligand can induce limited destruction of small tumor burden, but this treatment effect is not significant in mice with large tumor burden (i.e., tumors established for 14 d).
Figure 6.

Persistence and function of gp100 reactive T cells after adoptive transfer, vaccination, and administration of IL-2 in mice bearing subcutaneous B16 tumors. (A–C) Data derived from the same, representative experiment. (A) Representative treatment experiment using C57BL/6 mice bearing B16 tumors established for 3 d were treated with fresh pmel-1 T cells plus vaccination with rVVLacZ, rVVmgp100, or rVVhgp100 with or without IL-2. (B) Vaccine-induced specific T cells in peripheral blood. Numbers of CD8+ Tm+ cells isolated from PBL of groups from A are depicted as percentage of total CD8+ cells. (C) Effect of boost with vaccine and IL-2. Mice surviving to day 30 (continuation of B) were retreated by heterologous boosting with rFPVhgp100 plus IL-2. Numbers of CD8+Vβ13+ cells isolated from blood are depicted as percentage of total CD8+ cells. (D) Effect of IL-2 on the number and function of vaccine-induced gp100-specific T cells in tumor tissue. The absolute numbers used to calculate the ratios shown were obtained 7 d after treatment (14 d after tumor implantation) and are normalized for 300 mg of tumor tissue (the average weight of excised tumors on day 14). These numbers are as follows (IL-2/PBS): total lymphocyte gate (1,758:945), CD8+ T cells (1,271:619), CD8+Vβ13+ T cells (890:266), and CD8+Vβ13+ IFN-γ+ T cells (261:8). After a 4-h restimulation with 1 μM mgp10025–33 peptide, the number of CD8+Vβ13+ IFN-γ+ T cells measured from this group were (754:147) to give a ratio of 5:1 (not depicted).
We found that the effect of IL-2 on the levels of pmel-1 T cells, as a percentage of CD8+ T cells or total nucleated cells in blood, was modest (two- to fourfold, at most, compared with treatment groups receiving adoptively transferred pmel-1 cells and vaccination at any given time in multiple experiments; Fig. 6 B). When measured as either percentage of all nucleated cells in blood or absolute numbers in the spleen, administration of IL-2 increased the relative number of pmel-1 T cells maximally threefold. IL-2 did not influence the induction of persistent memory T cells that could be reactivated by booster vaccination 32 d after priming (Fig. 6 C).
We sought to understand the impact of IL-2 administration on absolute numbers of pmel-1 T cells, and to measure the effect it had on their localization and effector function. We found that administration of IL-2 after adoptive transfer and vaccination increased the total number of lymphocytes infiltrating into 300 mg of solid tumor by 1.9-fold 7 d after vaccination (Fig. 6 D). The number of Vβ13+CD8+ T cells was increased by 3.3-fold, corresponding to the modest increase in Vβ13+CD8+ T cell levels in blood (Fig. 6 D). Strikingly, the number of Vβ13+CD8+ T cells that produced IFN-γ directly ex vivo and without further stimulation was increased 34-fold by IL-2 administration. These data illustrate that in the absence of IL-2, T cells are present at the tumor site but not fully activated. Thus, IL-2 acted primarily as a costimulatory/activation factor rather than purely as a growth factor.
Histological analysis of B16 tumors 7 d after treatment of 7-d tumors with pmel-1 cells plus vaccination showed specific T cell infiltration but no marked tumor cell death or tissue destruction (Fig. 7 , left). By contrast, when the adoptive transfer of pmel-1 cells plus vaccination was followed by administration of IL-2, we observed extensive tumor cell death and loss of tissue integrity (Fig. 7, right). These data show that strong vaccination by itself can induce proliferation and tumor localization of antigen-specific T cells, but that these T cells do not effectively mediate tumor destruction unless provided with exogenous cytokine.
Figure 7.
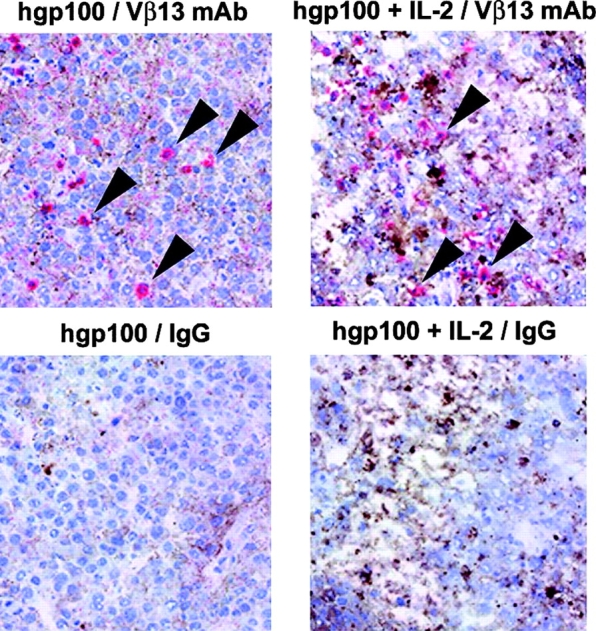
Histological analysis reveals presence of Vβ13+ T cells in tumors and demonstrates the activating effects of IL-2. T cells and tissue architecture within tumors were visualized after vaccination with or without IL-2. C57BL/6 mice bearing 14-d B16 tumors received 107 pmel-1 splenocytes and vaccination with hgp10025–33 peptide in IFA and anti-CD40 with or without IL-2. As was the case for vaccination with rVVs or rFPVs encoding hgp100, vaccination with hgp10025–33 peptide in IFA and anti-CD40 after the adoptive transfer of pmel-1 cells was greatly enhanced with the addition of IL-2 in repeated experiments (not depicted). Tumors were excised 7 d after the vaccination and 4-μm frozen sections were stained with anti-Vβ13 mAb (top, arrows) or IgG control (bottom), counterstained with hematoxylin, and photographed at an original magnification of 200. (left) pmel-1 cells plus hgp100 vaccination. (right) pmel-1 cells plus hgp100 vaccination followed by IL-2 administration.
Discussion
Strengths and Weaknesses of the Pmel-1 TCR Transgenic Model.
Here, we have investigated parameters that can be manipulated in vivo to transform a functionally tolerant transgenic T cell into an activated one that mediates the regression of large established tumors. As a tumor model, we used the poorly immunogenic B16 melanoma. This tumor failed to induce activation of adoptively transferred mgp100-specific pmel-1 T cells. Reports on the treatment of large, established tumors with systemic, rather than local therapies, remain rare, but not unprecedented in the scientific literature (39).
Most of the published mouse tumor models use prevention of tumor implantation and growth as the measure of success. When the treatment of established tumors is reported, treated tumors are generally significantly smaller than those treated in the present set of experiments. Furthermore, many of the existing tumor systems target model antigens that have been artificially inserted into the tumor genome, whereas the majority of human tumor–associated antigens used in current clinical efforts are nonmutated self-antigens. In studies focusing on adoptive immunotherapy, investigators generally report on the treatment of small pulmonary “metastases,” which are nodules created by the intravenous injection of tumor cells, which are treated with lymphocytes injected by the same route. The present paper describes the use of large, vascularized subcutaneous tumors.
One aspect of the current model that does not model the human tumor–bearing situation is the use of a transplantable tumor rather than spontaneous melanomas arising and progressing in each mouse, which could be subjected to treatment. Spontaneous tumor models can be more cumbersome and time-consuming to work with. However, the use of spontaneous tumors (40, 41) may more effectively model some aspects of tumor carcinogenesis and treatment. Efforts to validate the findings of the present paper in a model of spontaneous tumor are currently ongoing in the laboratory.
The Mere Presence of Tumor-reactive CD8+ T Cells Is Insufficient to Induce Tumor Regression.
The presence of overwhelming numbers of mgp100-specific T cells (20% of all splenocytes, >95% of all CD8+ T cells) in pmel-1 mice did not impact tumor growth. Apparently, large numbers of activated tumor-specific T cells are necessary but not sufficient to induce tumor destruction. This puzzling observation, observed several years ago in mice by Ohashi and colleagues (13), has been rediscovered in recent clinical trials (7–9, 42).
Provision of nonspecific systemic inflammation or “danger” signals by infection of mice with control vaccinia virus or injection of anti-CD40 mAb and IL-2 did not result in T cell activation or suppression of tumor growth. Furthermore, in vitro–activated specific T cells neither proliferated in response to tumor nor slowed the growth of even tiny subcutaneous tumors. This suggests that direct presentation of mgp100 antigen by tumor cells or its cross-presentation by activated professional APCs is insufficient to overcome peripheral tolerance and induce T cell activation and proliferation. A possible explanation for this poor presentation may reside in the low affinity of the mgp10025–33 peptide for H-2 Db and the resulting brief half-life of MHC–mgp10025–33 complexes on APCs (Fig. 2 C; references 31, 43). Indeed, vaccination with the self-antigen, mgp100, induced only marginal T cell proliferation and no control of tumor growth (Fig. 2, A and B; and Fig. 6, A and B).
When tumor-bearing recipients were vaccinated with a peptide epitope capable of binding H-2Db with 100-fold greater affinity than its mouse counterpart (31), we saw a greatly increased mgp100-specific T cell response (in which up to 80% of all CD8+ T cells in the blood can be mgp100-specific). It is possible that the use of an altered peptide ligand is not an absolute requirement in other cancer vaccines, particularly if the antigenic peptide of choice is, unlike the mgp100 peptide, highly capable of activating T cells. However, it is likely that T cells that have survived central and peripheral tolerance to self/tumor antigens generally target peptides with poor avidity. Our observations in mice seem analogous to those observed in human melanoma patients in which vaccination with the gp100209–217 peptide is inefficient, unless that peptide is altered to increase its avidity for the HLA-A*0201 molecule (9). Thus, the optimization of self/tumor peptides for binding to MHC molecules should be further explored in future cancer vaccine development.
One observation in mouse and in man is that therapeutic immunization designed to treat established tumor is generally ineffective. Where immunization can induce tumor antigen-specific T cells, the destruction of an established tumor is not reproducibly observed. In the present model, adoptive transfer of T cells was required in order for vaccine and IL-2 to reproducibly induce the destruction of tumor. There apparently are quantitative or qualitative differences between cells expanded ex vivo and used for adoptive transfer, and cells expanded in vivo as a result of immunization. Cells induced by vaccination are likely to be lower in number; furthermore, they may be quiescent or refractory to activation by tumor cells, perhaps in a way similar to the changes observed in T cells during chronic infection (44). Thus, we seek to use, in new clinical trials, the combined regimen of cells, IL-2, and vaccination with an altered peptide ligand described in this paper.
The use of lymphodepletion (nonmyeloablative conditioning) may enhance the function of adoptively transferred T cells as shown previously (45, 46). Lymphodepletion may facilitate immune responses by deletion of endogenous regulatory T cells, or by increasing the availability of endogenous immune activating cytokines such as IL-7 or IL-15. Results in the clinic are encouraging, but the consistent induction of complete responses in our patients with cancer remains the focus of our ongoing clinical efforts. We are now using the pmel-1 model to explore the immune-enhancing mechanisms of nonmyeloablative conditioning.
IL-2 Enhances CD8+ T Cell Function In Vivo.
IL-2 can be clinically successful but the mechanism responsible for the sometimes-dramatic clinical antitumor responses is unclear (47). We had found previously that IL-2 could augment the T cell response to a virus (38). These observations have been confirmed and expanded recently by others (48). In the present set of experiments, IL-2 seemed to function more as a T cell activation factor than as a growth factor: it not only boosted specific T cell numbers after immunization but also enhanced their ability to produce IFN-γ upon infiltration into tumors. It is possible that IL-2 increased both proliferation and death of pmel-1 T cells. We are currently investigating whether administration of IL-2 can be replaced by the induction of a productive CD4+ T cell response, the major source of IL-2 during a typical pathogen-specific immune response, or by other cytokines, including IL-15.
Autoimmune Consequences of Immunotherapy Targeted at the gp100 Self Antigen.
Vitiligo, the usually incomplete destruction of normal tissue, was observed in most mice where successful tumor destruction was observed (Fig. 4). Induction of vitiligo has been correlated with tumor protection in the induction of TRP-1–specific immune responses (5, 49, 50). In humans, vitiligo has previously been highly correlated with successful response to IL-2–based immunotherapy in patients with metastatic malignant melanoma (51).
Our observations in the pmel-1 model, and in human patients treated with T cells (45), are that CD8+ T cell activation results in the destruction of tumor cells at least as well as normal cells. Thus, we did not find evidence of a tumor-specific “barrier” that prevented T cell–mediated tumor destruction while allowing destruction of normal tissue (20). Both tumor and normal tissue were attacked and destroyed by the treatment regimen, with the tumor destruction generally being more complete in this model.
Conclusion.
Together, these observations have allowed us to identify three requirements that were all strictly necessary yet not individually sufficient for reversing the functionally tolerant state of adoptively transferred T cells resulting in strong tumor regression: (a) adoptive transfer of tumor-specific T cells; (b) T cell stimulation through antigen-specific vaccination using an altered peptide ligand rather than the native self-peptide for vaccination; and (c) coadministration of IL-2. When used in combination, these requirements defined the components of successful antitumor treatment. Based on these data, clinical trials that use adoptive cell transfer used in combination with IL-2 and vaccination with an altered peptide ligand are being initiated in patients with cancer.
Acknowledgments
We would like to thank D. Panicali, L. Gritz, and A. Gomez-Yafal from the Therion Biologics for providing recombinant poxviruses used in this work, T.N.M. Schumacher and M. Toebes for help with tetramer production, P. van den Berk for producing FGK45 anti-CD40 mAb, and J. Yang and P. Hwu for helpful discussions.
This work was supported in part by a grant (KWF 2001-2562) from the Dutch Cancer Society.
Abbreviations used in this paper: CM, culture media; rFPV, recombinant fowlpox virus; rVV, recombinant vaccinia virus.
W.W. Overwijk, M.R. Theoret, and S.E. Finkelstein contributed equally to this work.
References
- 1.Houghton, A.N., J.S. Gold, and N.E. Blachere. 2001. Immunity against cancer: lessons learned from melanoma. Curr. Opin. Immunol. 13:134–140. [DOI] [PubMed] [Google Scholar]
- 2.Weber, L.W., W.B. Bowne, J.D. Wolchok, R. Srinivasan, J. Qin, Y. Moroi, R. Clynes, P. Song, J.J. Lewis, and A.N. Houghton. 1998. Tumor immunity and autoimmunity induced by immunization with homologous DNA. J. Clin. Invest. 102:1258–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardoll, D.M. 2002. Spinning molecular immunology into successful immunotherapy. Nat. Rev. Immunol. 2:227–238. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg, S.A. 2001. Progress in human tumour immunology and immunotherapy. Nature. 411:380–384. [DOI] [PubMed] [Google Scholar]
- 5.Overwijk, W.W., D.S. Lee, D.R. Surman, K.R. Irvine, C.E. Touloukian, C.C. Chan, M.W. Carroll, B. Moss, S.A. Rosenberg, and N.P. Restifo. 1999. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc. Natl. Acad. Sci. USA. 96:2982–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Overwijk, W.W., and N.P. Restifo. 2000. Autoimmunity and the immunotherapy of cancer: targeting the “self” to destroy the “other.” Crit. Rev. Immunol. 20:433–450. [PMC free article] [PubMed] [Google Scholar]
- 7.Marchand, M., N. van Baren, P. Weynants, V. Brichard, B. Dreno, M.H. Tessier, E. Rankin, G. Parmiani, F. Arienti, Y. Humblet, et al. 1999. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int. J. Cancer 80:219–230. [DOI] [PubMed] [Google Scholar]
- 8.Nestle, F.O., S. Alijagic, M. Gilliet, Y. Sun, S. Grabbe, R. Dummer, G. Burg, and D. Schadendorf. 1998. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 4:328–332. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg, S.A., J.C. Yang, D.J. Schwartzentruber, P. Hwu, F. Marincola, S.L. Topalian, N.P. Restifo, M.E. Dudley, S.L. Schwarz, P.J. Spiess, et al. 1998. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 4:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen, L.T., A.R. Elford, K. Murakami, K.M. Garza, S.P. Schoenberger, B. Odermatt, D.E. Speiser, and P.S. Ohashi. 2002. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J. Exp. Med. 195:423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Touloukian, C.E., W.W. Leitner, R.E. Schnur, P.F. Robbins, Y. Li, S. Southwood, A. Sette, S.A. Rosenberg, and N.P. Restifo. 2003. Normal tissue depresses while tumor tissue enhances human T cell responses in vivo to a novel self/tumor melanoma antigen, OA1. J. Immunol. 170:1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Touloukian, C.E., W.W. Leitner, P.F. Robbins, Y.F. Li, X. Kang, R. Lapointe, P. Hwu, S.A. Rosenberg, and N.P. Restifo. 2002. Expression of a “self-”antigen by human tumor cells enhances tumor antigen-specific CD4(+) T-cell function. Cancer Res. 62:5144–5147. [PMC free article] [PubMed] [Google Scholar]
- 13.Speiser, D.E., R. Miranda, A. Zakarian, M.F. Bachmann, K. McKall-Faienza, B. Odermatt, D. Hanahan, R.M. Zinkernagel, and P.S. Ohashi. 1997. Self antigens expressed by solid tumors do not efficiently stimulate naive or activated T cells: implications for immunotherapy. J. Exp. Med. 186:645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Staveley-O'Carroll, K., E. Sotomayor, J. Montgomery, I. Borrello, L. Hwang, S. Fein, D. Pardoll, and H. Levitsky. 1998. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA. 95:1178–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spiotto, M.T., P. Yu, D.A. Rowley, M.I. Nishimura, S.C. Meredith, T.F. Gajewski, Y.X. Fu, and H. Schreiber. 2002. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 17:737–747. [DOI] [PubMed] [Google Scholar]
- 16.Wang, M., V. Bronte, P.W. Chen, L. Gritz, D. Panicali, S.A. Rosenberg, and N.P. Restifo. 1995. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J. Immunol. 154:4685–4692. [PMC free article] [PubMed] [Google Scholar]
- 17.Ochsenbein, A.F., S. Sierro, B. Odermatt, M. Pericin, U. Karrer, J. Hermans, S. Hemmi, H. Hengartner, and R.M. Zinkernagel. 2001. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 411:1058–1064. [DOI] [PubMed] [Google Scholar]
- 18.Tham, E.L., P. Shrikant, and M.F. Mescher. 2002. Activation-induced nonresponsiveness: a Th-dependent regulatory checkpoint in the CTL response. J. Immunol. 168:1190–1197. [DOI] [PubMed] [Google Scholar]
- 19.Hanson, H.L., D.L. Donermeyer, H. Ikeda, J.M. White, V. Shankaran, L.J. Old, H. Shiku, R.D. Schreiber, and P.M. Allen. 2000. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 13:265–276. [DOI] [PubMed] [Google Scholar]
- 20.Wick, M., P. Dubey, H. Koeppen, C.T. Siegel, P.E. Fields, L. Chen, J.A. Bluestone, and H. Schreiber. 1997. Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J. Exp. Med. 186:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melief, C.J., S.H. Van Der Burg, R.E. Toes, F. Ossendorp, and R. Offringa. 2002. Effective therapeutic anticancer vaccines based on precision guiding of cytolytic T lymphocytes. Immunol. Rev. 188:177–182. [DOI] [PubMed] [Google Scholar]
- 22.Dranoff, G., E. Jaffee, A. Lazenby, P. Golumbek, H. Levitsky, K. Brose, V. Jackson, H. Hamada, D. Pardoll, and R.C. Mulligan. 1993. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA. 90:3539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seliger, B., U. Wollscheid, F. Momburg, T. Blankenstein, and C. Huber. 2001. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res. 61:1095–1099. [PubMed] [Google Scholar]
- 24.Kawakami, Y., N. Dang, X. Wang, J. Tupesis, P.F. Robbins, R.F. Wang, J.R. Wunderlich, J.R. Yannelli, and S.A. Rosenberg. 2000. Recognition of shared melanoma antigens in association with major HLA-A alleles by tumor infiltrating T lymphocytes from 123 patients with melanoma. J. Immunother. 23:17–27. [DOI] [PubMed] [Google Scholar]
- 25.Sarma, S., Y. Guo, Y. Guilloux, C. Lee, X.F. Bai, and Y. Liu. 1999. Cytotoxic T lymphocytes to an unmutated tumor rejection antigen P1A: normal development but restrained effector function in vivo. J. Exp. Med. 189:811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colella, T.A., T.N. Bullock, L.B. Russell, D.W. Mullins, W.W. Overwijk, C.J. Luckey, R.A. Pierce, N.P. Restifo, and V.H. Engelhard. 2000. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen. Implications for tumor immunotherapy. J. Exp. Med. 191:1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Parijs, L., and A.K. Abbas. 1998. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 280:243–248. [DOI] [PubMed] [Google Scholar]
- 28.Irvine, K.R., M.R. Parkhurst, E.P. Shulman, J.P. Tupesis, M. Custer, C.E. Touloukian, P.F. Robbins, A.G. Yafal, P. Greenhalgh, R.P. Sutmuller, et al. 1999. Recombinant virus vaccination against “self” antigens using anchor-fixed immunogens. Cancer Res. 59:2536–2540. [PMC free article] [PubMed] [Google Scholar]
- 29.Slansky, J.E., F.M. Rattis, L.F. Boyd, T. Fahmy, E.M. Jaffee, J.P. Schneck, D.H. Margulies, and D.M. Pardoll. 2000. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 13:529–538. [DOI] [PubMed] [Google Scholar]
- 30.Dyall, R., W.B. Bowne, L.W. Weber, J. LeMaoult, P. Szabo, Y. Moroi, G. Piskun, J.J. Lewis, A.N. Houghton, and J. Nikolic-Zugic. 1998. Heteroclitic immunization induces tumor immunity. J. Exp. Med. 188:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Overwijk, W.W., A. Tsung, K.R. Irvine, M.R. Parkhurst, T.J. Goletz, K. Tsung, M.W. Carroll, C. Liu, B. Moss, S.A. Rosenberg, and N.P. Restifo. 1998. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 188:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhai, Y., J.C. Yang, P. Spiess, M.I. Nishimura, W.W. Overwijk, B. Roberts, N.P. Restifo, and S.A. Rosenberg. 1997. Cloning and characterization of the genes encoding the murine homologues of the human melanoma antigens MART1 and gp100. J. Immunother. 20:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kouskoff, V., K. Signorelli, C. Benoist, and D. Mathis. 1995. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods. 180:273–280. [DOI] [PubMed] [Google Scholar]
- 34.Overwijk, W.W., D.R. Surman, K. Tsung, and N.P. Restifo. 1997. Identification of a Kb-restricted CTL epitope of beta-galactosidase: potential use in development of immunization protocols for “self” antigens. Methods. 12:117–123. [DOI] [PubMed] [Google Scholar]
- 35.Earl, P.L., N. Cooper, and B. Moss. 1991. Preparation of cell cultures and vaccinia virus stocks. In Current Protocols in Molecular Biology. F.M. Asubel, R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, and L.A. Smith, editors. Greene Publishing Associates and Wiley Interscience, New York. 16.16.1–16.16.17.
- 36.Vyth-Dreese, F.A., H. Boot, T.A. Dellemijn, D.M. Majoor, L.C. Oomen, J.D. Laman, M. Van Meurs, R.A. De Weger, and D. De Jong. 1998. Localization in situ of costimulatory molecules and cytokines in B-cell non-Hodgkin's lymphoma. Immunology. 94:580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sotomayor, E.M., I. Borrello, E. Tubb, F.M. Rattis, H. Bien, Z. Lu, S. Fein, S. Schoenberger, and H.I. Levitsky. 1999. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 5:780–787. [DOI] [PubMed] [Google Scholar]
- 38.Bronte, V., K. Tsung, J.B. Rao, P.W. Chen, M. Wang, S.A. Rosenberg, and N.P. Restifo. 1995. IL-2 enhances the function of recombinant poxvirus-based vaccines in the treatment of established pulmonary metastases. J. Immunol. 154:5282–5292. [PMC free article] [PubMed] [Google Scholar]
- 39.Tamura, Y., P. Peng, K. Liu, M. Daou, and P.K. Srivastava. 1997. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science. 278:117–120. [DOI] [PubMed] [Google Scholar]
- 40.Kannan, K., N.E. Sharpless, J. Xu, R.C. O'Hagan, M. Bosenberg, and L. Chin. 2003. Components of the Rb pathway are critical targets of UV mutagenesis in a murine melanoma model. Proc. Natl. Acad. Sci. USA. 100:1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noonan, F.P., J.A. Recio, H. Takayama, P. Duray, M.R. Anver, W.L. Rush, E.C. De Fabo, and G. Merlino. 2001. Neonatal sunburn and melanoma in mice. Nature. 413:271–272. [DOI] [PubMed] [Google Scholar]
- 42.Lee, P.P., C. Yee, P.A. Savage, L. Fong, D. Brockstedt, J.S. Weber, D. Johnson, S. Swetter, J. Thompson, P.D. Greenberg, et al. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5:677–685. [DOI] [PubMed] [Google Scholar]
- 43.Gold, J.S., C.R. Ferrone, J.A. Guevara-Patino, W.G. Hawkins, R. Dyall, M.E. Engelhorn, J.D. Wolchok, J.J. Lewis, and A.N. Houghton. 2003. A single heteroclitic epitope determines cancer immunity after xenogeneic DNA immunization against a tumor differentiation antigen. J. Immunol. 170:5188–5194. [DOI] [PubMed] [Google Scholar]
- 44.Wherry, E.J., J.N. Blattman, K. Murali-Krishna, M.R. van der, and R. Ahmed. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dudley, M.E., J.R. Wunderlich, P.F. Robbins, J.C. Yang, P. Hwu, D.J. Schwartzentruber, S.L. Topalian, R. Sherry, N.P. Restifo, A.M. Hubicki, et al. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 298:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kjaergaard, J., L. Peng, P.A. Cohen, J.A. Drazba, A.D. Weinberg, and S. Shu. 2001. Augmentation versus inhibition: effects of conjunctional OX-40 receptor monoclonal antibody and IL-2 treatment on adoptive immunotherapy of advanced tumor. J. Immunol. 167:6669–6677. [DOI] [PubMed] [Google Scholar]
- 47.Overwijk, W.W., M.R. Theoret, and N.P. Restifo. 2000. The future of interleukin-2: enhancing therapeutic anticancer vaccines. Cancer J. Sci. Am. 6:S76–S80. [PMC free article] [PubMed] [Google Scholar]
- 48.Blattman, J.N., J.M. Grayson, E.J. Wherry, S.M. Kaech, K.A. Smith, and R. Ahmed. 2003. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 9:540–547. [DOI] [PubMed] [Google Scholar]
- 49.Trcka, J., Y. Moroi, R.A. Clynes, S.M. Goldberg, A. Bergtold, M.A. Perales, M. Ma, C.R. Ferrone, M.C. Carroll, J.V. Ravetch, and A.N. Houghton. 2002. Redundant and alternative roles for activating Fc receptors and complement in an antibody-dependent model of autoimmune vitiligo. Immunity. 16:861–868. [DOI] [PubMed] [Google Scholar]
- 50.Bowne, W.B., R. Srinivasan, J.D. Wolchok, W.G. Hawkins, N.E. Blachere, R. Dyall, J.J. Lewis, and A.N. Houghton. 1999. Coupling and uncoupling of tumor immunity and autoimmunity. J. Exp. Med. 190:1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenberg, S.A., and D.E. White. 1996. Vitiligo in patients with melanoma: normal tissue antigens can be targets for cancer immunotherapy. J. Immunother. Emphasis Tumor Immunol. 19:81–84. [PubMed] [Google Scholar]