

Abstract
The intracellular pathogen Brucella is the causative agent of brucellosis, a worldwide zoonosis that affects mammals, including humans. Essential to Brucella virulence is its ability to survive and replicate inside host macrophages, yet the underlying mechanisms and the nature of the replicative compartment remain unclear. Here we show in a model of Brucella abortus infection of murine bone marrow–derived macrophages that a fraction of the bacteria that survive an initial macrophage killing proceed to replicate in a compartment segregated from the endocytic pathway. The maturation of the Brucella-containing vacuole involves sustained interactions and fusion with the endoplasmic reticulum (ER), which creates a replicative compartment with ER-like properties. The acquisition of ER membranes by replicating Brucella is independent of ER-Golgi COPI-dependent vesicular transport. A mutant of the VirB type IV secretion system, which is necessary for intracellular survival, was unable to sustain interactions and fuse with the ER, and was killed via eventual fusion with lysosomes. Thus, we demonstrate that live intracellular Brucella evade macrophage killing through VirB-dependent sustained interactions with the ER. Moreover, we assign an intracellular function to the VirB system, as being required for late maturation events necessary for the biogenesis of an ER-derived replicative organelle.
Keywords: macrophage, Brucella, endoplasmic reticulum, type IV secretion, trafficking
Introduction
Pathogenic bacteria have evolved various strategies to counteract host defense mechanisms to create a safe niche of replication inside the host. In mammals, this involves surviving innate components of the immune response, such as killing by professional phagocytes. For instance, intracellular bacterial pathogens are capable of avoiding degradation along the endocytic pathway through various means (for review see reference 1), many mechanisms of which are yet to be understood.
Several of the microorganisms comprising the α-2 subgroup of proteobacteria have coevolved with their eukaryotic hosts, including bacteria of the genus Brucella. Others include the plant pathogen Agrobacterium tumefaciens, the plant symbiont Rhizobium meliloti, and the intracellular animal pathogens Bartonella spp. and Rickettsia spp. (2). Brucella is the causative agent of brucellosis, a worldwide zoonosis that affects a broad range of mammals, including livestock and humans (3). Human brucellosis is a chronic, debilitating disease with severe, and sometimes fatal, outcome (3). Brucella is considered a facultatively extracellular, intracellular pathogen that survives and replicates inside both phagocytic and nonphagocytic host cells (4). Intracellular survival and replication are key virulence features of Brucella because mutants defective for these attributes are also avirulent (5, 6). In infected hosts, Brucella resides in tissues of the reticuloendothelial system and survives intracellularly within macrophages, a feature essential to the establishment, development, and chronicity of the disease. However, the underlying molecular mechanisms of the bacteria's interaction with macrophages remain unclear.
It has been proposed, and partially demonstrated, that Brucella intracellular survival in macrophages results from an inhibition of fusion between the Brucella-containing vacuole (BCV) and lysosomes, through alteration of BCV maturation along the endocytic pathway (7, 8). Yet, the nature of the Brucella replicative compartment in macrophages is controversial (4). It has been suggested that Brucella replicate inside compartments with phagolysosomal characteristics (7), but this would imply a completed process of phagosome maturation, which contradicts with the inhibition of BCV fusion with lysosomes.
In HeLa cells, Brucella abortus and Brucella melitensis prevent lysosome fusion by escaping from the endocytic pathway before interacting with late endosomes. They transit through an intermediate vacuole with autophagic features, and ultimately replicate in a compartment characterized by the presence of ER markers (9–11). Consistently, early ultrastructural observations in Vero cells, or trophoblasts from infected pregnant goats, have shown the presence of Brucella spp. replicating in the ER (12, 13).
Attempts by several laboratories to identify bacterial genetic determinants involved in Brucella virulence recurrently led to the identification of the virB locus (5, 6, 11, 14–16). This region is homologous to the virB operon of A. tumefaciens, which encodes a type IV secretion system (14). Such machineries are dedicated to the secretion or export of nucleoprotein complexes or proteins, and many operate in pathogenic bacteria, such as A. tumefaciens, Bordetella pertussis, Helicobacter pylori, or the intracellular pathogen Legionella pneumophila (17). Various Brucella virB mutants are avirulent in a mouse model (5, 6, 16) and a functional VirB system is required for establishing and maintaining a persistent infection (5). Furthermore, this locus is required for intracellular survival in macrophages and HeLa cells (11, 14–16). Recent evidence suggests that the VirB system is involved in controlling the maturation of BCV into an organelle permissive for replication (11, 18). Thus, the current hypothesis is that VirB mediates translocation of bacterial effector molecule(s) into the host cell that controls BCV maturation (11, 18, 19). However, no VirB effector has been identified to date and the stage(s) of BCV maturation controlled by VirB is yet to be resolved.
Here we have performed a detailed analysis of B. abortus intracellular fate within murine bone marrow–derived macrophages (BMDM) to precisely define the nature, and understand the biogenesis of, the Brucella replicative compartment in professional phagocytes. In addition, we have also further defined the role of the VirB type IV secretion system in BCV maturation in macrophages. Our results clearly establish the essential role of the BCV interactions with the macrophage ER in bacterial intracellular survival.
Materials and Methods
Bacterial Strains.
The bacterial strains used in this study were the smooth virulent B. abortus strain 2308 (9) and an isogenic nonpolar mutant of the virB10 gene (16). Green fluorescent protein (GFP)-expressing derivatives were made by electroporating both strains with a pBBR1MCS-2 derivative (20) expressing the gfp-mut3 gene under the control of the lac promoter. Bacteria were grown in tryptic soy broth (TSB; Sigma-Aldrich) and kept frozen at −80°C in 15% glycerol-TSB medium. For infection, 2 ml TSB were inoculated with a single bacterial colony from a freshly streaked TSB agar plate and grown at 37°C for 15 h (early stationary phase) up to an optical density (OD600nm) of ∼2.0. In these growth conditions, the VirB type IV secretion system was expressed (unpublished data).
Antibodies and Reagents.
The primary antibodies used were: rabbit polyclonal anti–cation-independent mannose-6-phosphate receptor (CI-M6PR; 9); anti-Rab7 (21); anti–lysosomal-associated membrane glycoprotein 1 (LAMP-1; 22); anti–human cathepsin D (DakoCytomation); anti-calnexin (StressGen Biotechnologies); anti–human calreticulin (Affinity BioReagents, Inc.); anti-sec61β (9); rat anti–mouse LAMP-1 monoclonal antibody 1D4B (Developmental Studies Hybridoma Bank, National Institute of Child Health and Human Development, National Institutes of Health, University of Iowa); goat polyclonal anti–human EEA-1 (Santa Cruz Biotechnology, Inc.); and cow polyclonal anti–smooth B. abortus 2308 (9). The secondary antibodies used were: Alexa Fluor® 488–conjugated goat anti–cow, Alexa Fluor® 594–conjugated goat anti–rabbit, and Alexa Fluor® 594–conjugated donkey anti–goat IgG (Molecular Probes); and cyanin-5–conjugated donkey anti–rat (Jackson ImmunoResearch Laboratories). The fluorescent probe monodansylcadaverine (MDC; Sigma-Aldrich) was used to label autophagic compartments by incubating infected cells with 0.05 mM MDC in the culture medium for 30 min before processing for immunofluorescence. Proaerolysin was provided by G. van der Goot (University of Geneva, Geneva, Switzerland) and was used on infected cells at a concentration of 0.5 nM for the indicated time. Brefeldin A (BFA; Sigma-Aldrich) was used at a final concentration of 10 μg/ml.
BMDM Culture and Infection.
Bone marrow cells were isolated from femurs of 6–10-wk-old C57Bl/6 female mice and differentiated into macrophages as previously described (23). Infections were performed at a multiplicity of infection of 50:1 by centrifuging bacteria onto macrophages at 400 g for 10 min at 4°C, and then incubating the cells for 15 min at 37°C under 7% CO2 atmosphere. Macrophages were extensively washed with DMEM to remove extracellular bacteria and incubated for an additional 90 min in medium supplemented with either 100 μg/ml gentamicin (strain 2308) or 100 μg/ml streptomycin (strain virB10) to kill extracellular bacteria. Thereafter, the antibiotic concentrations were decreased to 10 μg/ml. At each time point, samples were washed three times with PBS before processing. To monitor Brucella intracellular survival, infected cells were lysed with 0.1% (vol/vol) Triton X-100 in H2O after PBS washing and serial dilutions of lysates were rapidly plated onto TSB agar plates to enumerate CFUs.
Immunofluorescence Microscopy.
Macrophages were fixed with 3% paraformaldehyde, pH 7.4, at 37°C for 10 min, and then processed for immunofluorescence staining as previously described (9). Specimens were observed either on a Leica DMRBE epifluorescence microscope for quantitative analysis, or a Zeiss LSM 510 laser scanning confocal microscope for image acquisition. Images of 1,024 × 1,024 pixels were acquired and assembled using Adobe Photoshop 7.0.
Transmission Electron Microscopy (EM).
Macrophages were processed as previously described (23). The detection of glucose-6-phosphatase (G6Pase) activity was performed by EM cytochemistry as previously described (24) with minor modifications. Cells were prefixed in 1.25% (vol/vol) glutaraldehyde in 0.1 M Pipes, pH 7.0, containing 5% (wt/vol) sucrose for 30 min on ice, washed three times for 3 min in 0.1 M Pipes, pH 7.0, containing 10% (wt/vol) sucrose, and then briefly in 0.08 M Tris-maleate buffer, pH 6.5. Cells were incubated in 0.06 M glucose-6-phosphate (Sigma-Aldrich), 0.1% lead nitrate (wt/vol) in 0.08 M Tris-maleate, pH 6.5, for 2 h at 37°C. After three washes in 0.08 M Tris-maleate buffer for 2 min and three washes in 0.1 M cacodylate buffer, pH 7.2, containing 0.1 M sucrose, 5 mM CaCl2, and 5 mM MgCl2, cells were postfixed in 1.25% (vol/vol) glutaraldehyde in the same buffer for 1 h at 4°C, and then with 1% OsO4 in the same buffer devoid of sucrose for 1 h at room temperature. Samples were then further processed as previously described (23).
Results
Survival and Replication of B. abortus inside Murine BMDM.
We first monitored the survival and replication of both the wild-type 2308 and the virB10 mutant strains by evaluating the number of CFUs retrieved from infected primary cultures of murine BMDM at different times after infection. When macrophages were infected with the wild-type strain, ∼90% of the ingested bacteria were rapidly killed by macrophages (Fig. 1 A) because a decrease of one order of magnitude in the number of intracellular live Brucella occurred within the first 4 h of infection (Fig. 1 A). Thereafter, some of the surviving wild-type Brucella were capable of replication as indicated by an increase in the number of intracellular bacteria after 4 h after infection (Fig. 1 A). When analyzing the fate of a virB10 mutant strain, a decrease of intracellular live bacteria comparable to that of the wild-type strain was observed until 4 h after infection (Fig. 1 A), indicating that a functional VirB system is not required for the short-term survival of Brucella inside macrophages. At later time points, the number of live virB10 mutants progressively decreased. Hence, the Brucella virB10 strain did not replicate, but rather was killed. To better define this time course of events, we monitored the overall levels of macrophage infection by scoring the number of intracellular bacteria for 2308- or virB10-infected BMDM. At 2 h after infection, for both strains the majority of BMDM contained less than five bacteria and the infection levels were similar (Fig. 1 B). By 8 h after infection, for both strains the percentage of BMDM with less than five bacteria decreased, whereas the number of uninfected macrophages significantly increased (Fig. 1 B). Thus, by 8 h after infection some macrophages cleared Brucella, consistent with CFU counts (Fig. 1 A). By 24 h after infection, 15.0 ± 6.1% of 2308-infected BMDM contained >10 bacteria, demonstrating that bacterial replication had occurred, but only within a limited number of macrophages. In contrast, no replication was observed for virB10 bacteria. Strikingly, the majority of the macrophage population at 24 h after infection was devoid of bacteria, for both 2308 and virB10 infections (Fig. 1 B), demonstrating a continuous killing of intracellular Brucella after 4 h of infection To confirm this, we analyzed by immunofluorescence the fusion events between BCVs and lysosomes by monitoring the colocalization of bacteria with the lysosomal luminal hydrolase cathepsin D (Fig. 1 C). Until 4 h after infection, a similar percentage of 2308- and virB10-containing vacuoles were cathepsin D+ (Fig. 1 C), demonstrating some fusion of wild-type and virB10 phagosomes with lysosomes at early stages of the infection. These results are consistent with the decreased number of intracellular bacteria observed for both strains over this time frame (Fig. 1 A). After 4 h of infection, coincident with the onset of bacterial replication, the percentage of cathepsin D+ 2308-containing vacuoles decreased (Fig. 1 C). By contrast, the percentage of cathepsin D+ virB10-containing vacuoles significantly increased with time (Fig. 1 C), consistent with bacterial killing as assessed by CFU counting (Fig. 1 A). Indeed, the majority of wild-type Brucella did not colocalize with cathepsin D at 24 h after infection, whereas most of the virB10-containing vacuoles were cathepsin D+ (Fig. 1 D). This indicates that although the virB10 mutants are capable of short-term survival, they cannot evade long-term degradation through fusion with lysosomes. Taken together, these results show that a fraction of intracellular Brucella that survive the initial macrophage killing are capable of further survival and replication, via a VirB-dependent prolonged evasion of fusion with lysosomes.
Figure 1.

Survival and replication of Brucella strains within BMDM. BMDM were infected with either the wild-type 2308 or the virB10 mutant Brucella strains for various times. CFUs were enumerated after lysis (A) or samples were fixed and processed for immunofluorescence (B–D). (A) Representative growth curves of wild-type 2308 (○) and virB10 (□) strains inside BMDM. (B) Quantitation of 2308- or virB10-infected BMDM containing either no bacteria (open bars), less than five bacteria (light gray bars), between 5 and 10 bacteria (dark gray bars), or >10 bacteria (solid bars) at 2, 8, and 24 h after infection. Data are means ± SD of three independent experiments. (C) Quantitation of cathepsin D acquisition by BCVs. Data are means ± SD of three independent experiments. (D) Representative confocal images showing colocalization of Brucella with cathepsin D in BMDM infected with either the wild-type 2308 or the virB10 mutant Brucella strains for 24 h. Arrows indicate colocalization and arrowheads show cathepsin D− BCVs. Bar, 5 μm.
Replicating BCVs Do Not Interact with Late Endocytic Compartments.
Next, we focused on defining the nature of the Brucella replicating compartment in macrophages. During our initial experiments, we observed that one (by 24 h after infection) to two thirds (by 48 h after infection) of the intracellular Brucella (detected by either 4′,6′-diamino-2-phenylindole or GFP expression) were not labeled by various Brucella-specific antisera (unpublished data), suggesting that significant surface changes on intracellular Brucella occur during replication. To overcome the shortcomings of the antisera staining and ensure we were analyzing the majority of the intracellular bacteria, we used GFP-expressing Brucella strains. Intramacrophagic GFP expression correlated well with intact bacteria because only a small fraction (10% at 4 h after infection and 13% at 24 h after infection) of intracellular GFP-2308 were accessible to propidium iodide staining after in vivo saponin permeabilization of macrophage membranes (unpublished data). Thus, we examined the BCV trafficking in BMDM by immunofluorescence. 5 min after entry, Brucella were found in compartments positive for the early endosomal antigen 1 EEA-1 (Fig. 2 A), indicating that early BCVs interact with early endosomes. This interaction was very transient because little to no colocalization was detected by 30 min after infection (Fig. 2 A). Concurrent with this decrease, a rapid acquisition of LAMP-1 was observed (Fig. 2 A) and the majority of BCVs remained LAMP-1+ until 4 h after infection (Fig. 2 C). Little to no colocalization with either the Rab7 GTPase, CI-M6PR, or cathepsin D was detected at the same time points, suggesting that LAMP-1 found on BCVs did not originate from interactions with late endosomes (Fig. 2 C). At times later than 4 h after infection, the percentage of LAMP-1+ BCVs progressively decreased to 9.7 ± 1.2% at 24 h after infection (Fig. 2, B and C), whereas no colocalization of Brucella with Rab7, CI-M6PR, or cathepsin D was detected (Fig. 2, B and C). Taken together, these results demonstrate that live Brucella interact with early but not late compartments of the endocytic pathway in primary macrophages, which excludes a late endosomal nature of the Brucella replicative compartment.
Figure 2.
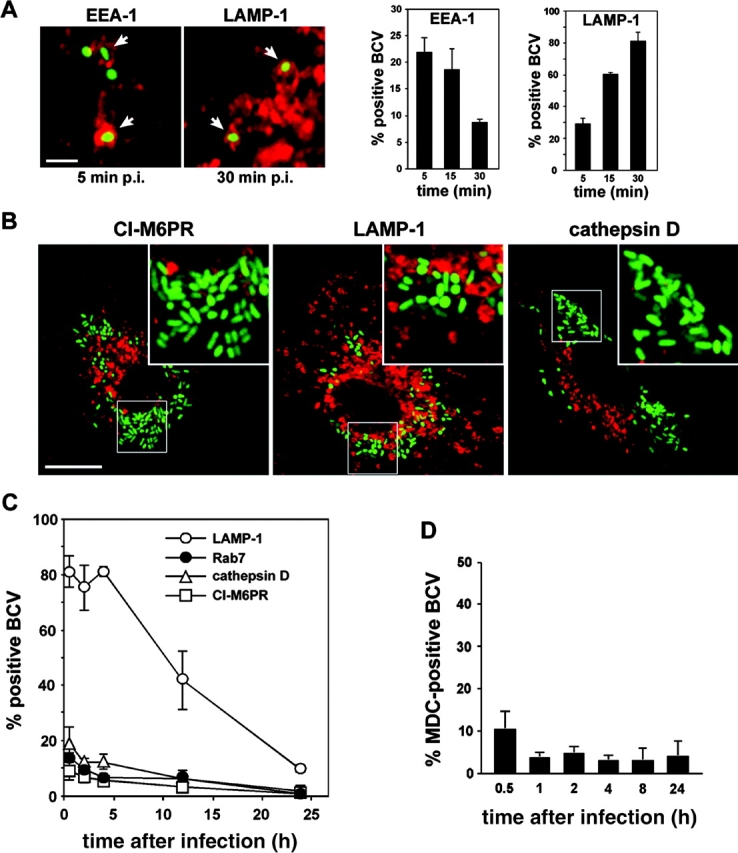
Live Brucella interact with early but not late endocytic compartments. BMDM were infected with the GFP-expressing Brucella wild-type strain GFP-2308 for various times. Samples were fixed and processed for immunofluorescence. (A) Confocal images and quantitation of EEA-1 or LAMP-1 acquisition by early BCVs. Arrowheads show intracellular Brucella within either EEA-1+ or LAMP-1+ vacuoles. Data are means ± SD of three independent experiments. (B) Confocal images of GFP-2308–infected BMDM at 24 h after infection. Replicating Brucella do not colocalize with either CI-M6PR, LAMP-1, or cathepsin D. (C) Quantitation of LAMP-1, Rab7, CI-M6PR, and cathepsin D acquisition by BCVs during maturation. Data are means ± SD of three to five independent experiments. (D) Quantitation of MDC accumulation within BCVs. Data are means ± SD of three independent experiments. Bars, 2 μm (A) and 10 μm (B).
BCVs Do Not Interact with Autophagic Compartments in Macrophages.
In epithelial cells, the autophagic pathway has been identified as an intermediate of Brucella intracellular trafficking (9, 10). By contrast, no significant colocalization of GFP Brucella with organelles positive for the autophageous probe MDC (25) in BMDM was observed at any time point examined (Fig. 2 D). Furthermore, all the MDC+ BCVs scored (a maximum of 10.3 ± 4.0% at 30 min after infection) contained bacteria with barely detectable levels of GFP expression (not depicted), hence likely degraded. Thus, Brucella do not seem to interact with autophagic compartments in BMDM, which was also previously described for J774 macrophage-like cells (7).
BCVs Acquire ER Markers during Maturation into a Replicative Organelle.
Given the interactions of intracellular Brucella with the ER of tissue culture epithelial cells (9, 11, 13) and trophoblasts from infected animals (12), we sought to determine whether such interactions also occurred in BMDM. Therefore, we analyzed the presence of specific markers of the ER on BCVs at various time points after infection, using immunofluorescence microscopy. A significant number of BCVs (a maximum of 35.7 ± 3.5% at 20 min after infection) were surrounded by ER+ structures as early as 10 min after entry (Fig. 3 B, inset), as described recently for phagocytosis of latex beads and some microbial pathogens (26). Thereafter, the number of calnexin+ BCVs slightly decreased and then increased again by 4 h after infection whereby 37.7 ± 6.7% of BCVs were positive (Fig. 3, A and B), suggesting sequential interactions with the ER. At later time points, such as 12 h after infection, 66.0 ± 6.0% of Brucella were tightly surrounded by calnexin labeling in a pattern different from earlier time points, which suggested a staining of the BCV itself (Fig. 3 A). By 24 h after infection, 91.0 ± 6.2% of BCVs were positive for calnexin (Fig. 3, A and B). Similar patterns were obtained when detecting the ER chaperone calreticulin or the translocator Sec61β (not depicted). The biphasic interactions observed do suggest that the early and late colocalizations of ER markers with BCVs are independent events. Additionally, these results indicate that BCVs interact with the ER during their maturation and acquire ER markers concomitantly with the biogenesis of the replicative compartment.
Figure 3.
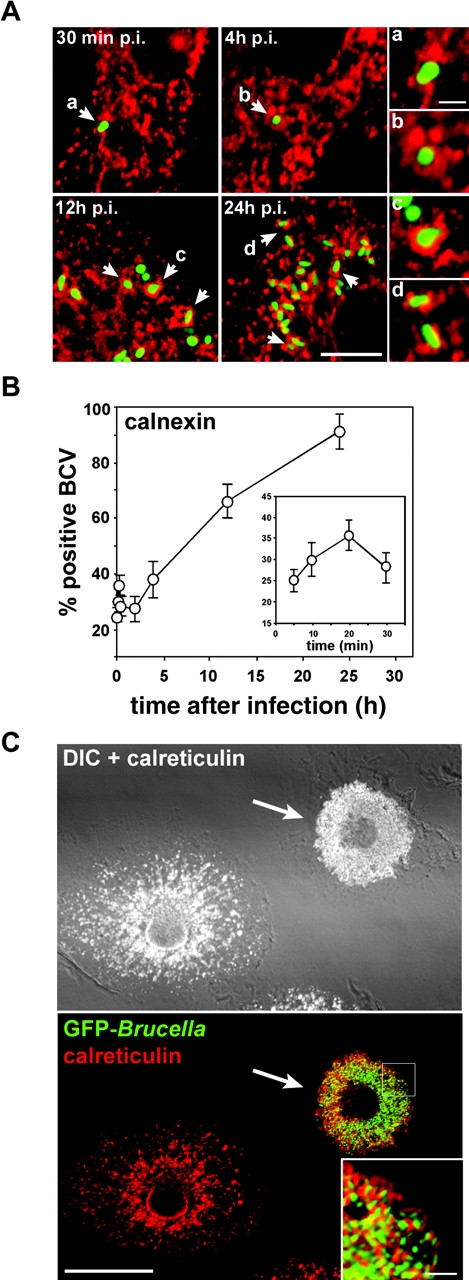
BCVs acquire ER markers during maturation. BMDM were infected with the GFP-expressing Brucella wild-type strain GFP-2308 for various times. Samples were fixed and processed for immunofluorescence. (A) Representative confocal images of a time course of GFP–Brucella interaction with ER structures. Arrowheads indicate calnexin+ BCVs. (B) Quantitation of BCV association with, and acquisition of, calnexin+ structures. Data are means ± SD of five independent experiments. (C) Overimposed confocal and differential interference contrast (DIC) images of BMDM infected with GFP Brucella for 48 h. The ER was labeled using rabbit anti–calreticulin, followed by Alexa Fluor® 594–conjugated goat anti–rabbit antibodies. The arrow indicates a Brucella-infected BMDM, whose ER is reorganized into or around BCVs. Bars, 5 μm (A), 1 μm (A insets), 20 μm (C), and 2 μm (C inset).
To test whether Brucella recruit ER membranes to their vacuole during maturation, we examined the ER structure of infected macrophages at 48 h after infection, when an extensive replication of Brucella has occurred. Compared with uninfected macrophages, Brucella-infected cells showed a dramatic reorganization of the ER, which was constrained to the bacterial replication area. No detectable ER structures were observed elsewhere in the infected cells (Fig. 3 C). This strongly suggests that the extensive replication of Brucella is linked to an accretion of ER, which could originate from ER membrane acquisition during BCV maturation.
Intermediate BCVs Harbor both LAMP-1 and ER Markers.
Given the striking presence of LAMP-1 on maturing BCVs (Fig. 2), we next assessed whether these LAMP-1+ BCVs acquire ER markers by examining the presence of both markers on bacterial vacuoles by confocal microscopy. During the first 4 h after infection, the majority of BCVs were positive only for LAMP-1 (Fig. 4 A). This population decreased thereafter to only 4.6 ± 2.3% of BCVs at 24 h after infection, indicating this LAMP-1+ BCV population is not persistent (Fig. 4 A) and corresponds to vacuoles that mature into either degradative or replicative compartments, hence either disappear or acquire ER markers. Interestingly, calnexin+/LAMP-1+ BCVs could be detected during the first 12 h after infection (Fig. 4 B), with the peak between 2 and 8 h after infection (28.4 ± 3.2% at 4 h after infection; Fig. 4 A). Nearly one third of the BCVs harbored both LAMP-1 and calnexin at times up to 8 h after infection, which demonstrates that a significant population of LAMP-1+ BCVs interacted with the ER (Fig. 4 B). Furthermore, ∼10% of BCVs were only positive for calnexin at 2 and 4 h after infection. This demonstrates that most calnexin+ BCVs detected up to 4 h after infection (Fig. 3 B) are also LAMP-1+ (Fig. 4 A), strongly suggesting these BCVs originate as LAMP-1+ vacuoles. At times later than 8 h after infection, the percentage of LAMP-1+/calnexin+ vacuoles decreased, whereas that of calnexin+ BCVs progressively increased (41.9 ± 7.5% at 12 h after infection and 87.7 ± 2.0% at 24 h after infection; Fig. 4, A and B). Taken together, these results demonstrate the occurrence of an early population of LAMP-1+/calnexin+ BCVs, which precedes the appearance of calnexin+ only vacuoles. Thus, BCV maturation is evidenced by an intermediate population of bacterial vacuoles that can mature into calnexin+ replicative compartments through the exclusion of LAMP-1, possibly by recycling LAMP-1+ membranes while interacting with the ER.
Figure 4.
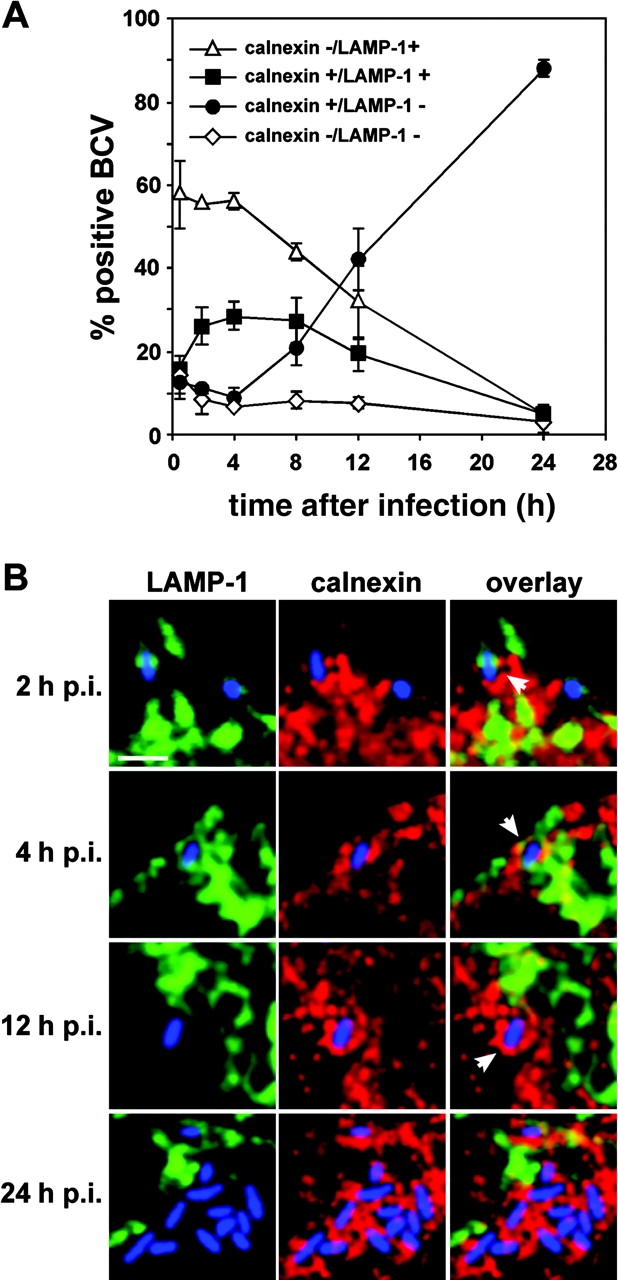
Some BCVs transiently harbor both LAMP-1 and calnexin. BMDM were infected with the wild-type GFP Brucella for various times. Samples were fixed and processed for immunofluorescence. Calnexin was detected using rabbit anti–calnexin, followed by Alexa Fluor® 594–conjugated goat anti–rabbit antibodies. LAMP-1 was detected using a rat anti–mouse LAMP-1 monoclonal antibody, followed by cyanin-5–conjugated donkey anti–rat antibodies. (A) Quantitation of calnexin−/LAMP-1+ (▵), calnexin+/LAMP-1+ (▪), calnexin+/LAMP-1− (•), and calnexin−/LAMP-1− (⋄) BCVs during a 24-h time course. BCVs were scored for LAMP-1 and calnexin presence from single 0.2-μm section images obtained by confocal microscopy. (B) Details from single confocal image sections showing representative BCVs at various times after infection. For clarity, the images have been artificially recolored to show bacteria in blue, LAMP-1 in green, and calnexin in red. Early BCVs (2 and 4 h after infection) harbor both markers (arrowheads) whereas late (12 h after infection) and replicative BCVs (24 h after infection) are only stained with calnexin (arrowhead). Bar, 2 μm.
BCVs Interact with the ER to Exchange Membranes during Maturation.
To address the possible acquisition of ER membranes by BCVs, we performed an ultrastructural analysis of the immediate environment of BCVs by EM. BCVs were found closely associated with some ER structures as early as 4 h after infection (Fig. 5 A). At 24 h after infection, replicating BCVs were surrounded by ER (Fig. 5 B), demonstrating a sustained association with this organelle. Of particular interest, these vacuoles were locally studded with ribosomes (Fig. 5 C) and intimate points of contact with the ER were detected (Fig. 5 D). This strongly suggested the occurrence of membrane exchange between BCVs and the ER. To confirm this, we examined the presence of the ER-specific G6Pase on BCVs, as a marker of BCV–ER fusion. At early time points (30 min after infection), no G6Pase was detected either inside or close to most BCVs (Fig. 5 E). By 2 h after infection, BCV contacted G6Pase+ structures and this was more accentuated at 4 h after infection (Fig. 5, F and G). However, no G6Pase was detected inside BCVs at these stages, showing that no major fusion events between BCV and ER had occurred. By contrast, at 24 h after infection, most BCVs were G6Pase+ (Fig. 5 H), and some were indeed found fusing with the ER (Fig. 5 I). Such results explain the different patterns of ER staining around or on the BCVs observed by immunofluorescence at early and late times after infection as sequential interaction events with the ER (Fig. 3 A). Together with our previous results, this clearly demonstrates that BCVs are capable of sustained interactions with the ER during maturation, and fuse with this organelle in a limited manner to acquire ER membranes.
Figure 5.
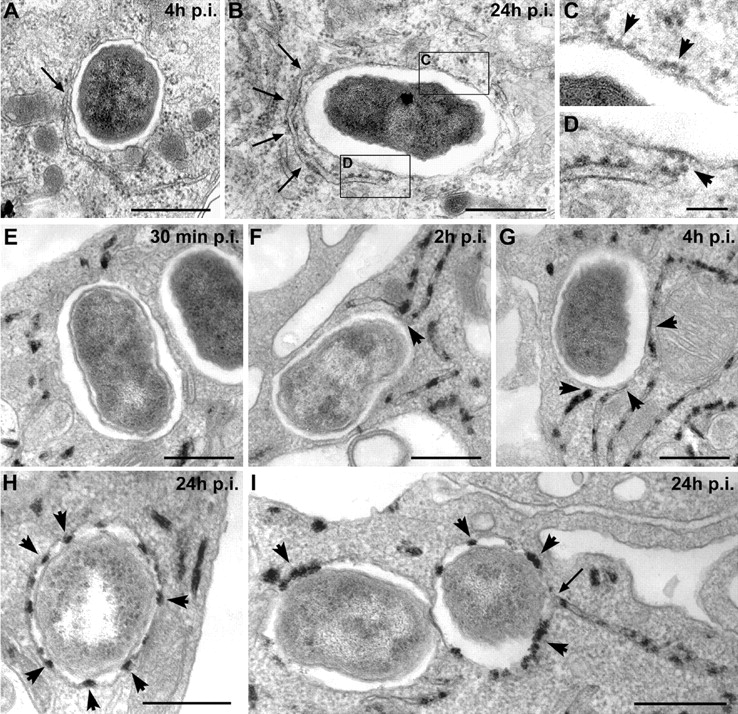
BCVs acquire ER membranes through limited fusion with the ER. BMDM were infected with the Brucella wild-type strain 2308 for various times. Samples were processed for conventional EM (A–D) or EM cytochemistry for G6Pase detection (E–I). (A) BCVs at 4 h after infection showing a close contact with the ER (arrow). (B) BCVs at 24 h after infection surrounded by ER (arrows). (C) Enlarged view of the vacuolar membrane from B showing ribosomes studding the membrane (arrowheads). (D) Enlarged view of the contact site between ER and the vacuolar membrane showing ribosomes on the vacuolar membrane (arrowhead). (E) BCVs at 30 min after infection showing no interaction with G6Pase+ ER. (F) BCVs at 2 h after infection in close contact with G6Pase+ ER (arrowheads). (G) BCVs at 4 h after infection in intimate contact with several G6Pase+ ER compartments (arrowheads). (H) Representative G6Pase+ late BCVs at 24 h after infection (arrowheads show the intravacuolar G6Pase reaction product). (I) G6Pase+ late BCVs at 24 h after infection. The right-hand side BCV is fusing with the ER (arrow). Bars, 1 μm (A and B), 0.2 μm (C and D), and 0.5 μm (E–I).
Late BCVs Are ER-derived Organelles.
To determine whether BCVs harbor ER functional properties, we reasoned that the toxin aerolysin from Aeromonas hydrophila, which vacuolates the ER (27), would also affect BCVs if they have acquired ER properties. BMDM infected with GFP Brucella for 36 h were left untreated or treated with 0.5 nM proaerolysin for 30 min before processing for immunofluorescence. In control macrophages, GFP Brucella were scattered in the perinuclear region in calnexin+ compartments (Fig. 6 A). By contrast, aerolysin treatment caused a dramatic reorganization of bacteria, which gathered inside calnexin+ giant vacuoles (Fig. 6 B). Because Brucella replicate in single bacterium-containing vacuoles in untreated macrophages (Fig. 6 C), the effect of aerolysin can be interpreted as triggering the fusion of individual BCVs. To confirm this, we monitored the action of aerolysin during the treatment using EM. 10 min after toxin addition to the infected cells, a typical swelling of the ER was observed (Fig. 6 D, arrows) and BCVs were found to fuse with the ER (Fig. 6 D, arrowhead). After 30 min, BCVs were fused together and covered with ribosomes, whereas ER structures were no longer detected (Fig. 6 E), demonstrating that aerolysin treatment had caused the fusion of BCVs together and with the ER. Thus, these results show that late BCVs not only harbor ER markers, but also display ER membrane properties, demonstrating that Brucella replicate in an ER-derived organelle inside macrophages.
Figure 6.
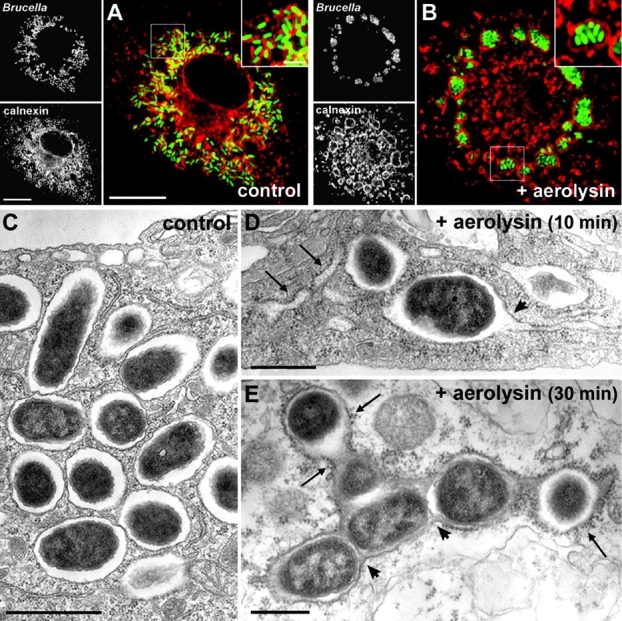
Late BCVs harbor ER properties. BMDM were infected with the wild-type GFP Brucella for 36 h, and then treated with 0.5 nM proaerolysin or mock treated for 10 or 30 min before processing for either immunofluorescence (A and B) or EM (C–E). (A) Confocal images of a control untreated macrophage showing replicating GFP Brucella scattered around the nucleus in calnexin+ vacuoles (red). The inset shows individual bacteria within calnexin+ vacuoles. (B) Confocal images of an aerolysin-treated macrophage showing a dramatic relocation of GFP Brucella into giant calnexin+ vacuoles. The inset shows multiple bacteria enclosed within a large calnexin+ vacuole. (C) EM picture of an untreated macrophage showing replicating Brucella in individual vacuoles. (D) EM picture of an infected macrophage treated with proaerolysin for 10 min. Typical aerolysin-induced swelling of the ER is visible (arrows) and BCVs start to fuse with the ER (arrowhead). (E) EM picture of an infected macrophage treated with proaerolysin for 30 min. BCVs have fused together (arrowheads) into a large compartment covered with ribosomes (arrows). Bars, 10 μm (A and B), 2 μm (insets in A and B), 1 μm (C), and 0.5 μm (D and E).
Biogenesis of the Brucella Replicative Vacuole Does Not Require ER-Golgi COPI-dependent Vesicular Transport.
The intracellular pathogen L. pneumophila replicates within macrophages in an ER-derived organelle (28). Recently, Kagan and Roy (29) showed that biogenesis of the Legionella replicative organelle depends upon ARF1/COPI-dependent ER-Golgi vesicular transport, and Legionella intracellular replication is inhibited when macrophages are pretreated with BFA. To examine whether Brucella replication is also affected by the inhibition of ER-Golgi COPI-dependent vesicular transport, BMDM were treated before or at various times after infection with BFA and CFU were enumerated at 1 and 24 h after infection. These treatments did not exceed 3 h to avoid cytotoxicity. BFA pretreatment did not impair Brucella uptake by macrophages, as determined by the number of intracellular bacteria at 1 h after infection (Fig. 7 A). Unlike Legionella, BFA treatment 30 min before infection did not affect Brucella intracellular replication, nor did BFA treatments at 30 min, 2, 5, or 8 h after infection (Fig. 7 A). To exclude that BFA blocked biogenesis of the BCV, yet vacuolar maturation resumed after the 3-h BFA treatments in a manner undetectable by CFU-based assays, we compared the net increase in calnexin+ BCVs in untreated and BFA-treated macrophages over various time periods. Compared with untreated controls, BFA treatment did not affect the rate of ER acquisition by BCVs, either from 30 min to 4 h after infection (Fig. 7 B), 4 to 8 h after infection (Fig. 7 C), or 8 to 12 h after infection (Fig. 7 D). Thus, the biogenesis of the Brucella replicative vacuole does not require ER-Golgi COPI-dependent vesicular transport, which indicates that the mechanisms of ER membrane acquisition by Brucella in macrophages are different to that of Legionella.
Figure 7.

Brucella intracellular replication does not require ER-Golgi COPI-dependent vesicular transport. (A) BMDM infected with the wild-type Brucella strain 2308 were left untreated or treated with 10 μg/ml BFA either 30 min before infection, or 30 min, 2, 5, or 8 h after infection, over a 3-h period. For each treatment, CFUs were enumerated after 1 and 24 h after infection. Data are from a representative experiment (n = 3 independent experiments). (B–D) BMDM infected with the wild-type Brucella strain GFP-2308 were left untreated or treated with 10 μg/ml BFA either from 30 min to 4 h after infection (B), 4–8 h after infection (C), or 8–12 h after infection (D). Samples were processed for immunofluorescence staining and calnexin+ BCVs were scored. Data are means ± SD of three independent experiments.
Maturation of BCVs into an ER-derived Organelle Depends upon a Functional VirB System.
Our previous results (Fig. 1) suggested a role for VirB in BCV late maturation events because the intracellular fate of wild-type and virB10 Brucella diverged after 4 h after infection. To clearly determine a role for VirB in BCV maturation in BMDM, we examined the vacuole maturation of the surviving GFP-expressing virB10 Brucella and compared their interactions with the ER to those of the wild-type GFP-2308. Using LAMP-1 as a marker of BCV maturation (Fig. 2 C), we observed that the majority of vacuoles containing either strain were LAMP-1+ until 4 h after infection (Fig. 8, A and B) . However, although the percentage of LAMP-1+ wild-type BCVs decreased after 4 h after infection, vacuoles containing the virB10 mutant remained positive (Fig. 8, A and B), as previously described in epithelial cells (18). This indicates that virB mutants are deficient in controlling vacuole maturation in BMDM after 4 h after infection.
Figure 8.

VirB mutant–containing vacuoles fail to sustain fusion-proficient interactions with the ER. BMDM were infected with either the wild-type Brucella strain GFP-2308 or the mutant strain GFP-virB10 for various times. Samples were processed for immunofluorescence (A–D) or EM staining for G6Pase (E–G). (A) Confocal images of infected BMDM showing acquisition of LAMP-1 on 2308 or virB10-containing vacuoles after 4 or 24 h of infection. (B) Quantitation of LAMP-1 acquisition by 2308 (○)- or virB10 (□)-containing vacuoles. Data are means ± SD of three independent experiments. (C) Confocal images of GFP Brucella–infected BMDM showing association with, or acquisition of, Sec61β+ structures on 2308 or virB10-containing vacuoles after 4 or 24 h of infection. (D) Quantitation of association of Sec61β1 structures with 2308 (○)- or virB10 (□)-containing vacuoles. Data are means ± SD of five independent experiments. (E) Staining for G6Pase in virB10-infected BMDM at 2 h after infection. The arrowhead shows a close association of BCVs with ER. (F) Staining for G6Pase in virB10-infected BMDM at 4 h after infection. The BCV is surrounded by ER (arrowheads). (G) Staining for G6Pase in virB10-infected BMDM at 8 h after infection Arrowheads show that the ER is no longer in the close vicinity of the BCV. G6Pase reaction product is not detectable inside the BCV. Bars, 5 μm (A and C), 1 μm (insets in A and C), and 0.5 μm (E–G).
We also compared the interaction of the virB10 mutant with ER structures using the translocator Sec61β as an ER marker. Until 4 h after infection, both the wild-type and virB10 mutant were surrounded by Sec61β+ structures (Fig. 8 C) to a comparable level (Fig. 8 D). Indeed, vacuoles containing the virB10 mutant were found in intimate contact with the ER (Fig. 8, E and F), like wild-type BCVs (Fig. 5, F and G). Thus, the absence of a functional VirB system does not prevent Brucella from initially contacting the ER. However, after 4 h of infection, although the percentage of Sec61β+ wild-type BCVs continued to increase (88.7 ± 6.4% positive at 24 h after infection; Fig. 8 D), that of the virB10 mutant–containing vacuoles stalled and then decreased to 25.7 ± 4.7% over the same time frame (Fig. 8 D). By EM, the few vacuoles containing intact bacteria we observed by 8 h after infection were no longer associated with the ER and no BCV–ER fusions were observed, as assessed by lack of G6Pase staining of bacterial vacuoles (Fig. 8 G). Hence, virB10 mutants are unable to control ER membrane acquisition to their vacuole. As a consequence, their compartment cannot mature into a replication-competent organelle and eventually fuses with lysosomes (Fig. 1). Taken together, these results demonstrate that the VirB type IV secretion system of Brucella is required for late maturation events of the vacuole, which correspond to the BCV–ER fusion necessary for the biogenesis of the Brucella ER-derived replicative vacuole.
Discussion
In this work, we have used an infection model of murine BMDM and identified some key aspects of Brucella intracellular survival and replication. We have shown that intracellular replication of B. abortus occurs within single bacterium-containing compartments segregated from the endocytic pathway, which possess properties of the host cell ER (Fig. 9) . Biogenesis of these organelles results from sustained interactions and limited fusion with the ER that depends upon a functional VirB type IV secretion system.
Figure 9.

Model for Brucella evasion of macrophage killing. After entry, intracellular Brucella resides within a vacuole (BCV) that interacts with early endosomes. These early BCVs avoid further interactions with the endocytic pathway, yet acquire LAMP-1 and are found surrounded by, or in close contact with, the ER within the first hours after infection (A). Such interactions are sustained over time and lead to limited fusion events (B), ultimately generating an ER-derived organelle permissive for Brucella replication (C). Vacuoles containing a virB-defective mutant fail to sustain fusion-proficient interactions with the ER (D) and ultimately fuse with lysosomes (E). The shaded area designates the VirB-dependent maturation events of BCVs described in this work. ER membrane acquisition by BCVs is illustrated by the localized presence of ribosomes.
Given their significant amounts of active hydrolytic enzymes (23) and the large number of late endosomes/lysosomes (30), BMDM constitute an adequate model to confront intracellular Brucella with highly bactericidal conditions, in which absence of fusion with lysosomes is not due to the scarcity of degradative organelles. Indeed, the majority of phagocytosed Brucella are rapidly degraded within BMDM. However, some bacteria do survive this initial killing event and some eventually replicate intracellularly. A mutant of the VirB type IV secretion system is initially killed to the same extent as the wild-type strain, indicating that Brucella initial survival is independent of VirB (Fig. 9). Similar results have been reported in human monocytes, using a Brucella suis virB9 mutant (31). Recently, Sun et al. (32) showed that the B. abortus VirB system is not involved in evading inducible nitric oxide synthase– or NADPH oxidase–mediated killing in vivo, which is in agreement with our results. In our system, VirB was only implicated in BCV trafficking events occurring after 4 h after infection, specifically the maturation events required for long-term survival and replication (Fig. 9). This is consistent with intracellular expression kinetics of the B. suis virB operon in J774 cells (19). A VirB-defective mutant, which cannot control BCV interactions with the ER, remains in an intermediate, immature compartment that ultimately fuses with lysosomes. Hence, the VirB-dependent maturation of the BCV into an ER-derived replicative compartment is essential to long-term survival and allows the creation of a safe intracellular niche.
Our results (Figs. 2 and 9) exclude a late endosomal nature for the Brucella replicative compartment in macrophages, as has been previously purported (7). Nonetheless, BCVs transiently interact with early but not late compartments of the endocytic pathway during intracellular trafficking. Furthermore, the acquisition of the late endosomal/lysosomal marker LAMP-1 suggests that BCVs interact with some LAMP-1+ compartments, either distinct from late endocytic organelles, or a subset of compartments devoid of CI-M6PR and cathepsin D. However, the absence of Rab7 recruitment, which controls fusion of late endocytic organelles, excludes BCV interactions with late endocytic compartments. Consistently, Brucella intracellular trafficking in nonphagocytic cells is independent of Rab7 (33). The nature and role of LAMP-1+ compartments in Brucella intracellular trafficking remain to be determined. These intermediate vacuoles likely mature into replicating compartments because a significant fraction of them are capable of interactions with the ER. Considered together, our results for BCV trafficking in BMDM are reminiscent of that described in epithelial cells (9), suggesting that Brucella uses similar mechanisms to control its intracellular survival and replication in diverse host cell types.
By replicating inside an ER-derived organelle in macrophages, Brucella displays an intracellular fate similar to that of Legionella (28). Biogenesis of Legionella-replicative compartments depends upon a rapid interception of COPI-dependent vesicular trafficking from endoplasmic ER exit sites, a process that can be blocked by BFA (29). In contrast, BFA did not have any inhibitory effect on Brucella intracellular replication, nor did it affect the rate of ER marker acquisition by BCVs, highlighting mechanistic differences between the maturation processes of these two replicative organelles. Indeed, we did not detect any docking of ribosome-lined, ER-derived vesicles on early BCVs, as is the case for the Legionella phagosome (34), but BCVs were shown to dock with the ER at later time points. Extensive Brucella replication at late time points is accompanied by a dramatic and nearly complete recruitment of ER to dividing BCVs. This indicates that the BCV–ER interactions are prolonged during bacterial replication and illustrates how important these interactions are for Brucella intracellular survival within macrophages. Altogether, these results clearly indicate that biogenesis mechanisms of the Brucella replicative organelle similarly involve the ER in both phagocytic and nonphagocytic cells, thus precluding previous interpretations on the differential maturation and nature of the BCVs in phagocytic versus nonphagocytic cells (7, 35).
How Brucella recruit and interact with the ER remains to be elucidated. Organelle membrane fusion after transport vesicle targeting is a complex multistage process that can be divided into (a) an initial loose or distant interaction of the vesicle with the target compartment, called tethering, and (b) a more intimate contact or docking, which leads to membrane fusion (36, 37). The initial tethering, which involves specific tethering molecules and Rab GTPases, is not sufficient for fusion (36). Subsequent docking and fusion events require proteins of the SNARE family (38). We have shown that BCVs contact ER structures in a VirB-independent manner, whereas the fusion-proficient, sustained interactions are dependent upon a functional VirB system. It is possible that the initial BCV–ER contact consists of a tethering of the BCV to the ER, or reciprocally, which is not sufficient for fusion. The subsequent fusion-proficient interactions would correspond to docking and fusion events, mediated by the action of the VirB type IV secretion system. We speculate that these docking and fusion events are controlled by a bacterial effector(s) translocated into the host cell or the vacuolar membrane.
In conclusion, we propose a model of the Brucella replicative organelle biogenesis through subversion of the macrophage ER (Fig. 9). Rapidly after entry, Brucella escape the endocytic pathway to be located in an intermediate, nonreplicative compartment, which is able to interact with the ER, possibly through tethering activities. Subsequent VirB-dependent maturation of BCVs consists of sustained, fusion-proficient interactions with the ER, which lead to ER membrane accretion by BCVs, to a stage where they acquire functional properties of this compartment. ER-derived organelles support Brucella replication in individual vacuoles through continual ER membrane accretion to provide the new input of membrane required during bacterial growth and division. Future studies are aimed at identifying the bacterial and host cell factors involved in these processes.
Acknowledgments
We wish to thank Leigh Knodler and Stéphane Méresse for critical reading of the manuscript. We are grateful to Dr. Gisou van der Goot for the kind gift of purified proaerolysin and Dr. Diego Comerci for providing the virB mutant and the pBBR-GFP plasmid.
This work was supported by institutional grants from INSERM and CNRS (France), by the research contract ICA4-CT-1999-10001 from the European Community (RTD project NOVELTARGETVACCINES), by the Ministerio de Ciencia y Tecnología/Consejo Nacional de Ciencia y Tecnología (Costa Rica), and by the Fondation pour la Recherche Médicale.
Abbreviations used in this paper: BCV, Brucella-containing vacuole; BFA, brefeldin A; BMDM, bone marrow–derived macrophages; CI-M6PR, cation-independent mannose-6-phosphate receptor; EM, electron microscopy; G6Pase, glucose-6-phosphatase; GFP, green fluorescent protein; LAMP-1, lysosomal-associated membrane glycoprotein 1; MDC, monodansylcadaverine.
J. Pizarro-Cerda's present address is Unité des Interactions Bactéries-Cellules, Institut Pasteur, 25, Rue du Docteur Roux, 75724 Paris, France.
References
- 1.Knodler, L.A., J. Celli, and B.B. Finlay. 2001. Pathogenic trickery: deception of host cell processes. Nat. Rev. Mol. Cell Biol. 2:578–588. [DOI] [PubMed] [Google Scholar]
- 2.Moreno, E., E. Stackebrandt, M. Dorsch, J. Wolters, M. Busch, and H. Mayer. 1990. Brucella abortus 16S rRNA and lipid A reveal a phylogenetic relationship with members of the alpha-2 subdivision of the class Proteobacteria. J. Bacteriol. 172:3569–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreno, E., and I. Moriyon. 2001. The genus Brucella. The Prokaryotes. M. Dorkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt, editors. Springer-Verlag, New York.
- 4.Gorvel, J.P., and E. Moreno. 2002. Brucella intracellular life: from invasion to intracellular replication. Vet. Microbiol. 90:281–297. [DOI] [PubMed] [Google Scholar]
- 5.Hong, P.C., R.M. Tsolis, and T.A. Ficht. 2000. Identification of genes required for chronic persistence of Brucella abortus in mice. Infect. Immun. 68:4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lestrate, P., R.M. Delrue, I. Danese, C. Didembourg, B. Taminiau, P. Mertens, X. De Bolle, A. Tibor, C.M. Tang, and J.J. Letesson. 2000. Identification and characterization of in vivo attenuated mutants of Brucella melitensis. Mol. Microbiol. 38:543–551. [DOI] [PubMed] [Google Scholar]
- 7.Arenas, G.N., A.S. Staskevich, A. Aballay, and L.S. Mayorga. 2000. Intracellular trafficking of Brucella abortus in J774 macrophages. Infect. Immun. 68:4255–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naroeni, A., N. Jouy, S. Ouahrani-Bettache, J.P. Liautard, and F. Porte. 2001. Brucella suis-impaired specific recognition of phagosomes by lysosomes due to phagosomal membrane modifications. Infect. Immun. 69:486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pizarro-Cerda, J., S. Meresse, R.G. Parton, G. van der Goot, A. Sola-Landa, I. Lopez-Goni, E. Moreno, and J.P. Gorvel. 1998. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect. Immun. 66:5711–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pizarro-Cerda, J., E. Moreno, V. Sanguedolce, J.L. Mege, and J.P. Gorvel. 1998. Virulent Brucella abortus prevents lysosome fusion and is distributed within autophagosome-like compartments. Infect. Immun. 66:2387–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delrue, R.M., M. Martinez-Lorenzo, P. Lestrate, I. Danese, V. Bielarz, P. Mertens, X. De Bolle, A. Tibor, J.P. Gorvel, and J.J. Letesson. 2001. Identification of Brucella spp. genes involved in intracellular trafficking. Cell. Microbiol. 3:487–497. [DOI] [PubMed] [Google Scholar]
- 12.Anderson, T.D., and N.F. Cheville. 1986. Ultrastructural morphometric analysis of Brucella abortus-infected trophoblasts in experimental placentitis. Bacterial replication occurs in rough endoplasmic reticulum. Am. J. Pathol. 124:226–237. [PMC free article] [PubMed] [Google Scholar]
- 13.Detilleux, P.G., B.L. Deyoe, and N.F. Cheville. 1990. Entry and intracellular localization of Brucella spp. in Vero cells: fluorescence and electron microscopy. Vet. Pathol. 27:317–328. [DOI] [PubMed] [Google Scholar]
- 14.O'Callaghan, D., C. Cazevieille, A. Allardet-Servent, M.L. Boschiroli, G. Bourg, V. Foulongne, P. Frutos, Y. Kulakov, and M. Ramuz. 1999. A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essential for intracellular survival of Brucella suis. Mol. Microbiol. 33:1210–1220. [DOI] [PubMed] [Google Scholar]
- 15.Foulongne, V., G. Bourg, C. Cazevieille, S. Michaux-Charachon, and D. O'Callaghan. 2000. Identification of Brucella suis genes affecting intracellular survival in an in vitro human macrophage infection model by signature-tagged transposon mutagenesis. Infect. Immun. 68:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sieira, R., D.J. Comerci, D.O. Sanchez, and R.A. Ugalde. 2000. A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence and intracellular multiplication. J. Bacteriol. 182:4849–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christie, P.J. 2001. Type IV secretion: intercellular transfer of macromolecules by systems ancestrally related to conjugation machines. Mol. Microbiol. 40:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Comerci, D.J., M.J. Martinez-Lorenzo, R. Sieira, J.P. Gorvel, and R.A. Ugalde. 2001. Essential role of the VirB machinery in the maturation of the Brucella abortus-containing vacuole. Cell. Microbiol. 3:159–168. [DOI] [PubMed] [Google Scholar]
- 19.Boschiroli, M.L., S. Ouahrani-Bettache, V. Foulongne, S. Michaux-Charachon, G. Bourg, A. Allardet-Servent, C. Cazevieille, J.P. Liautard, M. Ramuz, and D. O'Callaghan. 2002. The Brucella suis virB operon is induced intracellularly in macrophages. Proc. Natl. Acad. Sci. USA. 99:1544–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kovach, M.E., R.W. Phillips, P.H. Elzer, R.M. Roop, II, and K.M. Peterson. 1994. pBBR1MCS: a broad-host-range cloning vector. Biotechniques. 16:800–802. [PubMed] [Google Scholar]
- 21.Meresse, S., P. Andre, Z. Mishal, M. Barad, N. Brun, M. Desjardins, and J.P. Gorvel. 1997. Flow cytometric sorting and biochemical characterization of the late endosomal rab7-containing compartment. Electrophoresis. 18:2682–2688. [DOI] [PubMed] [Google Scholar]
- 22.Steele-Mortimer, O., S. Meresse, J.P. Gorvel, B.-H. Toh, and B.B. Finlay. 1999. Biogenesis of Salmonella typhimurium-containing vacuoles in epithelial cells involves interactions with the early endocytic pathway. Cell. Microbiol. 1:33–49. [DOI] [PubMed] [Google Scholar]
- 23.de Chastellier, C., C. Frehel, C. Offredo, and E. Skamene. 1993. Implication of phagosome-lysosome fusion in restriction of Mycobacterium avium growth in bone marrow macrophages from genetically resistant mice. Infect. Immun. 61:3775–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffiths, G., P. Quinn, and G. Warren. 1983. Dissection of the Golgi complex. I. Monensin inhibits the transport of viral membrane proteins from medial to trans Golgi cisternae in baby hamster kidney cells infected with Semliki Forest virus. J. Cell Biol. 96:835–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biederbick, A., H.F. Kern, and H.P. Elsasser. 1995. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol. 66:3–14. [PubMed] [Google Scholar]
- 26.Gagnon, E., S. Duclos, C. Rondeau, E. Chevet, P.H. Cameron, O. Steele-Mortimer, J. Paiement, J.J. Bergeron, and M. Desjardins. 2002. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 110:119–131. [DOI] [PubMed] [Google Scholar]
- 27.Abrami, L., M. Fivaz, P.E. Glauser, R.G. Parton, and F.G. van der Goot. 1998. A pore-forming toxin interacts with a GPI-anchored protein and causes vacuolation of the endoplasmic reticulum. J. Cell Biol. 140:525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy, C.R., and L.G. Tilney. 2002. The road less traveled: transport of Legionella to the endoplasmic reticulum. J. Cell Biol. 158:415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kagan, J.C., and C.R. Roy. 2002. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4:945–954. [DOI] [PubMed] [Google Scholar]
- 30.de Chastellier, C., T. Lang, A. Ryter, and L. Thilo. 1987. Exchange kinetics and composition of endocytic membranes in terms of plasma membrane constituents: a morphometric study in macrophages. Eur. J. Cell Biol. 44:112–123. [PubMed] [Google Scholar]
- 31.Rittig, M.G., M.T. Alvarez-Martinez, F. Porte, J.P. Liautard, and B. Rouot. 2001. Intracellular survival of Brucella spp. in human monocytes involves conventional uptake but special phagosomes. Infect. Immun. 69:3995–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun, Y.H., A.B. den Hartigh, R.L. Santos, L.G. Adams, and R.M. Tsolis. 2002. virB-mediated survival of Brucella abortus in mice and macrophages is independent of a functional inducible nitric oxide synthase or NADPH oxidase in macrophages. Infect. Immun. 70:4826–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaves-Olarte, E., C. Guzman-Verri, S. Meresse, M. Desjardins, J. Pizarro-Cerda, J. Badilla, J.P. Gorvel, and E. Moreno. 2002. Activation of Rho and Rab GTPases dissociates Brucella abortus internalization from intracellular trafficking. Cell. Microbiol. 4:663–676. [DOI] [PubMed] [Google Scholar]
- 34.Tilney, L.G., O.S. Harb, P.S. Connelly, C.G. Robinson, and C.R. Roy. 2001. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114:4637–4650. [DOI] [PubMed] [Google Scholar]
- 35.Kohler, S., V. Foulongne, S. Ouahrani-Bettache, G. Bourg, J. Teyssier, M. Ramuz, and J.P. Liautard. 2002. The analysis of the intramacrophagic virulome of Brucella suis deciphers the environment encountered by the pathogen inside the macrophage host cell. Proc. Natl. Acad. Sci. USA. 99:15711–15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfeffer, S.R. 1999. Transport-vesicle targeting: tethers before SNAREs. Nat. Cell Biol. 1:E17–E22. [DOI] [PubMed] [Google Scholar]
- 37.Guo, W., M. Sacher, J. Barrowman, S. Ferro-Novick, and P. Novick. 2000. Protein complexes in transport vesicle targeting. Trends Cell Biol. 10:251–255. [DOI] [PubMed] [Google Scholar]
- 38.Chen, Y.A., and R.H. Scheller. 2001. SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2:98–106. [DOI] [PubMed] [Google Scholar]