

Abstract
The prototranscription factor p100 represents an intersection of the NF-κB and IκB families, potentially serving as both the precursor for the active NF-κB subunit p52 and as an IκB capable of retaining NF-κB in the cytoplasm. NF-κB–inducing kinase (NIK) controls processing of p100 to generate p52, and thus NIK-deficient mice can be used to examine the biological effects of a failure in such processing. We demonstrate that treatment of wild-type osteoclast precursors with the osteoclastogenic cytokine receptor activator of NF-κB ligand (RANKL) increases both expression of p100 and its conversion to p52, resulting in unchanged net levels of p100. In the absence of NIK, p100 expression is increased by RANKL, but its conversion to p52 is blocked, leading to cytosolic accumulation of p100, which, acting as an IκB protein, binds NF-κB complexes and prevents their nuclear translocation. High levels of unprocessed p100 in osteoclast precursors from NIK−/− mice or a nonprocessable form of the protein in wild-type cells impair RANKL-mediated osteoclastogenesis. Conversely, p100-deficient osteoclast precursors show enhanced sensitivity to RANKL. These data demonstrate a novel, biologically relevant means of regulating NF-κB signaling, with upstream control and kinetics distinct from the classical IκBα pathway.
Keywords: mice knockout, bone remodeling/physiology, MAP kinase kinase kinases, cultured cells/physiology, murine RANKL
Introduction
Osteoclasts (OCs) are multinucleated cells derived from mononuclear myeloid bone marrow progenitors that express receptor activator of NF-κB (RANK), the receptor for the key osteoclastogenic cytokine RANK ligand (RANKL) (1). Interaction of RANK with RANKL in the presence of macrophage CSF (M-CSF) promotes OC differentiation. Thus, deletion of the RANK, RANKL, or M-CSF genes leads to an absence of OCs, causing osteopetrosis. Like other members of the TNF receptor superfamily, RANK potently activates the NF-κB pathway.
In the canonical NF-κB pathway (for reviews see reference 2, 3), ligation of a transmembrane receptor such as RANK activates the IκB kinase (IKK) complex, which phosphorylates NF-κB–associated IκBα, leading to its ubiquitination and proteosomal degradation. These events release NF-κB subunits in the cytosol, allowing them to travel to the nucleus where they enhance transcription of target genes. In mammals, the NF-κB family has five members: RelA (p65), RelB, c-Rel, NF-κB1, and NF-κB2. RelA, RelB, and c-Rel are synthesized as mature proteins, each bearing a transcriptional transactivation domain and a DNA-binding Rel homology domain. In contrast, the active forms of NF-κB1 and NF-κB2, p50 and p52, are derived from the full-length products of the nfkb1 and nfkb2 genes, p105 and p100, respectively. The generation of the transcriptionally active subunits p50 and p52 is dependent on phosphorylation of their precursors followed by ubiquitination and partial proteosomal degradation of their COOH termini (4, 5). The NH2-terminal Rel homology domain of p50 or p52, when cleaved from the COOH terminus, can form active NF-κB dimers with RelA, RelB, or c-Rel.
The COOH-terminal portions of p105 and p100, before processing to p50 and p52, contain IκB-like ankyrin repeats. Thus, similar to IκB, p105 and p100 are capable of retaining proteins with a Rel domain in the cytosol (6, 7). p105 is ubiquitously expressed, and its conversion to p50 appears to be largely constitutive, with only modest increases in levels of p50 in response to NF-κB activation (2). In contrast, p100 is expressed predominantly in lymphoid organs (8), and its processing is tightly controlled by a kinase cascade. NF-κB–inducing kinase (NIK) activates IKKα (a subunit of the IKK complex), which phosphorylates p100, targeting it for processing to p52 (9, 10).
The importance of the NIK/IKKα/p100/p52 pathway has been highlighted by several mouse models. The nfkb2 knockout mouse, which lacks both p100 and p52, exhibits abnormal B cell differentiation and function (11, 12). Radiation chimeras bearing IKKα2/− bone marrow or mice expressing nonactivatable IKKα have decreased p52 levels and similar defects in B cell maturation (10). Alymphoplasia (aly/aly) mice (13), which carry a mutation in the IKKα interaction domain of NIK (14, 15), also have diminished p52 levels (9, 16) and defective B cell maturation (16). Finally, animals lacking the COOH-terminal IκB-like domain of NFκB2, and which thus express unregulated p52, have increased lymphocyte proliferation and gastric mucosal hyperplasia (17). In sum, these models demonstrate that regulated expression of the active NF-κB subunit p52, via NIK- and IKKα-mediated processing of p100, is biologically relevant.
Despite these findings, the importance of the putative inhibitory effect of unprocessed p100 on global NF-κB activation, via its IκB activity, has not been established. In this study, we have used the NIK-deficient mouse (18) to examine the effects of a lack of p100 processing on OC differentiation and NF-κB signaling. Previous studies of the NIK/IKKα/p100 pathway in a physiological context have only been performed in the B cell lineage, which is dependent on p52 for full maturation (11, 12). The nfkb2 knockout mouse, which lacks both p100 and p52, has normal basal osteoclastogenesis in vivo (19), indicating that, in contrast to its role in B cell development, p52 itself is dispensable for OC differentiation. The fact that the NIK−/− mouse fails to process p100 to p52 positions us to examine the potential role of p100 in osteoclastogenesis, in the absence of a requirement for p52. Our studies show that when p100 levels are increased, as in NIK-deficient OC precursors, RANKL-stimulated osteoclastogenesis is impaired. Conversely, we find that OC differentiation is enhanced in the absence of p100 in vitro. These results demonstrate that stimulated osteoclastogenesis is regulated by a pathway involving RANKL, NIK, and p100.
Materials and Methods
Mice.
NIK−/− mice were generated by homologous recombination as described (18) on a 129Sv/Ev background. Mice were maintained by heterozygote matings in a specific pathogen-free facility.
Osteoclast Culture.
For whole marrow culture, unfractionated bone marrow cells were cultured in the presence of 1,25 dihydroxyvitamin D3 (2.5 × 10−8 M) to induce osteoclast differentiation (20). After 8 d, cultures were fixed and stained for tartrate-resistant acid phosphatase (TRAP), a marker of OC differentiation, according to manufacturer's instructions (Sigma-Aldrich). To obtain bone marrow–derived macrophages (Mφ), marrow cells were on Petri dishes in the presence of CMG14–12 supernatant containing the equivalent of 100 ng/ml recombinant M-CSF (21) for at least 5 d. Equal numbers of WT and NIK−/− Mφs were replated onto tissue culture plates in the presence of partially purified M-CSF (see M-CSF Preparation) equivalent to 20 ng/ml recombinant M-CSF and the indicated dose of GST-RANKL for 6 d and then fixed and stained for TRAP activity. For analysis of resorptive activity, Mφs were replated onto multiwell Osteologic slides (BD Biosciences), which are coated with inorganic hydroxyapatite matrix, in the presence of M-CSF and 150 ng/ml GST-RANKL. After 6 d, cells were fixed in neutral buffered formalin and stained with methylene blue.
In Vivo RANKL Treatment.
GST-RANKL was prepared as described (22). WT and NIK−/− mice (2–3 mo old) were injected subcutaneously with GST-RANKL (500 μg per day for 7 d) or PBS (sham). Calvaria were fixed overnight in 10% buffered formalin, decalcified in 14% EDTA, embedded in paraffin, sectioned, and stained for TRAP with a hematoxylin counterstain. The number of OC per 10 fields (100× magnification) for five mice in each group was counted. Statistics were performed with the unpaired Student's t test.
Antibodies.
mAbs to RelA and the NH2 terminus of NF-κB2 common to p100 and p52 were from Santa Cruz Biotechnology, Inc., as were the polyclonal antiserum directed against IκBα and RelB and the agarose conjugated anti-RelA polyclonal antibody used for immunoprecipitation.
Semiquantitative RT-PCR.
Total RNA was extracted using RNeasy (QIAGEN), and snf cDNA was generated with AMV-RT (Fisher Scientific) according to manufacturers' instructions using 1 μg total RNA and oligo-dT primers diluted to a final volume of 100 μl. Oligonucleotide primers used for PCR were: nfkb2 sense, 5′-TGGCTGGTGGGGACTTGG-3′; nfkb2 antisense, 5′-ACCCACCGAGATGTGGGC-3′; GAPDH sense, 5′-ACTTTGTCAAGCTCATTTCC-3′; GAPDH antisense, 5′-TGCAGCGAACTTTATTGATG-3′; calcitonin receptor (CTR) sense 5′-CATTCCTGTACTTGGTTGGC-3′; CTR antisense, 5′-AGCAATCGACAAGGAGTGAC-3′; matrix metaloproteinase 9 (MMP9) sense, 5′-CCTGTGTGTTCCCGTTCATCT-3′; MMP9 antisense, 5′-CGCTGGAATGATCTAAGCCCA-3′; β3 integrin sense, 5′-TTACCCCGTGGACATCTACTA-3′; and β3 integrin antisense, 5′-AGTCTTCCATCCAGGGCAATA-3′. For each reaction, 1–2 μl cDNA was used for the following PCR cycle numbers: nfkb2-24; GAPDH-24; CTR-34; MMP9-24; and β3 integrin-30.
M-CSF Preparation.
Supernatant was harvested from CMG14–12 cells grown in phenol red–free media, diluted into triethanolamine buffer, and loaded onto a Q sepharose FastFlow column (Amerhsam Biosciences). M-CSF was eluted first with a NaCl gradient (0–1.0 M), and then eluted from a second Q column by pH gradient (pH 7 to 4). Peak fractions were dialyzed against PBS and sterile filtered. Bioactivity was compared with recombinant murine M-CSF (R&D Systems) by assaying proliferation of M-NFS-60, an M-CSF–dependent cell line (21).
Subcellular Fractionation.
Cells were harvested by treatment with 10 mM EDTA in PBS for 15–30 min at 4°C followed by scraping. Nuclear and cytoplasmic fractions were isolated using the NuCLEAR kit (Sigma-Aldrich). Extracts were quantitated by a modified Coomassie method (Pierce Chemical Co.). For immunoblots, 5 μg of nuclear extract or 25 μg of cytoplasmic extract was used, and equivalent loading was verified by Ponceau red staining after transfer to PVDF membrane.
NF-κB DNA Binding Assays.
For electrophoretic mobility shift assay (EMSA), nuclear extracts (2 μg) were incubated with labeled double stranded κB3 oligonucleotide (5′-AAACAGGGGGCTTTCCCTCCTC-3′) at 4°C for 32P-labeled probes (see Figs. 4 and 5) or at room temperature for biotin-labeled probes (see Fig. 8). Protein–DNA complexes were resolved on nondenaturing 4–20% polyacrylamide gels (Invitrogen) in 0.5 × TBE. Radiolabeled gels were dried and subjected to autoradiography. For nonradioactive detection, the LightShift chemiluminescent system (Pierce Chemical Co.) was used, according to manufacturer's instructions, using a biotinylated version of the same oligo (Invitrogen). For RelA supershift, 2 μg polyclonal anti-RelA (Santa Cruz Biotechnology, Inc.) was added after formation of DNA–protein complexes at room temperature for 15 min. An ELISA-based NF-κB assay, the EZDetect p65 ELISA (Pierce Chemical Co.) was performed according to manufacturer instructions, using 2 μg nuclear extract per well, performed in triplicate. Background (extraction buffer alone, averaged for three wells) was subtracted from each sample. In this assay, nuclear extract is applied to wells precoated with specific NF-κB oligos followed by anti-p65 (RelA) antibody and an HRP-conjugated secondary. The assay is developed with chemiluminescent substrates and read on a luminometer/plate reader (SpectraFluor Plus; Tecan).
Figure 4.

NF-κB signaling in NIK−/− Mφs is intact. (A) Mφs were acutely stimulated with RANKL for the indicated times, and cytoplasmic (top) and nuclear (bottom) fractions were analyzed by immunoblot for IκBα and RelA, respectively. (B) NF-κB DNA binding activity was assessed at 0 and 15 min by EMSA using a κB3 oligonucleotide probe and nuclear extracts from A. NF-κB signaling is comparable in WT and NIK−/− Mφ cultures in response to RANKL.
Figure 5.

NF-κB does not accumulate in the nucleus of NIK−/− preOCs. (A) PreOCs, generated by treating Mφs with RANKL for 48 h followed by a 3 h starvation, were stimulated with RANKL for the indicated times, and cytoplasmic (top) and nuclear (bottom) fractions were isolated and analyzed by immunoblot for IκBα and RelA, respectively. Although IκBα gets degraded in both WT and NIK−/− cultures, RelA does not accumulate in the nucleus in the absence of NIK. (B) Nuclear extracts prepared as in A were analyzed for NF-κB DNA binding activity by EMSA. Whereas WT preOC cultures have detectable NF-κB activity at baseline and further induction by RANKL, NIK−/− preOC cultures show little binding activity with or without RANKL. (C) RelA-specific NF-κB DNA binding activity was assessed by ELISA (EZ Detect kit; Pierce Chemical Co.) using the same preOC nuclear extracts as in A. In this assay, NF-κB–specific oligos are bound to a plate. Nuclear extract is then applied followed by anti-p65 (RelA) antibody and an HRP-conjugated secondary. The assay is developed with chemiluminescent reagents and read on a luminometer/plate reader. Each sample was run in triplicate. Unmutated (UnMu) and mutant (Mu) soluble oligos were added to the WT 30-min sample to confirm specificity. The results of this assay mirror the EMSA in B and confirm lower baseline NF-κB activity and lack of RANKL-mediated induction in NIK−/− preOCs. Binding activity is abolished by addition of the unmutated oligo and unaffected by the mutant oligo (last two bars).
Figure 8.

Expression of a noncleavable form of p100 in WT Mφs inhibits NF-κB signaling and osteoclastogenesis. (A) WT Mφs were transduced with retrovirus bearing empty pMX or pMX-p100ΔGRR, a noncleavable form of p100, overnight and selected in puromycin for 3 d before lysis. Immunoblots for p100/p52 demonstrate expression of the retroviral driven p100ΔGRR (lane 2) at levels three to fourfold above endogenous p100 (lane 1), similar to that seen in RANKL-treated NIK−/− Mφs. (B) NF-κB activity was assessed by EMSA on Mφs transduced with pMX (control) or pMX-p100ΔGRR as above. Lanes 1–2, cells transduced with pMX, unstimulated (lane 1), or treated with RANKL for 15 min (lane2); lanes 3–4, cells transduced with pMX-p100ΔGRR, unstimulated (lane 3), or treated with RANKL for 15 min (lane 4); lane 5, RANKL-stimulated vector control, with unlabeled probe at 50-fold excess; and lane 6, RANKL-stimulated vector control supershifted with polyclonal anti-RelA antibody. The supershifted band is indicated (ss). Expression of p100ΔGRR in Mφs dramatically decreases NF-κB signaling by RelA in response to RANKL. (C) After the 3 d puromycin selection, retrovirally transduced Mφs were cultured in RANKL for 5 d and then stained for TRAP. Expression of p100ΔGRR inhibits osteoclastogenesis in these WT cultures. Bar, 300 μm.
Retroviral Transduction.
Mφ were transduced with retrovirus containing vector alone (pMX-puro) (23) or a noncleavable form of p100 (pMX-p100ΔGRR) for 24 h in the presence of CMG14–12 supernatant equivalent to 200 ng/ml rM-CSF and 4 μg/ml polybrene (Sigma-Aldrich) and then selected with puromycin for 3 d. Surviving Mφ were passaged on Petri dishes and grown in M-CSF overnight before the preparation of nuclear extracts, or passaged into 96-well plates, cultured in M-CSF (20 ng/ml) and GST-RANKL (150 ng/ml) for 5 d, and then fixed and stained for TRAP activity.
Results
Stimulated Osteoclastogenesis Is Blunted in NIK−/− Mice.
To determine if p100 processing has a role in OC differentiation, we placed NIK−/− (18) bone marrow cells in osteoclastogenic conditions and found they fail to generate multinucleated cells expressing TRAP, a marker of the OC phenotype (Fig. 1 A). In these whole marrow cultures, stromal cells provide the M-CSF and RANKL required for osteoclastogenesis. OCs can also be generated in vitro from pure populations of Mφs treated with soluble recombinant M-CSF and RANKL. As shown in Fig. 1 B, WT Mφs exposed to these cytokines form confluent sheets of large, spread OCs and exhibit exuberant resorptive activity (Fig. 1 C). In contrast, NIK−/− cultures, although rich in Mφs, contain only rare, poorly spread TRAP-positive cells (Fig. 1 B) with no detectable resorptive capacity (Fig. 1 C). The lack of osteoclastogenesis in NIK−/− cultures cannot be rescued by increasing RANKL by amounts up to 10-fold the optimal dose for WT cultures (Fig. 1 B, 1,000 ng/ml RANKL) or by doubling the time in culture (unpublished data). Given that these experiments were performed using only Mφs, the osteoclastogenic defect in NIK−/− whole marrow cultures resides in OC precursors and not in cytokine-producing stromal cells. The lack of morphological changes in NIK−/− cultures is accompanied by a failure, beyond day 1, to up-regulate transcripts characteristic of OCs, including β3 integrin, matrix metalloproteinase 9, and CTR (Fig. 1 D). However, NIK−/− cultures are not completely unresponsive to RANKL. At day 1, β3 integrin mRNA is induced in NIK−/− cultures at levels similar to WT. Additionally, NIK−/− Mφs show normal MAPK activation (unpublished data).
Figure 1.
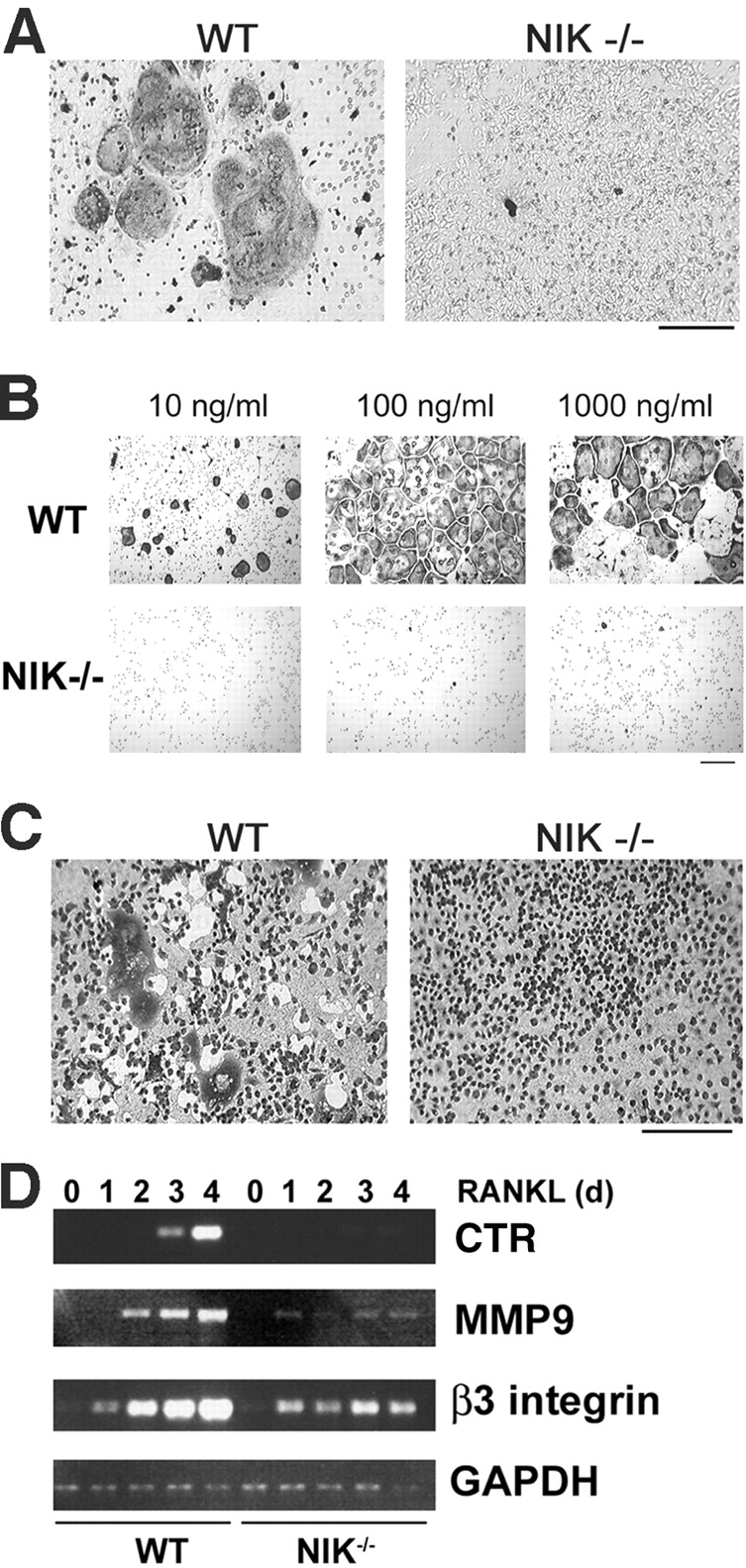
NIK−/− mice have defective RANKL-mediated osteoclastogenesis in vitro. (A) Equal numbers of WT and NIK−/− cells from unfractionated bone marrow were cultured for 8 d in the presence of vitamin D3 and then fixed and stained for TRAP, a marker of OC differentiation. Under these conditions, both stromal cells and OC precursors are present. NIK−/− cultures fail to form large, spread, TRAP-positive OCs. Bar, 300 μm. (B) Equal numbers of Mφs (expanded from bone marrow in high doses of M-CSF) from WT and NIK−/− mice were cultured in the presence of M-CSF and the indicated dose of RANKL for 6 d and then fixed and stained for TRAP. Very few, poorly spread TRAP-positive cells are seen in the NIK−/− cultures. Bar, 100 μm. (C) Osteoclastogenic cultures were generated as in B on an artificial hydroxyapatite matrix (Osteologic slides). On day 6, cultures were stained with methylene blue at basic pH. Resorbed matrix is seen as a clear white area around dark cells. NIK−/− cultures have negligible matrix resorption. Bar, 300 μm. (D) WT and NIK−/− Mφs were treated with RANKL for 0–4 d and then harvested for RNA extraction. Semiquantitative RT-PCR was performed for CTR, MMP9, and β3 integrin, all markers of OC differentiation, with GAPDH as control. Although β3 integrin levels are comparable at day 1, all three markers show a failure in induction from days 2–4 in NIK−/− cultures, confirming the morphological lack of differentiation.
Interestingly, in vivo basal osteoclastogenesis is unaltered in NIK−/− mice. Femurs of unmanipulated 3–4-mo-old WT and NIK−/− mice have similar cortical thickness, trabecular size and distribution, and number of osteoclasts (Fig. 2 A). On the other hand, and consistent with our in vitro observations, these mutants exhibit a blunted response to osteoclastogenic stimuli. Whereas WT mice generate a robust sevenfold increase in OC number along calvarial sinuses after 1 wk of RANKL treatment, NIK−/− mice show only a twofold rise (Fig. 2, B and C). Similarly, NIK−/− mice fail to show a significant osteoclastogenic response to a 3-d course of parathyroid hormone injection (unpublished data).
Figure 2.


NIK−/− mice have normal basal osteoclastogenesis and a blunted response to RANKL in vivo. (A) TRAP-stained sections of femurs from unmanipulated 3–4-mo-old WT and NIK−/− mice show no difference in trabecular architecture or OC number red-stained cells. Bar, 600 μm. (B) WT and NIK−/− mice were injected with 500 μg GST-RANKL or PBS daily for 7 d and killed on the 8th day. TRAP-stained coronal sections of calvaria adjacent to the sagittal suture show a dramatic increase in OCs along sutures and sinusoids only in WT RANKL-treated animals. Bar, 150 μm. (C) Quantitation of osteoclasts along inner (sinusoidal) calvarial surfaces for animals described in B confirms the significant difference in the response of WT and NIK−/− mice to RANKL injection. Error bars indicate SEM. *P < 0.0001 compared with WT sham. **P < 0.001 compared with NIK−/− sham and WT RANKL.
RANKL Induces Processing of p100 in WT but not NIK−/−, OC Precursors.
Because NIK has been shown to control p100 processing in response to other TNF family cytokines, we next assessed the effect of RANKL on p100 and p52 levels in WT and NIK−/− cultures. In unstimulated WT Mφs (Fig. 3 A, 0 h), p100 but not p52 is readily detected. Exposure to RANKL yields accumulation of p52 beginning at 4 h, with no change in p100 levels. In contrast, RANKL exposure for as long as 48 h fails to induce the appearance of p52 in NIK−/− OC precursors. Moreover, whereas basal p100 levels are comparable in WT and NIK−/− Mφs (0 h for WT and NIK−/− in Fig. 3 A), p100 is substantially enhanced (2.5–4-fold) in mutant cells by 8 h of RANKL exposure, and this increase persists for at least 48 h This effect is specific for RANKL, since exposure of WT Mφ to TNFα does not induce production of p52, although it does increase p100 levels (Fig. 3 B).
Figure 3.
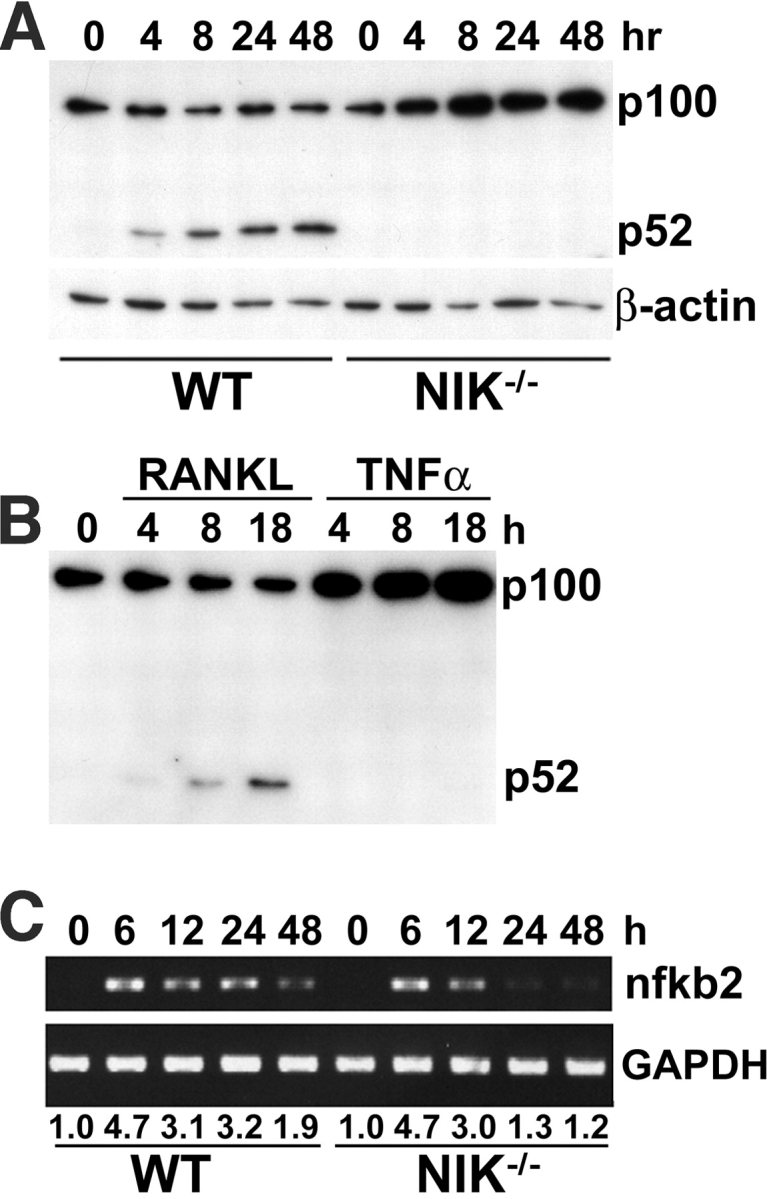
RANKL-induced p100 processing does not occur in NIK−/− OC precursors despite normal induction of nfkb2 transcription. (A) WT and NIK−/− Mφs were treated with RANKL for increasing times, and total lysates were analyzed by immunoblot to detect changes in p100 and p52 levels, using a monoclonal antibody recognizing the NH2 terminus common to p100 and p52. The blot was stripped and reprobed with anti-β actin as a loading control (bottom). In WT cultures, p100 levels remain steady, whereas p52 levels are increased by RANKL treatment. Parallel NIK−/− cultures fail to generate p52 at any time point and accumulate p100. (B) WT Mφs were treated with RANKL or TNFα for the indicated times, and p100/p52 levels were assessed by immunoblot as in A. p100 processing does not occur in response to TNFα. (C) WT and NIK−/− Mφs were treated with RANKL for increasing times, and RNA was harvested. Levels of nfkb2 mRNA were analyzed by semiquantitative RT-PCR using GAPDH as a control. Numbers beneath lanes indicate fold increase over WT 0 h baseline. Although WT and NIK−/− cultures show equivalent induction of nfkb2 at 6–12 h, levels are significantly lower in NIK−/− cultures thereafter.
Because the nfkb2 gene is itself regulated by NF-κB (24), we asked if increased transcription contributes to the accumulation of p100 in NIK-deficient cells. Semiquantitative RT-PCR shows that RANKL treatment up-regulates nfkb2 mRNA fourfold by 6 h in both WT and NIK−/− Mφ cultures (Fig. 3 C). Interestingly, nfkb2 mRNA levels are substantially lower in NIK−/− cells at 24 and 48 h. Because NF-κB transactivates the nfkb2 gene, this observation is consistent with a possible decrease in NF-κB signaling at these later times.
NF-κB Signaling Is Intact in NIK− /− Mφs but Blunted in NIK− /− preOCs.
To determine whether the differences in p100 processing between WT and NIK−/− cultures impacts NF-κB signaling, we assessed NF-κB activation in Mφs (time 0 in Fig. 3 A in which WT and NIK−/− cultures have equivalent p100 levels) and preOCs (48 h in Fig. 3 A in which NIK−/− cultures have more p100 and no p52). Acute stimulation of Mφs with RANKL led to normal degradation of IκBα, nuclear translocation of RelA, NF-κB DNA binding activity, and resynthesis of IκBα (an NF-κB–dependent event) in NIK−/− Mφ cultures (Fig. 4, A and B) consistent with the findings of Yin et al. (18). In contrast, RANKL stimulation of preOCs (generated by exposing Mφs to RANKL for 48 h, then starving of serum and cytokines for 3 h) has very different results in WT and NIK−/− cultures. Similar to its effects in Mφs (Fig. 4), RANKL stimulation of WT preOCs leads to IκBα degradation, nuclear translocation of RelA, and increased NF-κB DNA binding capacity (Fig. 5, A–C) . NIK−/− preOCs degrade IκBα in a manner similar to that of WT cells (Fig. 5 A), although basal levels are a bit lower than WT. On the other hand, RelA does not appear in nuclear extracts of the mutant cells (Fig. 5 A). Concordantly, both basal and stimulated NF-κB DNA binding activity is decreased as assessed by EMSA (Fig. 5 B) and an ELISA-based chemiluminescent assay for RelA-specific NF-κB activation (Fig. 5 C). In addition, resynthesis of IκBα is blunted in the absence of NIK (Fig. 5 A, 60 min).
To determine if the nuclear translocation of other NF-κB subunits is affected in NIK−/− cultures, we analyzed nuclear extracts of WT and NIK−/− Mφs and preOCs by immunoblot (Fig. 6) . As expected based on Fig. 3 A, within 60 min of RANKL stimulation, neither WT nor NIK−/− Mφs have nuclear p52. Only WT preOCs contain nuclear p52, and this is only slightly responsive to RANKL stimulation. Additionally, RelB is present at high levels in WT preOCs, at very low levels in WT Mφs, and absent in NIK−/− Mφs and preOCs.
Figure 6.

RelB and p52 fail to accumulate in the nucleus in NIK−/− preOCs. Immunoblot of nuclear extracts from Mφ and preOC cultures treated acutely with RANKL shows accumulation of p52 only in WT preOCs (top). Levels of nuclear RelB are highest in WT preOCs, with minimal acute induction by RANKL. WT Mφs show low levels of nuclear RelB, whereas NIK−/− Mφs and preOCs lack this NF-κB subunit. A background band (bkgd) in RelB immunoblots shows equivalent signal in all lanes.
These data indicate that, as in other cytokine signaling pathways, NIK is not involved in the degradation of IκBα in response to RANKL. NIK does, however, play a permissive role in NF-κB nuclear translocation in committed preOCs, suggesting increased activity of another IκB-like protein in NIK-deficient preOCs but not Mφ.
p100 Functions as an IκB Protein in NIK− /− preOCs.
To determine if the enhanced p100 appearing in RANKL-treated NIK−/− preOCs functions as an IκB, RelA was immunoprecipitated from resting or RANKL-stimulated preOCs, and its association with p100 and IκBα was assessed by immunoblot (Fig. 7) . As expected, RelA levels are similar in WT and NIK−/− cells, and association of the subunit with IκBα is decreased by RANKL treatment in both circumstances. On the other hand, although p100 does not associate with RelA in WT cells, it is readily detected in resting NIK−/− RelA immunoprecipitates, and this association increases in response to RANKL. These data suggest that, in NIK−/− OC precursors, exposure to RANKL results in accumulation of p100 because it cannot be processed to p52. Accumulated p100 would then bind NF-κB subunits as they are released from IκBα, preventing their nuclear translocation and the subsequent induction of gene transcription. This model predicts that expression of a nonprocessable form of p100 in WT OC precursors would inhibit osteoclastogenesis.
Figure 7.

Association of RelA and p100 is increased in NIK−/− preOCs. PreOCs (generated as in Fig. 5) were starved for 3 h and restimulated with RANKL for the indicated times before lysis. Total lysates were immunoprecipitated with anti-RelA antibody and analyzed by immunoblot for p100, RelA, and IκBα. In both WT and NIK−/−cultures, association of RelA and IκBα were reduced upon RANKL treatment. In WT cells, RelA associates with p100 only transiently. In contrast, RelA is bound to p100 in NIK−/− cultures before addition of RANKL, and this association is increased with RANKL treatment as IκBα is degraded.
To test this hypothesis, we used a p100 construct, p100ΔGRR, which lacks the glycine-rich region determining the site of proteosomal cleavage and hence cannot be processed to p52 (5). Using the pMX-puro retroviral vector (23), we expressed p100ΔGRR or vector alone as a control in WT Mφs (Fig. 8 A). Cells transduced with pMX or pMX-p100ΔGRR were stimulated with RANKL for 15 min, and NF-κB DNA binding activity was assessed by EMSA compared with unstimulated controls. Expression of p100ΔGRR markedly inhibits induction of NF-κB activity (Fig. 8 B). Transduced Mφs were also cultured in osteoclastogenic concentrations of RANKL plus M-CSF. Consistent with the role of unprocessed p100 as an NF-κB inhibitory protein, expression of p100ΔGRR inhibits formation of mature OCs (Fig. 8 C).
Finally, if, as we propose, p100 blunts RANKL-stimulated osteoclastogenesis, OC precursors lacking p100 should show enhanced sensitivity to RANKL. In fact, nfkb2−/− Mφs (which lack both p100 and p52) form OCs at lower doses of RANKL and with shorter exposure to the cytokine compared with WT controls (Fig. 9) . These data confirm that the nfkb2 gene product p100 has an inhibitory effect on RANKL-stimulated osteoclastogenesis, although p52, which potentially contributes to total NF-κB signaling, is dispensable.
Figure 9.

RANKL-mediated osteoclastogenesis in vitro is enhanced in the absence of p100. Mφs from WT (A) and nfkb2−/− (B) mice were cultured with increasing doses of GST-RANKL for 3, 4, or 5 d and then fixed and stained for TRAP. The number of mature OCs was counted for one 4× field per well, 4 wells per condition. At high doses of RANKL (50–150 ng/ml), WT OCs begin to form on day 4 and are confluent on day 5. In contrast, nfkb2−/− OCs begin to form on day 3 with as little as 25 ng/ml RANKL and are confluent on day 4. The peak numbers of OCs are slightly lower for nfkb2−/− cultures than for WT cultures because the nfkb2−/− OCs are larger. This is also the case for the nfkb2−/− cultures at 25 ng/ml RANKL on day 5 in which the OCs are smaller than at the higher doses. Thus, nfkb2−/− cultures, which lack both p100 and p52, show accelerated osteoclastogenesis at lower doses of RANKL compared with WT controls.
Discussion
OCs are uniquely capable of degrading both the organic and inorganic components of bone, and in concert with bone-building osteoblasts are required for maintenance of skeletal integrity and mineral homeostasis (1). Stimulated OC recruitment prompts the skeletal lesions of postmenopausal osteoporosis, rheumatoid arthritis, and osteolytic metastasis of malignant tumors. Unlike the mesenchyme-derived osteoblasts that survive for several months, bone marrow–derived OCs are short-lived with a lifespan of 1–2 wk (25). Therefore, tight control of OC differentiation is as critical as control of OC activity for skeletal homeostasis.
NF-κB is potently induced by RANKL in OC precursors (26), and the occurrence of osteopetrosis, with an early block in osteoclastogenesis, in nfkb1/nfkb2 double knockout mice established the importance of this signal (19, 27). Individual ablation of either nfkb1 or nfkb2 does not impact the skeleton of unmanipulated mice, indicating mutual compensation between p50 and p52. In vitro osteoclastogenesis, by coculture with osteoblasts, is also not impaired in either nfkb1- or nfkb2-deficient mice (19). nfkb1 −/− and nfkb2 −/− mice lack both the precursor (p105 or p100) and the active NF-κB subunit (p50 or p52). Due to the potential opposing functions of the precursors (as IκBs) and the products (as active NF-κBs), loss of both precursor and product may mask the function of either one. Thus, the impact of individual subunits on specific NF-κB targets, or on a physiological process such as osteoclastogenesis, is unresolved.
The conversion of p105 to p50 is predominantly constitutive, and no specific regulators of the process are in hand (4). In contrast, processing of p100 to p52 is tightly controlled by a known signaling pathway involving NIK (9, 10). Therefore, by manipulating this pathway it is possible to substantially alter the ratios of these two proteins. For instance, in the absence of processing, increased transcription of the nfkb2 gene can enhance p100 levels. When both synthesis and processing of p100 are stimulated, p52 levels rise while p100 levels remain stable, greatly increasing the ratio of p52 to p100. We have used the NIK−/− system in which p100 accumulates due to an absence of processing and have uncovered a requirement for the processing of p100 for RANKL-stimulated osteoclastogenesis.
NIK directly interacts with TRAFs (28), a family of adaptors that mediate NF-κB and JNK activation downstream of TNF receptor family members, including RANK, CD40, and LtβR. Although overexpression studies suggested that NIK is the kinase bridging this receptor/adaptor family and the classical NF-κB cascade (via its direct interaction with IKKs) (29), this role is not supported by data from the alymphoplasia and NIK−/− mice, which do not exhibit a global defect in NF-κB signaling (14, 18, 30). Furthermore, NIK, which binds to and activates IKKα, is specifically required for the processing of p100 to p52 (9). The similar defects in NIK-, IKKα-, and nfkb2-deficient mice indicate that the p100 to p52 pathway is critical for B cell maturation (10–12, 16, 18). BAFF, a TNF family member, has been shown recently to activate this pathway in B cells (31).
In the OC lineage, we find that p100 processing is initiated by RANKL and promotes osteoclastogenesis. Other RANKL-mediated signaling events, including activation of JNK and ERK and degradation of IκBα, occur within 5–15 min of cytokine–receptor interactions (32). NIK, IKKα, and p100 are all present in unstimulated OC precursors, but p100 processing is not detectable before 4–6 h after exposure to RANKL. Blockade of protein translation with cyclohexamide also prevents p100 processing (33, 34 and unpublished data). It is therefore likely that several intermediary events, including protein translation, occur between RANK activation and NIK-mediated p52 expression, leading to a several hour delay.
We and others find that induction of p100 processing is restricted to a subset of TNF family cytokines, including RANKL, BAFF, and CD40L but not TNFα or IL-1β (31, 33, 34 and this article). Although TNFα can synergize with RANKL to enhance osteoclastogenesis when added after 48 h or RANKL exposure, it inhibits OC differentiation if added simultaneously (35). The TNFα-mediated increase in p100 levels, without generation of p52, seen in Mφ (Fig. 3 B) may explain the initial inhibitory effect of TNF at early stages of osteoclastogenesis. The clear stimulatory effect of TNFα on OCs in vivo is not easily explained. However, other factors present in inflammatory conditions may counteract the p100 effect, allowing the many signals common to TNFα and RANKL, such as activation of the classical NF-κB pathway, MAPKs, and PI3K, to act unopposed.
The features of the NIK/IKKα/p100 pathway in the OC lineage distinguish it from all previous descriptions of the NF-κB pathway. We propose (Fig. 10) that RANKL induces degradation of IκBα, releasing NF-κB, predominantly as RelA/p50 dimers, to rapidly accumulate in the nucleus. After several hours, RANKL also stimulates NIK to process p100, thus releasing p52, which can then complex with RelA or with RelB and travel to the nucleus (36, 37). This transcriptionally active pool of nuclear NF-κB stimulates osteoclastogenesis. In the absence of NIK, NF-κB signaling in OC precursors is initially intact. However, after 1–2 d of RANKL stimulation sufficient p100 accumulates in the cytosol to bind RelA/p50 dimers as they are released from IκBα. This cytoplasmic retention of NF-κB combined with the lack of p52 reduces NF-κB–mediated transcription in the nucleus and inhibits osteoclastogenesis.
Figure 10.

Model for p100-mediated blockade of NF-κB signaling in NIK-deficient preOCs. See Discussion.
In vitro, the defect in RANKL-mediated osteoclastogenesis in NIK−/− cultures is profound. In vivo, NIK-deficient mice are resistant to stimulated osteoclastogenesis in response to injection of RANKL or parathyroid hormone but show no bone phenotype in the basal state. Alymphoplasia mice, like NIK−/− mice, have normal bone architecture and a striking defect in osteoclastogenesis in vitro (unpublished data). Mice lacking p100 (nfkb2 −/−) also show no basal bone phenotype, despite a pronounced enhancement of RANKL-mediated osteoclastogenesis in vitro. Two general possibilities present themselves to explain the apparent paradox in the in vivo and in vitro phenotypes. First, culture conditions may mirror stimulated but not basal osteoclastogenesis. Second, because of compensatory mechanisms operating in vivo, many knockout mice exhibit no phenotype until stressed. As regards to the present situation, although M-CSF and RANKL are necessary for osteoclastogenesis in vivo and sufficient for differentiation of pure OC precursors in vitro, other cytokines modulate osteoclastogenesis within the bone microenvironment. For example, TNFα, in the context of permissive levels of RANKL, enhances osteoclastogenesis in many inflammatory conditions (35, 38–40) and also supports a normal level of basal osteoclastogenesis via the p55 TNF receptor (41). TGFβ can also support RANKL-mediated osteoclastogenesis (42, 43). In fact, a combination of RANKL, TNFα, and TGFβ rescues the NIK−/− defect in osteoclastogenesis in vitro (unpublished data). We suspect that the presence of these factors, and perhaps others in the bone microenvironment, allows OC differentiation to occur at a normal basal rate in NIK−/− mice but cannot compensate for the inhibitory influence of accumulated p100 induced by exogenous RANKL or PTH, which exerts its osteoclastogenic effect through RANKL.
Our studies uncover a novel NF-κB pathway using RANKL-induced processing of p100 to control the pool of inhibitory IκB proteins. Whereas p100 processing is also regulated in B cells by NIK, it is increased p52 and not decreased p100 that is critical for B cell maturation (10, 17). Interestingly, p52 itself is dispensable for osteoclastogenesis, since the nfkb2-deficient mouse, which lacks both p100 and p52, has normal OC numbers in vivo (19) and increased OC numbers in vitro (Fig. 9). In addition, regulation of NF-κB signaling by p100 is distinct from the classical IκB pathway in both kinetics (hours versus minutes) and upstream regulation (IKK complex versus NIK/IKKα). Thus, these two distinct mechanisms for NF-κB control (IκBα degradation and p100 processing) allow for cell type and receptor-specific responses within the NF-κB pathway.
NIK−/− osteoclastogenesis in vivo is not impaired in the basal state but only in conditions of excess RANKL such as accompanies administration of PTH. This observation leads us to propose that the modulating effect of NIK/p100 on osteoclastogenesis is important in clinical conditions of increased osteoclastogenesis, such as osteoporosis, rheumatoid arthritis, and osteolytic bone metastasis, in which RANKL expression is enhanced. Most importantly, the fact that p100 arrests stimulated osteoclastogenesis makes it a potential therapeutic target in diseases of accelerated bone loss.
Acknowledgments
We thank W. Greene (University of California, San Francisco, San Francisco, CA) for the p100ΔGRR cDNA and F. Weih (Institute of Toxicology and Genetics, Karlsruhe, Germany) for nfkb2−/− mice.
This work was supported by grants from the National Institutes of Health: KO8 AR47846, 5T32-DK-07120, and P30 A448335 (to D. Veis Novack); R01CA43509 (to R.D. Schreiber); R01AR46852 and R01AR48812 (F.P. Ross); and R01AR32788, R01AR46523, and R01AR48853 (to S.L. Teitelbaum). Additional support was provided by grants from American Cancer Society (IRG-58-010-44), the Barnes-Jewish Hospital Foundation (to D. Veis Novack), and the Cancer Research Institute (to L. Yin).
Abbreviations used in this paper: CTR, calcitonin receptor; EMSA, electrophoretic mobility shift assay; IKK, IκB kinase; Mφ, bone marrow–derived macrophages; M-CSF, macrophage CSF; MMP9, matrix metalloproteinase 9; NIK, NF-κB–inducing kinase; OC, osteoclast; RANK, receptor activator of NF-κB ligand; RANKL, RANK ligand; TRAP, tartrate-resistant acid phosphatase.
References
- 1.Teitelbaum, S.L. 2000. Bone resorption by osteoclasts. Science. 289:1504–1508. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh, S., M.J. May, and E.B. Kopp. 1998. NF-κB and REL proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16:225–260. [DOI] [PubMed] [Google Scholar]
- 3.Rothwarf, D.M., and M. Karin. 1999. The NF-κB activation pathway: a paradigm in information transfer from membrane to nucleus. Sci. STKE. 1999:RE1. [DOI] [PubMed]
- 4.Palombella, V.J., O.J. Rando, A.L. Goldberg, and T. Maniatis. 1994. The ubiquitin-proteasome pathway is required for processing of the NF-κB1 precursor protein and the activation of NF-κB. Cell. 78:773–785. [DOI] [PubMed] [Google Scholar]
- 5.Heusch, M., L. Lin, R. Geleziunas, and W.C. Greene. 1999. The generation of nfkb2 p52: mechanism and efficiency. Oncogene. 16:6201–6208. [DOI] [PubMed] [Google Scholar]
- 6.Rice, N.R., M.L. MacKichan, and A. Israel. 1992. The precursor of NF-κ p50 has IκB-like functions. Cell. 71:243–253. [DOI] [PubMed] [Google Scholar]
- 7.Scheinman, R.I., A.A. Beg, and J. Baldwin, A.S. 1993. NF-κB p100 (Lyt-10) is a component of H2TF1 and can function as an IκB-like molecule. Mol. Cell. Biol. 13:6089–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paxian, S., S. Liptay, A. Alder, H. Hameister, and R.M. Schmid. 1999. Genomic organization and chromosomal mapping of mouse nuclear factor kappa B2 (NFKB2). Immunogenetics. 49:743–750. [DOI] [PubMed] [Google Scholar]
- 9.Xiao, G., E.W. Harhaj, and S.-C. Sun. 2001. NF-κB–inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell. 7:401–409. [DOI] [PubMed] [Google Scholar]
- 10.Senftleben, U., Y. Cao, G. Xiao, F.R. Greten, G. Krahn, G. Bonizzi, Y. Chen, Y. Hu, A. Fong, S.-C. Sun, et al. 2001. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 293:1495–1499. [DOI] [PubMed] [Google Scholar]
- 11.Caamano, J.H., C. Rizzo, S.K. Durham, D.S. Barton, C. Raventos-Suarez, C.M. Snapper, and R. Bravo. 1998. Nuclear factor (NF)-κB (p100/p52) is required for normal splenic microarchitecture and B cell–mediated immune responses. J. Exp. Med. 187:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franzoso, G., L. Carlson, L. Poljak, E.W. Shores, S. Epstein, A. Leonardi, A. Grinberg, T. Tran, T. Scharton-Kersten, M. Anver, et al. 1998. Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J. Exp. Med. 187:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyawaki, S., Y. Nakamura, H. Suzuka, M. Koba, R. Yasumizu, S. Ikehara, and Y. Shibata. 1994. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur. J. Immunol. 24:429–434. [DOI] [PubMed] [Google Scholar]
- 14.Matsushima, A., T. Kaisho, P.D. Rennert, H. Nakano, K. Kurosawa, D. Uchida, K. Takeda, S. Akira, and M. Matsumoto. 2001. Essential role of nuclear factor (NF)-κB–inducing kinase and inhibitor of κB (IκB) kinase α in NF-κB activation through lymphotoxin β receptor, but not through tumor necrosis factor receptor I. J. Exp. Med. 193:631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinkura, R., K. Kitada, F. Matsuda, K. Tashiro, K. Ikuta, M. Suzuki, K. Kogishi, T. Serikawa, and T. Honjo. 1999. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat. Genet. 22:74–77. [DOI] [PubMed] [Google Scholar]
- 16.Yamada, T., T. Mitani, K. Yorita, D. Uchida, A. Matsushima, K. Iwamasa, S. Fujita, and M. Matsumoto. 2000. Abnormal immune function of hemopoietic cells from alymphoplasia (aly) mice, a natural strain with mutant NF-kappa B-inducing kinase. J. Immunol. 165:804–812. [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa, H., D. Carrasco, E. Claudio, R.-P. Ryseck, and R. Bravo. 1997. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J. Exp. Med. 186:999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin, L., L. Wu, H. Wesche, C.D. Arthur, J.M. White, D.V. Goeddel, and R.D. Schreiber. 2001. Defective lymphotoxin-β receptor-induced NF-κB transcriptional activity in NIK-deficient mice. Science. 291:2162–2165. [DOI] [PubMed] [Google Scholar]
- 19.Iotsova, V., J. Caamano, J. Loy, Y. Yang, A. Lewin, and R. Bravo. 1997. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat. Med. 3:1285–1289. [DOI] [PubMed] [Google Scholar]
- 20.Kitazawa, R., R.B. Kimble, J.L. Vannice, V.T. Kung, and R. Pacifici. 1994. Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. J. Clin. Invest. 94:2397–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeshita, S., K. Kaji, and A. Kudo. 2000. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J. Bone Miner. Res. 15:1477–1488. [DOI] [PubMed] [Google Scholar]
- 22.McHugh, K.P., K. Hodivala-Dilke, M.H. Zheng, N. Namba, J. Lam, D. Novack, X. Feng, F.P. Ross, R.O. Hynes, and S.L. Teitelbaum. 2000. Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Invest. 105:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Onishi, M., T. Nosaka, K. Misawa, A.L.-F. Mui, D. Gorman, M. McMahon, A. Miyajima, and T. Kitamura. 1998. Identification and characterization of a constitutive active STAT5 mutant that promotes cell proliferation. Mol. Cell. Biol. 18:3871–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liptay, S., R.M. Schmid, E.G. Nabel, and G.J. Nabel. 1994. Transcriptional regulation of NF-κB2: evidence for κB-mediated positive and negative autoregulation. Mol. Cell. Biol. 14:7695–7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parfitt, A.M., G.R. Mundy, G.D. Roodman, D.E. Hughes, and B. Boyce. 1996. A new model for the regulation of bone resorption, with particular reference to the effects of bisphonates. J. Bone Miner. Res. 11:150–159. [DOI] [PubMed] [Google Scholar]
- 26.Jimi, E., S. Akiyama, T. Tsurukai, N. Okahashi, K. Kobayashi, N. Udagawa, T. Nishihara, N. Takahashi, and T. Suda. 1999. Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J. Immunol. 163:434–442. [PubMed] [Google Scholar]
- 27.Franzoso, G., L. Carlson, L. Xing, L. Poljak, E.W. Shores, K.D. Brown, A. Leonardi, T. Tran, B.F. Boyce, and U. Siebenlist. 1997. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 11:3482–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song, H.Y., C.H. Regnier, C.J. Kirschning, D.V. Goeddel, and M. Rothe. 1997. Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-kappaB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc. Natl. Acad. Sci. USA. 94:9792–9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malinin, N.L., M.P. Boldin, A.V. Kovalenko, and D. Wallach. 1997. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature. 385:540–544. [DOI] [PubMed] [Google Scholar]
- 30.Garceau, N., Y. Kosaka, S. Masters, J. Hambor, R. Shinkura, T. Honjo, and R.J. Noelle. 2000. Lineage-restricted function of nuclear factor kappaB-inducing kinase (NIK) in transducing signals via CD40. J. Exp. Med. 191:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claudio, E., K. Brown, S. Park, H. Wang, and U. Siebenlist. 2002. BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat. Immunol. 3:958–965. [DOI] [PubMed] [Google Scholar]
- 32.Wei, S., M.W. Wang, S.L. Teitelbaum, and F.P. Ross. 2001. Interleukin-4 reversibly inhibits osteoclastogenesis via inhibition of NF-κB and MAP kinase signaling. J. Biol. Chem. 21:6622–6630. [DOI] [PubMed] [Google Scholar]
- 33.Coope, H.J., P.G.P. Atkinson, B. Huhse, M. Belich, J. Janzen, M.J. Holman, G.G.B. Klaus, L.H. Johnston, and S.C. Ley. 2002. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J. 21:5375–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mordmuller, B., D. Krappmann, M. Esen, E. Wegener, and C. Scheidereit. 2003. Lymphotoxin and lipopolysaccharide induce NF-κB-p52 generation by a co-translational mechanism. EMBO Rep. 4:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam, J., S. Takeshita, B.J.E., O. Kanagawa, F.P. Ross, and S.L. Teitelbaum. 2000. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Invest. 106:1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duckett, C.S., N.D. Perkins, T.F. Kowalik, R.M. Schmid, E.S. Huang, A.S. Baldwin, Jr., and G.J. Nabel. 1993. Dimerization of NF-KB2 with RelA(p65) regulates DNA binding, transcriptional activation, and inhibition by an I kappaB-alpha (MAD3). Mol. Cell. Biol. 13:1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobrzanski, P., R.P. Ryseck, and R. Bravo. 1993. Both N- and C-terminal domains of RelB are required for full transactivation: role of the N-terminal leucine zipper-like motif. Mol. Cell. Biol. 13:1572–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cenci, S., M.N. Weitzmann, C. Roggia, N. Namba, D. Novack, and R. Pacifici. 2000. Estrogen deficiency induces bone loss by enhancing T cell production of TNFα. J. Clin. Invest. 106:1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keffer, J., L. Probert, H. Cazlaris, S. Georgopoulos, E. Kaslaris, D. Kioussis, and G. Kollias. 1991. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10:4025–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birkedal-Hansen, H. 1993. Role of cytokines and inflammatory mediators in tissue destruction. J. Periodontal Res. 28:500–510. [DOI] [PubMed] [Google Scholar]
- 41.Abu-Amer, Y., F.P. Ross, J. Edwards, and S.L. Teitelbaum. 1997. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its p55 receptor. J. Clin. Invest. 100:1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuller, K., J.M. Lean, K.E. Bayley, M.R. Wani, and T.J. Chambers. 2000. A role for TGFβ1 in osteoclast differentiation and survival. J. Cell Sci. 113:2445–2453. [DOI] [PubMed] [Google Scholar]
- 43.Kaneda, T., T. Nojima, M. Nakagawa, A. Ogasawara, H. Kaneko, T. Sato, H. Mano, M. Kumegawa, and Y. Hakeda. 2000. Endogenous production of TGF-β is essential for osteoclastogenesis induced by a combination of receptor activator of NF-κB ligand and macrophage-colony-stimulating factor. J. Immunol. 165:4254–4263. [DOI] [PubMed] [Google Scholar]