

Abstract
Epithelial tissues in which carcinomas develop often contain systemically derived T cell receptor (TCR)αβ+ cells and resident intraepithelial lymphocytes that are commonly enriched in TCRγδ+ cells. Recent studies have demonstrated that γδ cells protect the host against chemically induced cutaneous malignancy, but the role of αβ T cells has been enigmatic, with both protective and tumor-enhancing contributions being reported in different systems. This study aims to clarify the contributions of each T cell type to the regulation of squamous cell carcinoma induced in FVB mice by a two-stage regimen of 7,12-dimethylbenz[a]anthracene initiation followed by repetitive application of the tumor promoter 12-O-tetradecanoylphorbol 13-acetate. This protocol permits one to monitor the induction of papillomas and the progression of those papillomas to carcinomas. The results show that whereas γδ cells are strongly protective, the nonredundant contributions of αβ T cells to the host's protection against papillomas are more modest. Furthermore, at both high and low doses of carcinogens, αβ T cells can contribute to rather than inhibit the progression of papillomas to carcinomas. As is likely to be the case in humans, this study also shows that the contribution of T cells to tumor immunosurveillance is regulated by modifier genes.
Keywords: carcinogenesis, squamous cell carcinoma, TCR γδ, TCR αβ, immunogenetics
Introduction
Nonmelanoma skin cancer (NMSC) is the most prevalent form of malignancy reported among Caucasians, and the extent to which its development might be regulated by the immune system is of considerable interest. In support of such regulation is the fact that iatrogenically immunosuppressed organ transplant recipients show >24-fold susceptibility to the development of cutaneous squamous cell carcinoma (SCC), by far the highest alteration of tumor incidence reported in cases of clinical immunosuppression (1). Such data drive intense interest in the prospect of immunotherapy as a treatment for SCC and other NMSCs.
Nonetheless, it is also reported that the incidence of SCC is decreased by nonsteroidal antiinflammatory reagents, such as inhibitors of cyclooxygenase-2, which would be predicted to decrease immune regulation of tumor tissue (2, 3). An hypothesis to accommodate the enhanced susceptibility of immunosuppressed individuals and the protective effects of antiinflammatory agents is that there are qualitatively distinct components of the immune system that de facto inhibit and contribute to tumor development, respectively. Therefore, one would predict that pharmacological antiinflammatory agents that preferentially inhibit lymphoid cells contributing to tumor development, would on aggregate display antitumor efficacy.
The concept of different immunological components responding to tumors finds support in the observation that many of the epithelia in which carcinomas develop harbor distinct subsets of T cells, which comprise resident, intraepithelial lymphocytes (IELs) and infiltrating systemic T cells, respectively (4, 5). Considering the large surface area of epithelia, it has been proposed that IELs comprise somewhere between one quarter and one half of the body's T cells. When cross compared, IELs and systemic T cells display highly distinct gene expression profiles and T cell receptor usage (6, 7). For example, whereas the murine and human systemic T cell compartments are dominated by TCRαβ+ cells, the IEL repertoires are disproportionately enriched in TCRγδ+ cells (5).
Murine skin is a striking example of IEL and systemic T cell coexistence. The constitutive IEL repertoire comprises a dendritic epidermal T cell (DETC) subset of which >95% of cells express a single, homogeneous γδ TCR (8). At the same time, the epidermis harbors tissue-associated DCs, known as Langerhans cells, which process antigens encountered in the skin and migrate to the local LNs, where they prime and provoke skin infiltration by antigen-reactive systemic αβ T cells. Recently, we reported that TCRγδ+ DETCs actively suppress the migration into the skin of αβ T cell–dependent cellular infiltrates (9). This example of functional interaction between the “local” and systemic components of the immune system suggests that DETCs can protect epithelial integrity from internal disruption. An additional protection of epithelial integrity by DETCs is suggested by reports that DETCs produce locally active keratinocyte growth factor (10).
Relative to their immunocompetent counterparts, TCRδ−/− mice were also demonstrated to be highly susceptible to the development of SCC induced by any of three regimens (11). As a potential underlying mechanism, isolated DETCs were shown to lyse SCC cells dependent on the engagement of two γδ cell surface receptors, TCRγδ and NKG2D (11). Although TCRαβ−/− mice were likewise susceptible to some regimens of SCC induction (11), this was not universal, raising the possibility that the distinct contribution of different T cell subsets to tumor regulation is manifest in murine skin. Consistent with this, Siegel et al. (12) reported that mice expressing a transgenic TCRαβ showed an increased incidence of skin papillomas when the αβ T cells were preactivated by immunization with antigenic peptide. Likewise, it was recently reported that CD4+ αβ T cells, apparently responding to bacterial infection, can cause tissue disruption that promotes the progression of skin tumors induced by transgenic oncogenes (13). The concept that some T cell responses to chemical carcinogenesis might promote tumor development is tested more generally in this paper, in which substantial numbers of mice are subjected to a two-stage chemical carcinogenesis regimen in which the induction of papillomas and the progression to carcinomas are separately monitored. The FVB mouse strain used in this paper has previously been shown to be highly susceptible to chemically induced SCC (14). The tumors that develop are entirely syngeneic, theoretically limiting conventional, MHC-restricted αβ T cell recognition to the expression of tumor antigen–derived peptides. We find that γδ cell deficiency causes a profound increase in the incidence of papillomas and carcinomas. By contrast, there was a less pronounced effect of αβ T cells on papilloma development, and most strikingly, clear evidence that the presence of αβ T cells promotes the progression of papillomas to carcinomas. Additionally, we show that the immunosurveillance of tumors by γδ cells is under genetic regulation.
Materials and Methods
Animals.
TCRδ−/− and TCRβ−/− C57BL/6 mice (The Jackson Laboratory) were backcrossed to FVB mice (Taconic) for 11 generations each. FVB.β−/−δ−/− mice were produced by first crossing FVB.β−/− with FVB.δ−/− mice to obtain double heterozygotes, and then intercrossing these animals to obtain the double knockouts. For genetic studies, FVB.δ−/− mice were mated with C57BL/6.δ−/− mice to produce both (FVB × C57BL/6)F1.δ−/− and (C57BL/6 × FVB)F1.δ−/− offspring. F1.δ−/− mice were then intercrossed to produce F2.δ−/− offspring, and were backcrossed to FVB.δ−/− to produce (F1 × FVB)BC.δ−/− and (FVB × F1)BC.δ−/− animals. Mice were kept in filter-topped cages with sterilized food and water, and autoclaved corncob bedding, which was changed at least once weekly. The facility is accredited by the American Association for Accreditation of Laboratory Animal Care.
Chemical Carcinogens.
Chemicals were obtained from Sigma-Aldrich. 7,12-dimethylbenz[a]anthracene (DMBA) was dissolved in acetone at a concentration of 200 nmol per 100 μl. 12-O-tetradecanoylphorbol 13-acetate (TPA) was dissolved in 100% ethanol at a concentration of 5 nmol per 100 μl (0.05 mM), or 40 nmol per 100 μl (0.4 mM).
Fetal Liver Hematopoetic Stem Cell (FLHSC) Reconstitution.
Newborn TCRβ−/− mice were reconstituted with peripheral αβ T cell progenitors from FLHSCs of FVB mice as follows: day 13.5 fetal livers were harvested and gently pressed between the frosted edges of two glass slides, releasing the cells into ice-cold HBSS containing 4 mM Hepes buffer and antibiotics. Fetal liver cells were filtered through nylon mesh (Nytex cloth 88/42; Tetko Inc.) to remove debris and give single cell suspensions, washed twice with HBSS, and resuspended at appropriate concentrations in sterile PBS before intraperitoneal injection at 3 × 106 cells in 30 μl PBS per newborn TCRβ−/− recipient.
Two-Stage Chemical Carcinogenesis Protocol.
Initiation was performed by pipette application of 200 nmol of DMBA, followed by weekly treatment with 5 or 40 nmol of the tumor promoter TPA in acetone onto the back skin of 8-wk-old mice, 1 wk after shaving hair with electric clippers followed by depilatory cream. (For the experiment shown in Fig. 2 C, a hand razor alone was used to remove the hairs on all mice).
Figure 2.
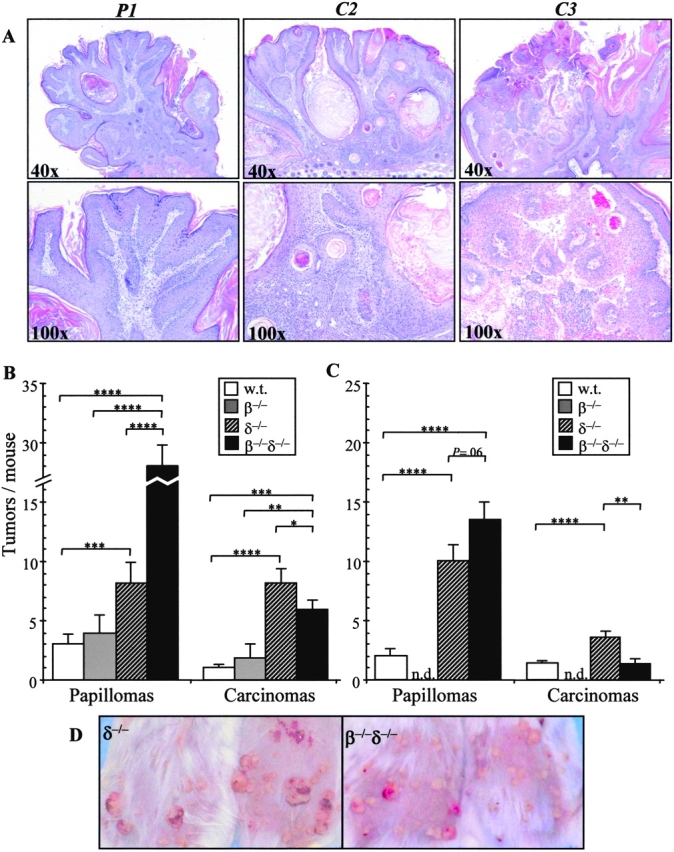
Morphologic assessment of tumor development in T cell–deficient mice. Tumors that developed under two-stage chemical carcinogenesis (refer to Fig. 1) were scored as clinically apparent papillomas (P) or carcinomas (C), as described in Materials and Methods. (A) Hematoxylin and eosin–stained sections of representative tumors were obtained and typical examples are shown. This papilloma (P1; see text for scoring index) exhibited a symmetric outline, acanthosis, and papillomatosis (×40), with uniform keratinocytes with moderately abundant cytoplasm and small round nuclei (×100). This early carcinoma (C2) exhibited an asymmetric outline and expansion of neoplastic follicle-like structures composed of atypical epithelium (×40). There was haphazard infiltration of the dermis by pleomorphic keratinocytes with increased nuclear/cytoplasmic ratio (×100). This later stage carcinoma (C3) exhibited a poorly organized, asymmetric pattern of growth (×40) comprised of atypical keratinocytes aggregated around vascular connective tissue cores (×100). (B) Papilloma and carcinoma development for the experiment depicted in Fig. 1 at 17 wk. *, P < 0.05; **, P < 0.01; ***, P < 0.005; ****, P < 0.001. (C) Papilloma and carcinoma development in a second independent experiment at study termination. n.d., not done. (D) Tumor appearance of two representative mice from each of the TCRδ−/− and TCRβ−/−δ−/− groups.
Tumor Scoring and Histology.
Mice were assessed every 1–2 wk for tumor development, and tumors were counted, measured, and scored as clinically apparent papillomas (typically well-demarcated, symmetrical, pedunculated, or dome-shaped papules, without erosion or ulceration), or clinically apparent carcinomas (poorly demarcated, asymmetrical, nonpedunculated, or dome-shaped papules with erosion or ulceration). Tumors were evaluated by visual inspection by an observer blinded to the experimental group. To assess the validity of the clinical scoring system, three tumors from each type (papilloma and carcinoma) and size category (>1 mm2, ≥4 mm2, ≥9 mm2, and ≥16 mm2; see Results) were submitted for histologic review by a dermatopathologist blinded to the clinical score. Of 18 tumors reviewed, there was 100% concordance between the clinical and histological assessments (representatives are depicted in Fig. 2 A). At the conclusion of experiments, tumors were excised, formalin fixed, paraffin embedded, and 5-μm sections were hematoxylin and eosin stained and examined (by E. Glusac) for histologic findings.
Statistical Analysis.
Statistical significance was evaluated by two-tailed, unpaired Student's t test, or nonparametric analysis if standard deviations were significantly different between the two compared groups.
Results
Tumor Incidence Is Increased and Tumor Onset Is More Rapid in TCRδ−/− and TCRβ−/−δ−/− Mice.
To induce cutaneous SCC, FVB mice were painted with a single initiating application of 200 nmol DMBA, followed by weekly treatment with a low dose (5 nmol) of the tumor promoter TPA. Each experimental group comprised 15 mice, in which the development of palpable tumors (>1 mm) was recorded over time. Tumor size was classified as T1 > 1 mm2, T2 ≥ 4 mm2, T3 ≥ 9 mm2, and T4 ≥ 16 mm2 (Table I). In WT mice, tumors first became visible between 11 and 13 wk after initiation (Fig. 1) , and then the incidence increased slowly but steadily until 17 wk, when the experiment was curtailed for humane reasons. By that time, 13 out of 15 mice (87%) recorded 60 tumors (mean 4.0 ± 0.9/mouse; Fig. 1 and Table I.
Table I.
Tumor Sizes at Weeks 13, 15, and 17 after DMBA Initiation
| WK 13
|
WK 15
|
WK17
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T1 | T2 | T3 | T4 | Percent Lg. | T1 | T2 | T3 | T4 | Percent Lg. | T1 | T2 | T3 | T4 | Percent Lg. | |
| WT | 21 | 7 | 1 | 0 | 27.6 | 36 | 14 | 1 | 0 | 29.4 | 36 | 18 | 4 | 2 | 40.0 |
| β−/− | 33 | 19 | 2 | 0 | 38.9 | 31 | 29 | 5 | 3 | 54.4 | 30 | 38 | 6 | 12 | 65.1 |
| δ−/− | 72 | 69 | 13 | 2 | 53.2 | 33 | 130 | 17 | 3 | 80.7 | 57 | 142 | 36 | 10 | 76.7 |
| β−/−δ−/− | 183 | 144 | 11 | 0 | 45.9 | 113 | 262 | 11 | 9 | 73.2 | 181 | 282 | 33 | 13 | 64.4 |
Percent Lg., percent tumors ≥T2; where 1 mm2 < T1 < 4 mm2 ≤ T2 < 9 mm2 ≤ T3 < 16 mm2 ≤ T4.
Figure 1.

Rapid tumor onset and increased tumor incidence in TCRδ−/− and TCRβ2/2δ2/2 mice. Under a two-stage chemical carcinogenesis protocol of initiation with 200 nmol DMBA and weekly application of (low dose) 5 nmol TPA, WT (w.t.) and T cell–deficient mice were followed for the development of all cutaneous tumors (papillomas or carcinomas >1 mm; see Fig. 2 and Table II for breakdown). Although TCRβ−/− mice (deficient in only αβ T cells) showed no significant difference in tumorigenesis relative to WT mice, TCRδ−/− mice (deficient in γδ T cells) and TCRβ−/−δ−/− mice (deficient in both αβ and γδ T cells) exhibited an earlier onset and marked increase in tumor development. *, P ≤ 0.0001 for β−/−δ−/− and δ−/− versus WT or β−/−.
In TCRβ−/− mice, two tumors appeared before any were detected in WT mice, but thereafter tumor development was comparable in the two strains. By 17 wk, 10 out of 15 mice collectively bore 86 tumors, yielding an average of 5.7 ± 1.8 tumors per mouse, which was not significantly different from WT (P < 0.08; Fig. 1 and Table I). By contrast, a substantially greater susceptibility to tumor induction was shown by TCRδ−/− mice. Tumors first appeared ∼1 mo before one was detected in WT mice, and then tumor incidence increased steeply between 11 and 13 wk, after which the increase was steady but less dramatic (Fig. 1). By 17 wk, 100% of TCRδ−/− mice bore 245 tumors (mean 16.3 ± 2.3 tumors per mouse, greater than fourfold higher incidence than WT; P [δ−/− vs. WT] < 0.0000064, P [β−/− vs. δ−/−] < 0.00015). These data are consistent with and expand the reported high susceptibility of TCRδ−/− mice to SCC (11) and emphasize the inability of the αβ T cells in TCRδ−/− mice to effectively control chemically induced tumor incidence in the absence of γδ cells.
The highest susceptibility to tumor development was shown by TCRβ−/−δ−/− mice lacking both γδ cells and αβ T cells (Fig. 1). The appearance of tumors occurred no earlier than that in TCRδ−/− mice, but the rise in tumor development between 11 and 13 wk was even steeper. By 17 wk, 100% of TCRβ−/−δ−/− mice collectively bore 509 tumors (mean 33.9 ± 1.8 per mouse; Fig. 1 and Table I. This incidence (greater than eightfold increase over WT; P < 10−10) significantly exceeded that in all other strains.
Collectively, these data demonstrate that murine γδ cells play a significant role in down-regulating chemically induced cutaneous tumorigenesis, both in terms of time of onset and cumulative tumor incidence. Indeed, at every time point, the fraction of tumors that were large in size was greatest in TCRδ−/− mice (Table I and see below). The prolific occurrence of tumors in TCRβ−/−δ−/− mice lacking all T cells, relative to the incidence in TCRδ−/− mice, seemingly demonstrates that αβ T cells can also down-regulate tumor development, despite the fact that the TCRβ−/− mouse is not significantly more susceptible to tumor development than its WT FVB counterpart. One interpretation would be that in TCRβ−/− mice, γδ cells readily compensate for most of the antitumor activities of αβ T cells, whereas in the TCRδ−/− mice there is some capacity of αβ T cells to substitute for some of the roles of γδ cells. Hence, because it lacks any T cell activity, the TCRβ−/−δ−/− mouse shows an exaggerated defect in tumor surveillance. This capacity for one T cell subset to compensate to varying degrees for the absence of another was previously evident in reports of aggressive, transplantable B cell lymphomas that develop in fas-deficient mice lacking all T cells, but not in fas-deficient mice that lack only a single T cell subset (15).
Morphologic Tumor Progression Is Inhibited by γδ Cells and Enhanced by αβ T Cells.
Developing papillomas may regress, increase in size while retaining their overall phenotype, or show progression to irregularly shaped carcinomas that penetrate the basement membrane, invading the dermis. The third of these outcomes constitutes the most severe pathology, yet the factors that regulate the respective outcomes are poorly understood. To determine whether there is an immunological contribution to tumor progression, the relative numbers of papillomas versus carcinomas were scored in each experimental group. Tumors scored as papillomas were well-demarcated, symmetrical, pedunculated, or dome-shaped papules, without erosion or ulceration. Tumors scored as carcinomas were poorly demarcated, asymmetric, nonpedunculated, or dome-shaped papules, with erosion or ulceration (Fig. 2 A). To be consistent with the tumor size classification (Table I), carcinomas were classified as: C2 ≥ 4 mm2, C3 ≥ 9 mm2, and C4 ≥ 16 mm2 (no carcinoma was <4 mm2; Table II).
Table II.
Tumor Morphologies at Weeks 13, 15, and 17 after DMBA Initiation
| WK 13
|
WK 15
|
WK 17
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Papa | C2 | C3 | C4 | C/Pb | Papa | C2 | C3 | C4 | C/Pb | Papa | C2 | C3 | C4 | C/Pb | |
| WT | 29 | 0 | 0 | 0 | 0.00 | 42 | 8 | 1 | 0 | 0.21 | 45 | 9 | 4 | 2 | 0.33 |
| β−/− | 53 | 0 | 1 | 0 | 0.02 | 53 | 7 | 5 | 3 | 0.28 | 59 | 9 | 6 | 12 | 0.46 |
| δ−/− | 152 | 0 | 2 | 2 | 0.03 | 110 | 56 | 14 | 3 | 0.66 | 122 | 77 | 36 | 10 | 1.01 |
| β−/−δ−/− | 328 | 2 | 8 | 0 | 0.03 | 332 | 43 | 11 | 9 | 0.19 | 421 | 45 | 30 | 13 | 0.21 |
Pap, total papillomas.
C/P, carcinoma/papilloma ratio.
By 13 wk, WT mice displayed 29 papillomas and 0 carcinoma, whereas by 17 wk, the same mice displayed 45 papillomas and 15 carcinomas, giving a “carcinoma/papilloma ratio” = 0.33 (Table II and Fig. 2 B). In TCRβ−/− mice, the progression to carcinomas was similar to that in WT mice. At 13 wk, there were 53 papillomas and 1 carcinoma (Table II), whereas by 17 wk, there were 59 papillomas and 27 carcinomas (carcinoma/papilloma ratio = 0.46; Table II and Fig. 2 B.
By contrast, the greatly enhanced susceptibility of TCRδ−/− mice to tumor development was also manifest in an increased progression of tumors to carcinomas. By 15 wk, ∼40% (73 out of 183) of tumors were carcinomas, compared with ∼20% in WT mice and TCRβ−/− mice (Table II). By 17 wk, carcinomas (123) were as frequent as papillomas (122; Table II and Fig. 2 B). The resulting carcinoma/papilloma ratio (=1.01) was the only instance in which the ratio exceeded 1. Of note, although TCRβ−/− δ−/− mice developed many more tumors than other strains, they primarily remained as papillomas (Table II and Fig. 2 B). At 15 wk, the carcinoma/papilloma ratio (=0.19) was similar to that in WT and TCRβ−/− mice, and by 17 wk, it remained at a similar level (0.21), approximately fivefold less than that in TCRδ−/− mice (Table II and Fig. 2 B). The contrasting appearance of tumor development in TCRδ−/− mice and TCRβ−/−δ−/− mice is depicted in Fig. 2 D.
Collectively, the data demonstrate that γδ cells act both to prevent tumor development (i.e., papilloma formation) and inhibit progression to carcinoma, whereas the absence of αβ T cells in TCRβ−/−δ−/− mice reduces tumor progression relative to that in TCRδ−/− mice. To test this further, an additional experiment was undertaken, this time comparing tumor development and progression over a 15-wk period in 15 mouse groups of WT, TCRδ−/−, and TCRβ−/−δ−/− strains. Certain parameters were different in the second experiment (Fig. 2 C). Both males and females were used (whereas in the first experiment age-matched females were used), a different stock of DMBA was used, and a hand razor was used for the preparation of the skin for carcinogen application (whereas in the first experiment an electric razor plus depilatory cream was used). For all or any of these reasons, the overall incidence of tumors was less, but despite these differences, the pattern of the results obtained was identical. The susceptibility to papilloma formation was again in the order WT << TCRδ−/− < TCRβ−/− δ−/− mice, whereas the progression of tumors to carcinomas in TCRβ−/−δ−/− mice was again less than that in TCRδ−/− mice (Fig. 2 C). The carcinoma/papilloma ratios were TCRδ−/− = 0.33 and TCRβ−/−δ−/− = 0.07. Thus, the ratio in TCRβ−/−δ−/− mice was approximately fivefold lower than that in TCRδ−/− mice in both experiments.
Tumor Regulation by αβ T Cells at Higher Doses of Carcinogens.
To examine further the regulation of tumor progression by αβ T cells, experiments were undertaken in which tumor promotion was provoked by weekly application of 40 nmol (rather than 5 nmol) of TPA to WT and TCRβ−/− mice. Under such conditions, WT mice develop substantially more tumors than when lower doses of TPA are used. Under these conditions, the tumor incidence in TCRβ−/− mice was substantially lower than that in WT mice (12.7 ± 1.7 vs. 20.9 ± 1.9 at 15 wk; P = 0.0015; not depicted), and represented a markedly smaller tumor area per mouse (68.2 ± 9.5 mm2 vs. 203.1 ± 25.2; P < 0.001; Fig. 3 A). Furthermore, TCRβ−/− mice were approximately fivefold less susceptible to tumor progression to carcinomas (2.35 ± 0.5 per mouse vs. 11.8 ± 1.1 cm2; P < 0.0001) at 15 wk (Fig. 3 B). The contrasting appearance of tumor development in WT and TCRβ−/− mice is depicted in Fig. 3 C.
Figure 3.
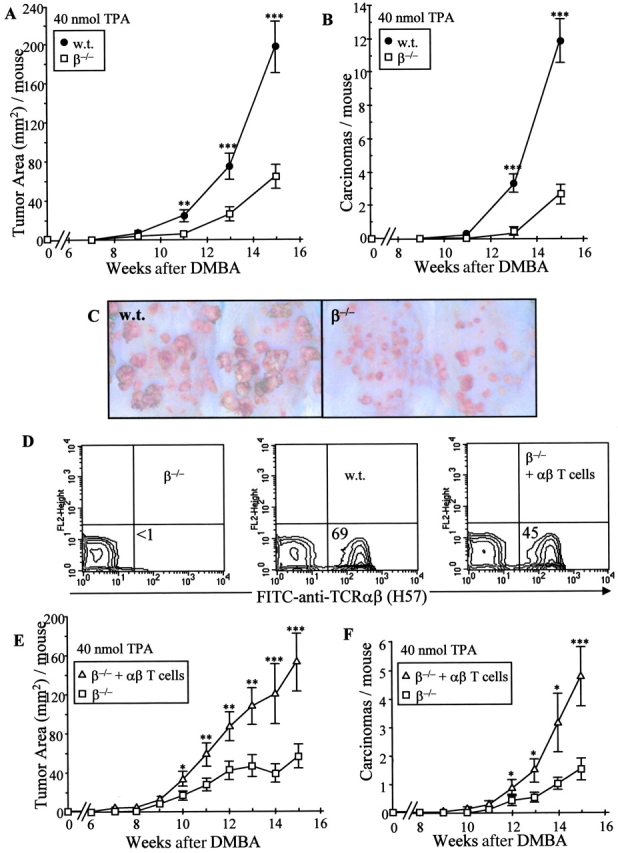
Marked decrease in tumor burden and progression in TCRβ−/− mice. Under a two-stage chemical carcinogenesis protocol of initiation with 200 nmol DMBA and weekly application of (high dose) 40 nmol TPA, WT (w.t.) and TCRβ−/− mice were followed for the development of tumors (>1 mm). TCRβ−/− mice deficient in αβ T cells demonstrated a markedly lower (A) tumor area and (B) carcinoma development. (C) Clinical tumor appearance of two representative mice from each of the WT and β−/− groups. (D) Representative flow cytometric analysis for the presence of αβ+ (H57) T cells in the spleens of 6-wk-old TCRβ−/− mice, WT mice, and TCRβ−/− mice reconstituted at birth by intraperitoneal injection of day 13.5 FLHSCs. (E and F) Reconstituted TCRβ−/− mice (β−/− + αβ T cells) demonstrated a higher tumor area (E) and greater rate of carcinoma formation (F) than TCRβ−/− mice. *, P ≤ 0.02; **, P ≤ 0.01; ***, P ≤ 0.005.
Although the TCR mutant mice used in this study were extensively backcrossed to the FVB strain, it remained important to show that the heightened incidence of tumor progression in the TCRβ−/− mouse was due primarily to their lack of αβ+ T cells. Therefore, TCRβ−/− mice were reconstituted at birth with day 13.5 FLHSCs from WT FVB donors. Flow cytometry of the spleens of the reconstituted mice (n = 13) showed substantial repopulation of peripheral αβ T cell compartment at 6 wk of age (mean 43.0 ± 5.2%; Fig. 3 D), whereupon the mice were subjected to two-stage carcinogenesis using 40 nmol TPA. Compared with mock-reconstituted TCRβ−/− mice, the αβ T cell–reconstituted TCRβ−/− demonstrated an approximately threefold higher tumor area (147.1 ± 32.4 vs. 43.3 ± 10.1 mm2; P = 0.0025; Fig. 3 E) and threefold greater number of carcinomas per mouse (4.6 ± 1.0 vs. 1.4.1 ± 0.4 cm2; P = 0.0025; Fig. 3 F) at 15 wk after initiation. These data show that mice that harbor αβ T cells are more susceptible to tumor progression than mice of identical genotype that lack αβ T cells.
Genetic Modifiers Regulate Tumor Surveillance by γδ Cells.
Having established that γδ T cell deficiency increases the susceptibility of FVB mice to SCC, we examined whether this observation might be used to reveal additional genetic modifiers of tumor development. Therefore, the DMBA TPA (5 nmol) regimen for SCC induction was applied to a total of 483 WT and TCRδ−/− FVB mice, WT and TCRδ−/− C57BL/6 (B6) mice, and intercrosses thereof. The data confirmed that FVB is more susceptible than B6 to chemical induction of SCC (Fig. 4 ; reference 14). They also confirmed the greater susceptibility of the FVB.TCRδ−/− mouse. However, this increased susceptibility was not apparent in either B6.TCRδ−/− mice or in (B6 × FVB)F1.TCRδ−/− mice (Fig. 4), in both of which tumor resistance is high. Thus, the powerful contribution of γδ cells to SCC regulation is a feature of FVB animals that are highly susceptible to SCC. Because no B6.TCRδ−/− mice showed more than two tumors, we operationally defined enhanced susceptibility as mice bearing more than two 2 tumors. Provocatively, 26.2% of (B6 × FVB) F2.TCRδ−/− mice and 46.3% of (B6 × FVB)F1.TCRδ−/− mice backcrossed to FVB showed enhanced susceptibility. In the case that a single gene segregating between B6 and FVB mice regulates the susceptibility to γδ deficiency, the expected frequencies would be 25% in the F2 mice and 50% in the backcross mice. Although it is likely that modifier genes might be influencing carcinogenesis in this system via diverse mechanisms (e.g., susceptibility to mutagenesis by DMBA, susceptibility to irritancy by TPA), future attempts to map those genes may elucidate factors that compensate for local T cell immunosurveillance in highly tumor-resistant hosts.
Figure 4.

Tumor development in crosses of susceptible FVB.TCRδ2/2 with resistant C57BL/6.TCRδ2/2 mice. FVB.TCRδ−/− (FVB.δ−/−) and C57BL/6.TCRδ−/− (B6.δ−/−) mice were bred to produce F1.δ−/−, F2.δ−/−, and (F1 × FVB) backcross (BC).δ−/− offspring. Under a two-stage chemical carcinogenesis protocol of initiation with 200 nmol DMBA and weekly application of (low dose) 5 nmol TPA, mice were followed for the development of all cutaneous tumors (papillomas or carcinomas >1 mm) at week 16 after DMBA initiation. The mean number ± standard deviation of total tumors per animal for each group is represented. Although none of the B6.δ−/− mice developed more than two tumors, 46.3% of the BC.δ−/− and 26.2% of the F2.δ2/2 mice developed more than two tumors.
Discussion
This extensive study of cutaneous malignancy induced at each of two doses of chemical carcinogens in WT and T cell–deficient FVB mice has demonstrated that the immune surveillance of tumor development can be classified into qualitatively distinct immunologic components. Whereas γδ cells are protective against chemically induced SCC, the nonredundant contribution of αβ T cells to protection against low doses of carcinogens is more modest (Fig. 1). Moreover, at higher doses of carcinogens, TCRβ−/− mice are more resistant than WT mice to papillomas (Fig. 3). This failure of αβ T cells to contribute to host protection was most obvious when scoring the conversion of papillomas to carcinomas, which was de facto promoted by αβ T cells, particularly under conditions in which the number of precursor lesions (i.e., papillomas) was high. This was seen in four experiments: two independent experiments in which slightly different protocols were used to apply low doses of carcinogens to TCRβ−/−δ−/− mice versus TCRδ−/− mice (Fig. 2, B and C), one experiment in which high dose carcinogens were applied to TCRβ−/− mice versus WT mice (Fig. 3 B), and one experiment in which high dose carcinogens were applied to TCRβ−/− mice versus TCRβ−/− mice reconstituted with αβ T cells (Fig. 3, E and F).
Clearly, αβ T cells often display protective antitumor activity, as in the antigen-specific targeting of tumor-associated antigens in human melanoma (16, 17), and the decreased susceptibility to methylcholanthrene-induced NMSC in mice lacking αβ T cells (11). Therefore, it will be important to determine the factors that regulate whether on aggregate αβ T cells inhibit or promote tumor development. One factor might be the immunogenicity of a tumor. Melanomas, for example, might be sufficiently immunogenic to offer myriad targets to systemic αβ T cells. By contrast, SCC induced in syngeneic hosts by chemicals that act with two-stage kinetics might be less immunogenic. Indeed, Williams et al. (18) reported that such tumors failed to activate protective systemic T cell responses, even after experimental up-regulation of costimulator molecules. In such cases, αβ T cell involvement might be tumor antigen nonspecific, for example, as a response to local inflammation, and might promote tumor growth by the secretion of large amounts of cytokines and metalloproteinases (13, 19). Regardless of the mechanisms involved, the data presented here may aid immunotherapy strategies by emphasizing that enhanced systemic T cell activation might be most effective against tumors at the early stages of their development. As a tumor progresses, treatments designed to enhance systemic T cell responses may show more variable efficacy.
At the same time, it would seem important not to ignore the contribution to tumor surveillance of γδ cells, and possibly other locally acting, nonconventional T cells that may resemble them (5, 6). The efficacy of γδ cells in inhibiting SCC may reflect the fact that the tumors initiate in a region rich in TCRγδ+ IELs. Thus, the anatomical site of tumor development will probably dictate the nature of the immune response best equipped to attack the tumor. A corresponding protective T cell compartment in human epidermis has not been reported, although a unique TCRγδ+ repertoire in human dermis was recently described (20). Moreover, the human gut is known to be rich in both systemic T cells and IELs, including γδ cells, which have been reported to attack human bowel carcinomas expressing the MHC class IB molecule, MICA (21). Thus, the protective effects of local T cells described here may provide a tractable animal model for the surveillance of human carcinomas. The capacity of TCRγδ+ IELs to target MHC class IB molecules such as MICA, might be particularly effective against tumors that have lost expression of conventional MHC antigens, a common clinical problem.
As well as targeting tumors directly, the effects of local T cells might be multifaceted, because the fact that DETC are potent suppressors of αβ T cell–mediated inflammation of the skin (9) strongly suggests that local T cells might reduce the potential for αβ T cells to promote carcinoma formation. It is likewise possible that locally resident and/or infiltrating T cell subsets may directly modify (e.g., augment or reduce) the proinflammatory effects of the applied TPA. The potentiation of environmental carcinogens might be one mechanism by which T cells regulate carcinogenesis in humans, although evidence that αβ T cells may promote tumor development has additionally been reported in systems that do not involve chemical carcinogens (12, 13, 19).
Many genes influence skin carcinogenesis in mice (as is the case in humans; 14), and we have shown in this paper that the increased susceptibility to tumor development seen in TCRδ−/− mice is dependent on modifier genes found on the FVB background versus the C57BL/6 background. Those genes can now be identified. Nonetheless, their existence raised the formal possibility that genes that cosegregated with the disrupted TCRβ locus during the backcrossing of the TCR mutant mice to FVB may have contributed to the observed decrease in tumor progression in TCRβ−/− FVB mice relative to WT FVB (Fig. 3). This possibility was excluded by showing that the rates of tumor progression in TCRβ−/− mice could be substantially increased by reconstitution with peripheral αβ T cells without affecting any other genes. Hence, in this model, the presence of αβ T cells is a primary determinant of enhanced tumor progression.
In sum, the design and application of appropriate immunotherapy regimens might usefully be informed by classifying types of tumors according to their anatomical site, their immunogenicity, their stage of development, and the genetics of the host. Indeed, effective immunosurveillance might be most relevant in hosts who are genetically predisposed to tumor susceptibility, whereas in more resistant hosts, the action of other genes may substitute for immunosurveillance. Such genetic influences are modeled in this paper by the different dependence of B6 and FVB mice, respectively, on tumor surveillance by TCRγδ cells. The ongoing studies to identify the gene(s) underlying this difference may prove useful in elucidating factors that underpin the significance of immunosurveillance in humans.
Acknowledgments
We thank our lab colleagues for many helpful discussions.
This work is supported by the National Institutes of Health (NIH) and American Skin Association (M. Girardi), the NIH-awarded Skin Diseases Research Core Center (R.E. Tigelaar), and the Wellcome Trust (A. Hayday).
Abbreviations used in this paper: DETC, dendritic epidermal T cell; DMBA, 7,12-dimethylbenz[a]anthracene; FLHSC, fetal liver hematopoetic stem cell; IEL, intraepithelial lymphocyte; NMSC, nonmelanoma skin cancer; SCC, squamous cell carcinoma; TPA, 12-O-tetradecanoylphorbol 13-acetate.
References
- 1.Peto, J. 2001. Cancer epidemiology in the last century and the next decade. Nature. 411:390–395. [DOI] [PubMed] [Google Scholar]
- 2.Pentland, A.P., J.W. Schoggins, G.A. Scott, K.N. Khan, and R. Han. 1999. Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis. 20:1939–1944. [DOI] [PubMed] [Google Scholar]
- 3.Orengo, I.F., J. Gerguis, R. Phillips, A. Guevara, A.T. Lewis, and H.S. Black. 2002. Celecoxib, a cyclooxygenase 2 inhibitor as a potential chemopreventive to UV-induced skin cancer: a study in the hairless mouse model. Arch. Dermatol. 138:751–755. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson, A. 1977. Intraepithelial lymphocytes of the small intestine. Gut. 18:921–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayday, A., E. Theodoridis, E. Ramsburg, and J. Shires. 2001. Intraepithelial lymphocytes: exploring the Third Way in immunology. Nat. Immunol. 2:997–1003. [DOI] [PubMed] [Google Scholar]
- 6.Shires, J., E. Theodoridis, and A.C. Hayday. 2001. Biological insights into TCR gamma delta+ and TCR alpha beta+ intraepithelial lymphocytes provided by serial analysis of gene expression (SAGE). Immunity. 15:419–434. [DOI] [PubMed] [Google Scholar]
- 7.Fahrer, A.M., Y. Konigshofer, E.M. Kerr, G. Ghandour, D.H. Mack, M.M. Davis, and Y.H. Chien. 2001. Attributes of gamma delta intraepithelial lymphocytes as suggested by their transcriptional profile. Proc. Natl. Acad. Sci. USA. 98:10261–10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asarnow, D.M., W.A. Kuziel, M. Bonyhadi, R.E. Tigelaar, P.W. Tucker, and J.P. Allison. 1988. Limited diversity of gamma delta antigen receptor genes of Thy-1+ dendritic epidermal cells. Cell. 55:837–847. [DOI] [PubMed] [Google Scholar]
- 9.Girardi, M., J. Lewis, E. Glusac, R.B. Filler, L. Geng, A.C. Hayday, and R.E. Tigelaar. 2002. Resident skin-specific gamma delta T cells provide local, nonredundant regulation of cutaneous inflammation. J. Exp. Med. 195:855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boismenu, R., and W.L. Havran. 1994. Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science. 266:1253–1255. [DOI] [PubMed] [Google Scholar]
- 11.Girardi, M., D.E. Oppenheim, C.R. Steele, J.M. Lewis, E. Glusac, R. Filler, P. Hobby, B. Sutton, R.E. Tigelaar, and A.C. Hayday. 2001. Regulation of cutaneous malignancy by gamma delta T cells. Science. 294:605–609. [DOI] [PubMed] [Google Scholar]
- 12.Siegel, C.T., K. Schreiber, S.C. Meredith, G.B. Beck-Engeser, D.W. Lancki, C.A. Lazarski, Y.X. Fu, D.A. Rowley, and H. Schreiber. 2000. Enhanced growth of primary tumors in cancer-prone mice after immunization against the mutant region of an inherited oncoprotein. J. Exp. Med. 191:1945–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daniel, D., N. Meyer-Morse, E.K. Bergsland, K. Dehne, L.M. Coussens, and D. Hanahan. 2003. Immune enhancement of skin carcinogenesis by CD4+ T cells. J. Exp. Med. 197:1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hennings, H., A.B. Glick, D.T. Lowry, L.S. Krsmanovic, L.M. Sly, and S. Yuspa. 1993. FVB/N mice: an inbred strain sensitive to the chemical induction of squamous cell carcinomas in the skin. Carcinogenesis. 14:2353–2358. [DOI] [PubMed] [Google Scholar]
- 15.Peng, S.L., M.E. Robert, A.C. Hayday, and J. Craft. 1996. A tumor-suppressor function for Fas (CD95) revealed in T cell–deficient mice. J. Exp. Med. 184:1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg, S.A., J.C. Yang, D.J. Schwartzentruber, P. Hwu, F.M. Marincola, S.L. Topalian, N.P. Restifo, M.E. Dudley, S.L. Schwarz, P.J. Spiess, et al. 1998. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 4:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banchereau, J., A.K. Palucka, M. Dhodapkar, S. Burkeholder, N. Taquet, A. Rolland, S. Taquet, S. Coquery, K.M. Wittkowski, N. Bhardwaj, et al. 2001. Immune and clinical responses in patients with metastatic melanoma CD34+ progenitor-derived dendritic cell vaccine. Cancer Res. 61:6451–6458. [PubMed] [Google Scholar]
- 18.Williams, I.R., R.J. Ort, D. Daley, L. Manning, T. Karaoli, R.L. Barnhill, and T.S. Kupper. 1996. Constitutive expression of B7-1 (CD80) on mouse keratinocytes does not prevent development of chemically induced skin papillomas and carcinomas. J. Immunol. 156:3382–3388. [PubMed] [Google Scholar]
- 19.Coussens, L.M., C.L. Tinkle, D. Hanahan, and Z. Werb. 2000. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 103:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holtmeier, W., M. Pfander, A. Hennemann, T.M. Zollner, R. Kaufmann, and W.F. Caspary. 2001. The TCR-delta repertoire in normal human skin is restricted and distinct from the TCR-delta repertoire in the peripheral blood. J. Invest. Dermatol. 116:275–280. [DOI] [PubMed] [Google Scholar]
- 21.Groh, V., R. Rhinehart, H. Secrist, S. Bauer, K.H. Grabstein, and T. Spies. 1999. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc. Natl. Acad. Sci. USA. 96:6879–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]