

Abstract
Limited oxygen delivery to tissues (hypoxia) is common in a variety of disease states. A number of parallels exist between hypoxia and acute inflammation, including the observation that both influence vascular permeability. As such, we compared the functional influence of activated polymorphonuclear leukocytes (PMN) on normoxic and posthypoxic endothelial cells. Initial studies indicated that activated PMN preferentially promote endothelial barrier function in posthypoxic endothelial cells (>60% increase over normoxia). Extension of these findings identified at least one soluble mediator as extracellular adenosine triphosphate (ATP). Subsequent studies revealed that ATP is coordinately hydrolyzed to adenosine at the endothelial cell surface by hypoxia-induced CD39 and CD73 (>20-and >12-fold increase in mRNA, respectively). Studies in vitro and in cd39-null mice identified these surface ecto-enzymes as critical control points for posthypoxia-associated protection of vascular permeability. Furthermore, insight gained through microarray analysis revealed that the adenosine A2B receptor (AdoRA2B) is selectively up-regulated by hypoxia (>5-fold increase in mRNA), and that AdoRA2B antagonists effectively neutralize ATP-mediated changes in posthypoxic endothelial permeability. Taken together, these results demonstrate transcription coordination of adenine nucleotide and nucleoside signaling at the vascular interface during hypoxia.
Keywords: adenosine, ectonucleotidase, endothelium, neutrophil, inflammation
Introduction
The study of physiologic adaptation and pathophysiologic response to hypoxia is presently an area of intense investigation. Recent reports suggest that both transcriptional and nontranscriptional hypoxia pathways may contribute to a broad range of diseases, and that a number of parallels exist between tissue responses to hypoxia and to acute inflammation (1). For example, during episodes of hypoxia, PMN are mobilized from the intravascular space to the interstitium, and such responses may contribute significantly to tissue damage during consequent reperfusion injury (2–4). Moreover, myeloid cell migration to sites of inflammation are highly dependent on hypoxia-adaptive pathways (5). As such, emigration of PMN through the endo- and epithelial barrier may lead to a disruption of such tissue barriers (6–8) and such a setting creates the potential for extravascular fluid leakage and subsequent edema formation (9, 10). However, with some exceptions, most episodes of hypoxia and/or ischemia-reperfusion are self-limiting, suggesting that endogenous protective mechanisms may exist to fortify the vascular barrier during such insults.
Previous studies have indicated that activated PMN release a number of soluble mediators, which regulate vascular permeability during transmigration. For example, PMN have been demonstrated to actively release both glutamate (3) and adenine nucleotides (in the form of AMP, which through a one step metabolic conversion liberates adenosine at the vascular surface; reference 11). Glutamate and adenosine liberated by this process have been shown to promote microvascular endothelial barrier, and as such, may provide innate pathways to reestablish endothelial cell-cell contact following PMN transmigration (11–13).
In the present studies, we sought to determine whether hypoxia might differentially influence vascular permeability in response to activated PMN. Somewhat surprisingly, initial results revealed that posthypoxic endothelia demonstrated increased protective responses to activated PMN. Studies aimed at isolating potentially novel factors in this pathway revealed that PMN-derived ATP was, at least in part, responsible for the increase observed in the posthypoxic endothelium. Indeed, enhanced extracellular metabolism and signaling after hypoxia were explained by transcriptional increases in functional surface apyrase (CD39), nucleotidase (CD73), and adenosine A2B-receptor (AdoRA2B), respectively. Such studies identify a coordinated, barrier-protective response initiated by endothelial preexposure to hypoxia.
Materials and Methods
Endothelial Cell Isolation and Culture.
Human microvascular endothelial cells (HMEC-1) were a gift of Francisco Candal, Centers for Disease Control, Atlanta, GA (14) and were harvested and cultured by a modification of methods described previously (3, 11). In brief, HMEC-1 were harvested with 0.1% trypsin and incubated at 37°C in 95% air/5% CO2. Culture medium was supplemented with heat-inactivated fetal bovine serum, penicillin, streptomycin, l-glutamine, epidermal growth factor, and hydrocortisone. Where indicated, primary human microvascular endothelial cells (HMVEC; Cascade Biologics) or bovine aortic endothelial cells (BAE; American Type Culture Collection) were cultured as described previously (11, 12). For preparation of experimental monolayers, confluent endothelial cells were seeded at ∼105 cells/ cm2 onto either permeable polycarbonate inserts or 100-mm Petri dishes. Endothelial cell purity was assessed by phase microscopic “cobblestone” appearance and uptake of fluorescent acetylated low-density lipoprotein. For hypoxic exposure, confluent endothelial monolayers were subjected to indicated periods of hypoxia (pO2 20 torr) as described previously (3).
Isolation of Human Neutrophils.
PMN were freshly isolated from whole blood obtained by venipuncture from human volunteers and anticoagulated with acid citrate/dextrose (2). Platelets, plasma, and mononuclear cells were removed by aspiration from the buffy coat following centrifugation through Histopaque®-1077 at 400 g for 30 min at 25°C. Erythrocytes were removed using a 2% gelatin sedimentation technique. Residual erythrocytes were removed by lysis in cold NH4Cl buffer. Remaining cells were >97% PMN as assessed by microscopic evaluation. PMN were studied within 2 h of their isolation.
Preparation of Activated PMN Supernatants.
Freshly isolated PMN (108 cells/ml in HBSS with 10−6 M FMLP) were incubated end-over-end for 1 min at 37°C (based on pilot experiments; unpublished data). PMN were then immediately pelleted (1,000 g for 20 s, 4°C) and supernatants filtered (0.45 μm; Phenomenex). For initial experiments isolating active PMN fractions, 108 PMN/ml were activated, cells were removed by pelleting and supernatants were filtered (0.2 μm). Resultant cell-free supernatants were resolved by high-performance liquid chromatography (model 1050; Hewlett-Packard) with an HP 1100 diode array detector by reverse-phase on an HPLC column (Luna 5-μm C18, 150 × 4.60 mm; Phenomenex) with 100% H20 mobile phase. Ultraviolet absorption spectra were obtained throughout. 1 ml fractions were collected, evaporated to dryness by speed-vac, reconstituted in HBSS (20-fold concentrated) and bioactivity was determined by permeability assay.
In experiments measuring supernatant concentrations of ATP, 100-μl samples were taken from PMN suspensions, immediately spun (1,000 g for 20 s, 4°C), filtered (0.45 μm), and analyzed via HPLC. ATP was measured with a H2O:CH3CN 96:4 mobile phase (1 ml/min). E-ATP and E-AMP were measured with a 0–50% methanol/H2O gradient (10 min) mobile phase (2 ml/min). Absorbance was measured at 260 nm. UV absorption spectra were obtained at chromatographic peaks. ATP and adenine nucleotides were identified by their chromatographic behavior (retention time, UV absorption spectra, and coelution with standards).
To measure the time course of ATP release from PMN, 107 PMN/ml were activated for indicated periods of time, supernatants were collected, and ATP content was quantified using CHRONO-LUME reagent (Crono-log Corp.). Luciferase activity was assessed on a luminometer (Turner Designs Inc.) and compared with internal ATP standards.
Endothelial Macromolecule Paracellular Permeability Assay.
Using a modification of methods previously described (11), HMEC-1 on polycarbonate permeable inserts (0.4-μm pore, 6.5-mm diam; Costar Corp.) were studied 7–10 d after seeding (2–5 d after confluency). Inserts were placed in HBSS-containing wells (0.9 ml), and HBSS (alone or with PMN, PMN supernatant, or ATP) was added to inserts (100 μl). At the start of the assay (t = 0), FITC-labeled dextran 70 kD (concentration 3.5 μM) was added to fluid within the insert. The size of FITC-dextran, 70 kD, approximates that of human albumin, both of which have been used in similar endothelial paracellular permeability models (15, 16). Fluid from opposing well (reservoir) was sampled (50 μl) over 60 min (t = 20, 40, and 60 min). Fluorescence intensity of each sample was measured (excitation, 485 nm; emission, 530 nm; Cytofluor 2300; Millipore Corp., Waters Chromatography) and FITC-dextran concentrations were determined from standard curves generated by serial dilution of FITC-dextran. Paracellular flux was calculated by linear regression of sample fluorescence (11).
Immunoprecipitation.
Confluent cells were labeled with biotin, lysed, and cell debris removed by centrifugation. Lysates were precleared with 50 μl preequilibrated protein G-Sepharose (Amersham Biosciences). Immunoprecipitation was performed with mouse mAb to human CD39 (Research Diagnostics, Inc.; 5 μg/ml), CD73 with mAb 1E9 (5 μg/ml, a gift from Dr. Linda Thompson, Oklahoma Medical Research Foundation, Oklahoma City, OK) or AdoRA2B with goat polyclonal (Santa Cruz Biotechnology, Inc.) followed by addition of 50 μl preequilibrated protein G-Sepharose and overnight incubation. Washed immunoprecipitates were boiled in reducing sample buffer (2.5% SDS, 0.38 M Tris, pH 6.8, 20% glycerol, and 0.1% bromophenol blue), separated by SDS-PAGE, transferred to nitrocellulose, and blocked overnight in blocking buffer. Biotinylated proteins were labeled with streptavidin-peroxidase and visualized by enhanced chemiluminescence (ECL; Amersham Biosciences).
Transcriptional Analysis.
Semiquantitative RT-PCR was used to verify endothelial CD39 mRNA regulation, as described previously (17). The PCR reaction contained 1 μM each of the sense primer 5′-AGC AGC TGA AAT ATG CTG GC-3′ and the antisense primer 5′-GAG ACA GTA TCT GCC GAA GTC C-3′. The primer set was amplified using increasing numbers of cycles of 94°C for 1 min, 60°C for 2 min, 72°C for 4 min, and a final extension of 72°C for 7 min. The PCR transcripts were visualized on a 1.5% agarose gel containing 5 μg/ml of ethidium bromide. Human β-actin (sense primer, 5′-TGA CGG GGT CAC CCA CAC TGT GCC CAT CTA-3′; and antisense primer, 5′-CTA GAA GCA TTT GCG GTG GAC GAT GGA GGG-3′) in identical reactions was used to control for the starting template.
In subsets of experiments, the transcriptional profile of endothelial cells subjected to normobaric hypoxia (12 h) was compared in RNA derived from control or hypoxic endothelium using quantitative genechip expression arrays (Affymetrix, Inc.) as described before (18). Where indicated, mRNA was also quantified by real-time PCR (iCycler; Bio-Rad Laboratories Inc.), as described previously (19). The primer sets contained 1 μM sense and 1 μM antisense containing SYBR Green I (Molecular Probes Inc.) in the reaction mixture. Primer sets (sense sequence, antisense sequence, and transcript size, respectively) for the following genes were used: CD39 (5′-AGC AGC TGA AAT ATG CTG GC-3′, 5′-GAG ACA GTA TCT GCC GAA GTC C-3′, 199 bp); CD73 (5′-ATT GCA AAG TGG TTC AAA GTC A-3′, 5′-ACA CTT GGC CAG TAA AAT AGG G-3′, 123 bp); adenosine A1-receptor (AdoRA1) (5′-GTT CAC AGT TTT TTA TTA GTC AC-3′,5′-AAC ATG AGT GTC AAC TCC-3′, 109 bp); adenosine A2A-receptor (AdoRA2A) (5′-CTT GGG TTC TGA GGA AGC AG-3′, 5′-CAG CAG CTC CTG AAC CCT AG-3′, 253 bp); AdoRA2B (5′-ATC TCC AGG TAT CTT CTC-3′, 5′-GTT GGC ATA ATC CAC ACA G-3′, 322 bp); adenosine A3-receptor (AdoRA3) (5′-CTT GAT TAC TTC CAC TGA GGT GG-3′, 5′-CAA CAT CTT CTA GGC ATC CTC C-3′, 334 bp); and β-actin (5′-GGT GGC TTT TAG GAT GGC AAG-3′, 5′-ACT GGA ACG GTG AAG GTG ACA G-3′, 162 bp) in identical reactions was used to control for starting template. Transcript levels and fold change in mRNA were determined as described previously (20). In addition, real-time PCR was performed from RNA isolations of human saphenous vein after ex vivo exposure to hypoxia. After approval by the institutional review board, saphenous vein material was obtained from patients undergoing aorta-coronary bypass surgery. Equal portions of dissected vein tissue were subjected to hypoxia (pO2 20 torr) immediately after the operation for different time points (0, 2, 8, and 24 h). After hypoxic exposure, total RNA was isolated from the tissues and real-time PCR was performed as described above.
Measurement of Surface Enzyme Activity of CD39.
We assessed CD39 surface enzyme activity as described previously (18) by quantifying the conversion of etheno-ATP (E-ATP) to etheno-AMP (E-AMP). Briefly, HBSS (with addition of the CD73 inhibitor αβ-methylene-ADP [10 μM] to prevent further metabolism of E-AMP to E-adenosine) was added to endothelial monolayers on a 60 mm Petri dish. After 10 min, E-ATP (final concentration 100 μM) was added. Samples were taken at indicated time-points, removed, acidified to pH 3.5 with HCl, spun (10,000 g for 20 s, 4°C), filtered (0.45 μm), and frozen (–80°C) until analysis via reverse phase HPLC. Ratio of E-ATP (Molecular Probes Inc.) and E-AMP (Sigma-Aldrich) was measured with a 0–50% methanol/H2O gradient mobile phase (2 ml/min over 10 min). Absorbance was measured at 260 nm, and ultraviolet absorption spectra were obtained at chromatographic peaks. CD39 activity was expressed as percent E-ATP conversion in this time frame.
CD39 Suppression with RNA Interference.
HMEC-1 were either grown on inserts or in 100 mm Petri dishes. SiRNA directed against human CD39 (sense strand 5′-GAA UAU CCU AGC CAU CCU UdTdT-3′ and antisense strand 5′-dTdT CUU AUA GGA UCG GUA GGA A-3′) were designed using standard molecular tools and synthesized by Dharmacon®. A nonspecific control ribonucleotide (sense strand 5′-ACU CUA UCU GCA CGC UGA CdTdT-3′ and antisense strand 5′-dTdT UGA GAU AGA CGU GCG ACU G-3′, Dharmacon®) was used under identical conditions. HMEC-1 loading was accomplished using standard conditions of Polyfect (QIAGEN) transfection (450 ng/monolayer cm2) when cells had reached 40–60% confluence. After 12 h loading, cells were incubated in either normoxia or hypoxia, as indicated.
In Vivo Hypoxia Model.
Mice deficient in cd39 on the C57BL/6/129 svj strain were generated, validated, and characterized as described previously (21). Control mice were matched according to sex, age, and weight. Total organ vascular permeability was quantified by intravascular administration of Evans blue as described previously (22). For the purpose of quantifying vascular permeability, 0.2 c.c. of Evans blue (0.5% in PBS) were injected intravenously. Animals were then exposed to normobaric hypoxia (8% O2, 92% N2) or room air for 4 h (n = 4 animals per condition). After hypoxia/normoxia exposure the animals were killed and the colon, muscle, kidney, brain, liver, and lungs were harvested. Organ Evans blue concentrations were quantified after formamide extraction (55°C for 2 h) by measuring absorbances at 610 nm with subtraction of reference absorbance at 450 nm. This protocol was in accordance with NIH guidelines for use of live animals and was approved by the Institutional Animal Care and Use Committee at Brigham and Women's Hospital.
Data Analysis.
CD39 and CD73 bioactivity and paracellular permeability data were compared by two-factor ANOVA, or by Student's t test where appropriate. Values are expressed as the mean ± SD from at least three separate experiments.
Results
Enhanced Barrier Protection to Activated PMN in Posthypoxic Endothelial Cells.
We have previously demonstrated that during the process of transmigration, activated PMNs release soluble mediators which “reseal” endothelial monolayers after transit (11, 12, 23). It was further shown that leukocyte derived AMP is at least one leukocyte-derived factor responsible for this increase in barrier function (measured as a decrease in paracellular permeability; reference 11). Given the association of hypoxia and acute inflammation (1, 24) we questioned whether posthypoxic endothelia might differentially respond to activated PMN. As such, we compared the influence of activated PMN on permeability of normoxic and posthypoxic (pO2 20 torr for 48 h) HMEC-1. Consistent with previous work (11), increasing numbers of activated PMN elicited a concentration-dependent increase in endothelial barrier (measured as a decrease in paracellular flux rate of 70 kD FITC-labeled dextran; Fig. 1 A, P < 0.025 compared with no PMN). As shown in Fig. 1 A, parallel analysis of posthypoxic HMEC-1 revealed a significant increase in endothelial barrier protection to activated PMN, relative to normoxia (P < 0.01 by ANOVA). Indeed, fewer PMN were required to elicit an equivalent increase in posthypoxic endothelial barrier, with an approximate 10-fold increase in endothelial sensitivity to PMN after hypoxia. Similar results were obtained using nonimmortalized BAE as an endothelial source (unpublished data). Such results suggest that the posthypoxic endothelium is functionally distinct in this regard and that barrier protective pathways are enhanced after exposure to hypoxia.
Figure 1.
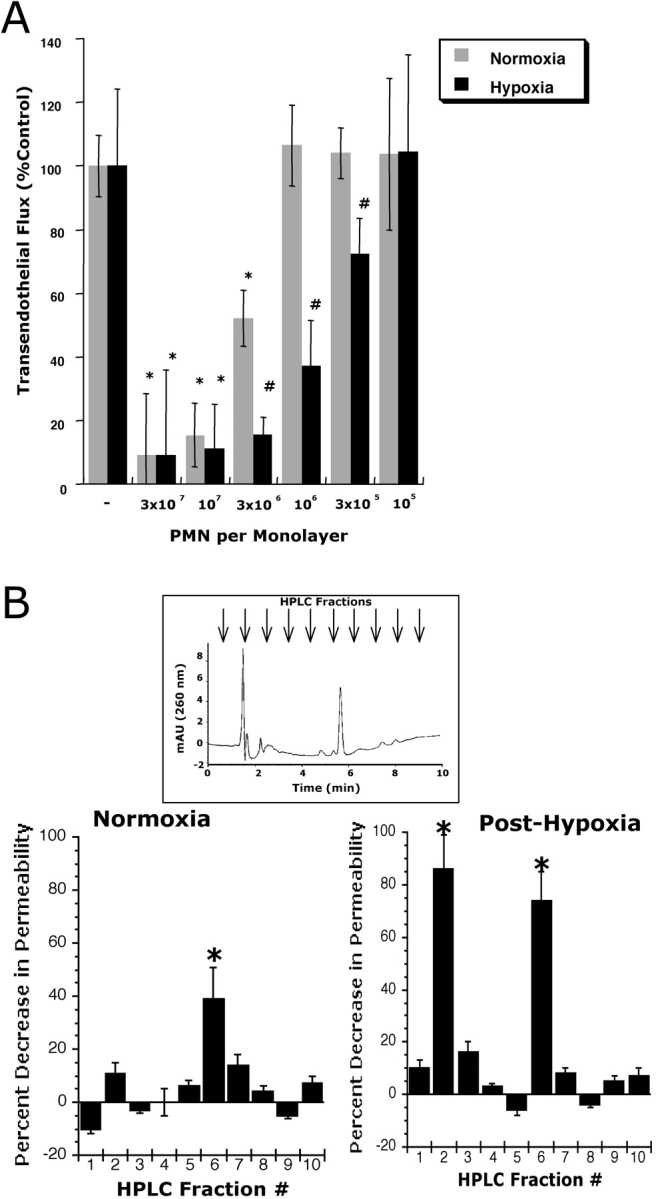
Influence of PMN on permeability of normoxic and posthypoxic endothelia. (A) Activated PMN (10−6 M FMLP) at indicated concentrations were added to the apical surface of confluent normoxic (48 h exposure to pO2 147) or posthypoxic (48 h exposure to pO2 20 torr) HMEC-1 and permeability to FITC-dextran (70 kD) was quantified. Transendothelial flux was calculated by linear regression (4 samples over 60 min) and normalized as percent of control (HBSS). Data are derived from six monolayers in each condition. Data are expressed as mean ± SD of percent control flux with HBSS only. Asterisk (*) indicates significant differences from baseline (P < 0.05) and a cross (#) indicates differences from baseline and from normoxia (P < 0.05). (B) Supernatants derived from activated PMN (10−6 M FMLP for 10 min) were fractionated by HPLC (H20 mobile phase at 1 ml/min, 1 ml samples; inset indicates representative UV spectra at 260 nm and indicated samples collected at individual arrows). Samples were concentrated 20-fold and tested for bioactivity on normoxic or posthypoxic HMEC-1 in a model of paracellular permeability. Fraction #6 promotes barrier function in normoxic and posthypoxic endothelial cells, whereas fraction #2 is relatively selective for posthypoxic endothelia (*, P < 0.01 compared with HBSS).
Identification of PMN-derived ATP as a Mediator of Permeability Changes in Posthypoxic Endothelial Cells.
We next sought to determine the existence of soluble mediators from activated PMN that might contribute to enhanced barrier protection after hypoxia. To do this, we used HPLC to fractionate supernatants derived from activated PMN (10−6 M for 1 min, based on pilot experiments), and tested bioactivity of individual fractions on normoxic and posthypoxic endothelium. As shown in Fig. 1 B, of 10 fractions that were isolated, fraction 6 increased endothelial barrier function in both posthypoxic and normoxic endothelial cells. In contrast, fraction 2 was associated with an increase in barrier function only in posthypoxic endothelial cells. Parallel spectral scans from eluted samples revealed dominant, single UV peaks at 260 nm for fractions 2 and 6 (see inset Fig. 2 A), a spectral profile consistent with nucleotides (25).
Figure 2.

Activated PMN rapidly release ATP. (A) Chromatographic identification of ATP in supernatants derived from activated PMN. Shown here is a representative overlay chromatogram of activated PMN supernatant (bold line) and authentic ATP (narrow line). Inset represents an overlay UV spectra derived from fraction #2 (see Fig. 1) and of authentic ATP with one dominant peak at 260 nm. (B) Time course of ATP release from activated PMN (107 PMN/ml activated with 10−6 M FMLP). PMN were activated for indicated periods of time and supernatant ATP was quantified by luciferase assay. Data represent mean ± SD ATP/107 PMN for three separate experiments. (C) Authentic ATP selectively promotes endothelial barrier function in posthypoxic endothelia. Indicated concentrations of ATP were added to HMEC-1 preexposed to normoxia or hypoxia. As indicated, ATP influenced endothelial permeability only in posthypoxic endothelial cells (*, P < 0.025 compared with no ATP). In contrast, the addition of adenosine (Ado, 100 μM) was associated with an increase in barrier function in both normoxic and posthypoxic endothelial cells (#, P < 0.01 compared with buffer alone). The addition of 8-phenyl-theophylline (8-PT at 10 μM, a nonselective adenosine receptor antagonist) obviated the barrier effect of 100 μM ATP in posthypoxic endothelial cells. Data are derived from six monolayers in each condition. Data are expressed as mean ± SD of percent control flux with HBSS only.
We had previously shown that PMN-derived 5′-adenosinemonophosphate (AMP) promotes barrier function in endothelial permeability assays (11). We therefore compared retention times of authentic 5-AMP with the retention times of the HPLC fractions that were tested in this experiment. The retention time of AMP was identical to that of fraction 6 and coinjection with authentic AMP revealed a single peak (unpublished data), thus identifying fraction 6 as AMP. Further analysis of comparative nucleotides identified fraction 2 as ATP (based on retention time, UV absorption spectra, and coelution with internal ATP standards). Indeed, UV spectra analysis demonstrated similar absorption characteristics of fraction 2 and authentic ATP (Fig. 2 A). To verify ATP release from activated PMN, we used standard luminometric ATP detection assays. As shown in Fig. 2 B, ATP rapidly accumulated in supernatants derived from fMLP-activated PMN (maximal levels 215 ± 16 nmoles/107 PMN). Measurable levels of ATP rapidly dissipated and approached basal levels within 5 min. These results indicate that activated PMN rapidly release ATP, and that such extracellular ATP preferentially promotes barrier function in posthypoxic endothelial cells.
ATP Promotes Barrier Function Selectively in Posthypoxic Endothelial Cells.
Having demonstrated that activated PMN release ATP, we next examined the influence of ATP on normoxic and posthypoxic endothelial paracellular permeability. As shown in Fig. 2 C, the addition of ATP (range 1–100 μM) did not influence normoxic endothelial permeability. In striking contrast, following a 48 h exposure to hypoxia, endothelial flux rates were significantly decreased in an ATP concentration-dependent fashion (P < 0.01 by ANOVA), and were similar to responses observed with 100 μM adenosine (Fig. 2 C). Similar results were obtained using posthypoxic, nonimmortalized BAE as an endothelial source (60 ± 8% decrease in permeability with 100 μM ATP, P < 0.01, n = 3). Such findings suggest that ATP selectively promotes endothelial barrier of the posthypoxic endothelium.
To gain insight into potential mechanisms of ATP regulation of posthypoxic endothelial permeability, we reasoned that ATP may directly activate surface ATP receptors, or alternatively, may be metabolized to adenosine at the endothelial surface. Thus, we determined the relative influence of adenosine in this response by using the nonspecific adenosine receptor antagonist 8-phenyltheophylline (8-PT). As shown in Fig. 2 C, the permeability changes associated with the addition of authentic ATP (100 μM) on posthypoxic endothelial cells was neutralized by 8PT (10 μM, P = not significant compared with buffer alone), indicating that the barrier protective influence observed with ATP reflects adenosine receptor activation.
Prominent Role for CD39 in ATP-modulated Permeability of Posthypoxic Endothelial Cells.
Metabolism of extracellular ATP is predominantly determined by CD39, an ecto-apyrase which metabolizes ATP/ADP to 5′-AMP (26, 27). Our previous work indicated that the ecto-nucleotidases CD39 and CD73 (metabolizes extracellular AMP to adenosine) are readily induced in epithelia by hypoxia (18), thus providing the possibility that extracellular metabolism of ATP to adenosine is enhanced in the posthypoxic endothelium. We therefore pursued the hypothesis that relative levels of CD39 and CD73 are coordinately induced by hypoxia. First, we examined the relative impact of hypoxia on HMEC-1 CD39. Semiquantitative RT-PCR analysis (comparison of CD39 and control β-actin with increasing PCR cycle numbers) was employed to determine the general features of this response. As shown in Fig. 3 A this assessment revealed prominent induction of CD39 mRNA expression after 18 and 24 h of exposure to hypoxia (13 ± 2-fold increase of integrated band density in cells exposed to hypoxia compared with those exposed to normoxia, P < 0.01), suggesting that CD39 is induced by hypoxia. Moreover, comparison of HMEC-1 mRNA levels by real-time PCR revealed that CD39 (Fig. 3 A) is prominently induced by hypoxia. Similar results of CD39 inducibility were found when human saphenous vein tissues were subjected to hypoxia, indicating that such findings are not limited to cultured endothelium. Real time PCR analysis of RNA preparations of saphenous vein revealed induction of CD39 as early as 2 h of ex vivo exposure to hypoxia (Fig. 3 C, P < 0.025 by ANOVA).
Figure 3.

Induction of functional CD39 is by hypoxia. (A) Confluent HMEC-1 monolayers were exposed to normoxia (pO2 147 torr, 18 h) or hypoxia (pO2 20 torr, 18 h). Total RNA was isolated, and CD39 mRNA levels were determined by RT-PCR using semiquantitative analysis (increasing cycle numbers, as indicated). As shown, β-actin transcript was determined in parallel and used as a control. (B) Real-time PCR was employed to confirm hypoxia inducibility of CD39 in cultured endothelial cells (HMEC-1). Data were calculated relative to internal housekeeping gene (β-actin) and are expressed as fold increase over normoxia ± SD at each indicated time. Results are derived from three experiments in each condition. (C) Human saphenous vein was obtained from patients undergoing aorto-coronary bypass surgery and exposed ex vivo to ambient normoxia (pO2 147 torr, 24 h) or hypoxia (pO2 20 torr for 2, 8, or 24 h). After total RNA isolation, real-time PCR was performed to investigate CD39 inducibility by hypoxia. Data were calculated relative to internal control (β-actin) and are expressed as fold increase over normoxia ± SD at each indicated time. Results are derived from three experiments in each condition. (D) Increase in surface CD39 surface protein with hypoxic exposure. Confluent HMEC-1 monolayers were exposed to indicated periods of hypoxia, monolayers were washed, surface proteins were biotinylated, and cells were lysed. CD39 was immunoprecipitated with mAb directed against human CD39. Immunoprecipitates were resolved by SDS-PAGE, and resultant Western blots were probed with avidin-peroxidase. A representative experiment of three is shown. (E) Validation etheno-ATP (E-ATP) and etheno-AMP (E-AMP) resolution by HPLC. Shown is a representative tracing indicating resolution of definitive peaks at UV 260 nm. (F) Functional increase in CD39 surface activity by hypoxia. Endothelial monolayers were exposed to 48 h hypoxia or normoxia, washed, and surface CD39 activity was determined by HPLC analysis of E-ATP conversion to E-AMP in the presence of the CD73-inhibitor αβ-methylene-ADP (10 μM, to prevent further metabolism of E-AMP to E-adenosine). Data are derived from five to seven monolayers in each condition, and results are expressed as E-AMP: E-ATP ratio ± SD.
We extended these findings to determine if CD39 is functionally relevant. Initially, we determined whether CD39 protein was induced by hypoxia. As shown in Fig. 3 D, immunoprecipitation of CD39 from total surface biotinylated protein revealed time-dependent induction of CD39 (range 12–48 h), with maximal protein levels observed at 48 h (Fig. 3 D; no additional increase at 72 h; data not shown). We then assessed whether hypoxia induced CD39 was functional. To do this, we adopted an HPLC method to compare the relative amounts of E-ATP metabolism to E-AMP (in the presence of the CD73 inhibitor α,β-methylene-ADP [10 μM] to inhibit further metabolism of E-AMP to E-adenosine, see method validation in Fig. 3 E). As shown in Fig. 3 F, exposure of HMEC-1 to hypoxia (48 h) induced an increase in functional CD39 (ANOVA, P < 0.01 relative to normoxia), and represented a 22 ± 5-fold increase in the rate of E-ATP metabolism with preexposure to hypoxia. Together, these data suggest a transcriptional induction of CD39 by hypoxia, which manifests as readily measurable increases in surface enzyme activity.
CD39 Hypoxia Induction Is Necessary for ATP Elicited Increases of Barrier Function in Posthypoxic Endothelial Cells.
After having shown that functional CD39 is induced by hypoxia, we examined if CD39 induction by hypoxia is necessary for the observed increase of barrier function in posthypoxic endothelial cells upon ATP exposure. As no specific CD39 inhibitors have come available (28), we used RNA interference to suppress CD39 induction by hypoxia and studied the functional influence on permeability. Thus, we loaded HMEC-1 with specific CD39-siRNA, and exposed the cells to hypoxia. Immunoprecipitation of biotinylated surface CD39-protein showed an induction of CD39 in untreated controls and in HMEC-1 loaded with control oligonucleotide (Fig. 4 A). In contrast, normoxic and posthypoxic CD39 levels were decreased after loading with specific CD39-siRNA. Moreover, CD73 induction by hypoxia was not affected by CD39 siRNA loading, and as such, provided a specificity control for these experiments.
Figure 4.
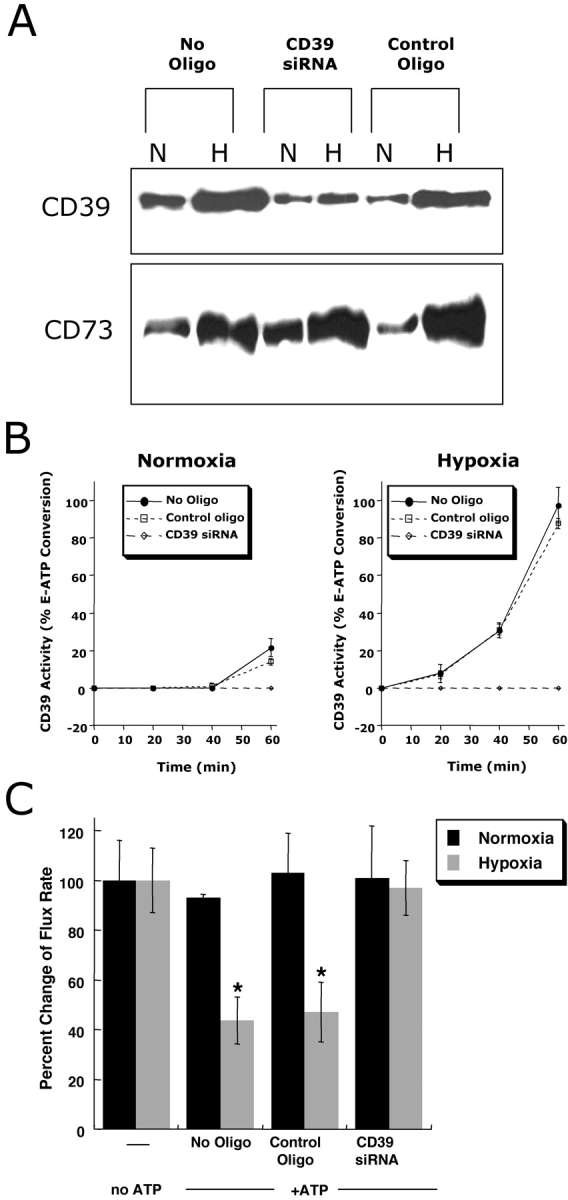
Role of CD39 in ATP-elicited changes of posthypoxic endothelial permeability. (A) HMEC-1 were loaded with CD39-specific siRNA, control ribonucleotide or mock treated (control) and exposed to hypoxia or normoxia (48 h). Monolayers were washed, surface protein was biotinylated, and cells were lysed. CD39 was immunoprecipitated and resolved by SDS-PAGE, and resultant Western blots were probed with avidin-peroxidase. As a control for specificity, CD73 protein induction by hypoxia was assessed in parallel. (B) Influence of CD39 suppression by siRNA on functional surface protein. HMEC-1 were loaded with CD39-specific siRNA, control-ribonucleotide, or mock treated and CD39 activity was determined by HPLC analysis of E-ATP conversion to E-AMP (in the presence of the CD73-inhibitor αβ-methylene-ADP). Data are derived from five to seven monolayers in each condition, and results are expressed as E-AMP: E-ATP ratio ± SD. (C) Influence of CD39 suppression by siRNA on endothelial barrier. HMEC-1 were loaded with CD39-specific siRNA, control ribonucleotide or mock treated (control), exposed to hypoxia or normoxia (48 h), and permeability to 70 kD FITC in the presence or absence of ATP (100 μM) was assessed (*, P < 0.01 compared with no ATP). Data are derived from six monolayers in each condition, and data are expressed as mean ± SD of percent control flux.
As additional insight, we determined whether siRNA directed against CD39 was functional. First, we measured enzymatic conversion of E-ATP to E-AMP in siRNA-mediated CD39 deficient cells. In controls and in HMEC-1 loaded with control oligonucleotide, a 48 h exposure to hypoxia was associated with a significant increase in CD39 functional activity (ANOVA, P < 0.01; Fig. 4 B). In contrast, loading with specific CD39-siRNA obviated a functional induction of CD39 enzyme activity, indicating that such RNA suppression was effective at the functional protein level. To determine the relative role of CD39 in ATP-mediated changes in endothelial barrier function, we investigated the influence of ATP on barrier function in normoxic and posthypoxic endothelial cells following inhibition of CD39 with specific siRNA. In controls and in HMEC-1 loaded with a control oligonucleotide, ATP elicited an approximate 60% decrease in permeability in posthypoxic endothelial cells, but not in normoxic cells (Fig. 4 C). In contrast, no significant ATP-induced change in barrier was observed in either normoxic or posthypoxic endothelial cells after suppression of CD39 with siRNA (P < 0.01). These results indicate that selective targeting of CD39 by siRNA effectively neutralizes the influence of ATP on changes in endothelial barrier.
Inhibition of CD73 Blocks ATP-mediated Changes in Posthypoxic Endothelial Permeability.
We next addressed the role of CD73 in ATP-mediated changes in permeability. Our previous studies indicated that CD73 is important for the maintenance of barrier function during interactions with PMN or soluble PMN mediators (e.g., AMP; reference 23). Similarly, it was previously shown that AMP enhances epithelial barrier recovery via activation of surface AdoRA2B receptors (11). As confirmation of our previous findings in epithelia (18), we observed a time-dependent induction of CD73 by hypoxia in endothelia (see Fig. 5 A). To determine the functional role of CD73 in this response, we addressed whether the specific CD73 inhibitor α,β-methylene ADP might block ATP-induced changes in the posthypoxic endothelium. As shown in Fig. 5 B, the addition of increasing concentrations of α,β-methylene ADP in combination with ATP (100 μM) resulted in a concentration-dependent inhibition of ATP-mediated changes in permeability selectively in posthypoxic endothelia (P < 0.01). These findings suggest that the increment of CD73 induced by hypoxia is relevant to endothelial functional responses (permeability).
Figure 5.
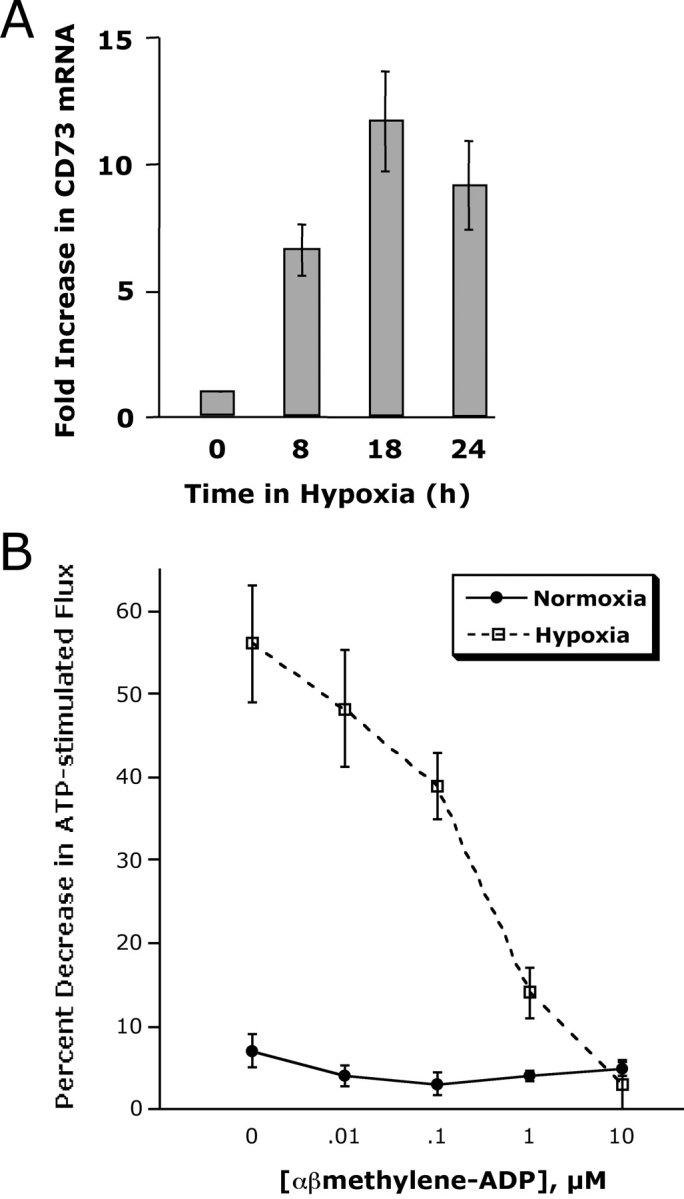
Induction of CD73 by hypoxia is necessary for ATP-elicited increases in posthypoxic endothelial barrier function. (A) Induction of endothelial CD73 by hypoxia. Real-time PCR was employed to confirm hypoxia inducibility of CD73 in cultured endothelial cells (HMEC-1). Data were calculated relative to internal control genes (β-actin) and are expressed as fold increase over normoxia ± SD at each indicated time. Results are derived from three experiments in each condition. (B) Influence of CD73-inhibitor αβ-methylene-ADP on ATP-elicited changes of endothelial permeability. Indicated concentrations of αβ-methylene-ADP were added to HMEC-1 monolayers that were cultured under normoxic or hypoxic conditions (48 h) and stimulated with 100 μM ATP. In normoxic HMEC-1, neither the addition of ATP, nor αβ-methylene-ADP were associated with changes in paracellular flux. In contrast, ATP-elicited changes in endothelial flux in posthypoxic HMEC-1 were obviated in a concentration-dependent fashion (P < 0.01 by ANOVA). Data are derived from six monolayers in each condition. Data are expressed as mean ± SD of percent control flux with HBSS only.
Hypoxia Induces AdoRA2B: Role in Coordinated Vascular Permeability Regulation.
Having demonstrated a functional role for hypoxia-regulated CD39 and CD73 in endothelial permeability, we next addressed how endpoint signaling (i.e., adenosine receptor activation) is amplified in posthypoxic endothelia. First, we profiled the relative expression of adenosine receptors in normoxic and hypoxic (12 h exposure to pO2 20 torr) endothelial cells by microarray analysis. For these experiments, nonimmortalized HMVECs were used. Interestingly, these experiments demonstrated that the AdoRA2B was selectively induced by hypoxia (see Fig. 6 A, P < 0.05 by ANOVA), and that other isoforms were either not changed (AdoRA3) or significantly down-regulated (AdoRA1 and AdoRA2A). As shown in Fig. 6 B, these microarray results were verified in HMEC-1 by real-time PCR in RNA derived from endothelial cells exposed to a time course of hypoxia, and consistently revealed that the AdoRA3 isoform remained unchanged, both AdoRA1 and AdoRA2A levels were decreased at later time points of hypoxia (18 and 24 h, P < 0.025), and that the AdoRA2B was increased by as much as 4 ± 05-fold (P < 0.01 compared with normoxia). Identical results were obtained with HMVEC as an endothelial source (unpublished data). Extensions of these findings revealed that surface protein levels of AdoRA2B were similarly increased by exposure of HMEC-1 to hypoxia (Fig. 6 C).
Figure 6.
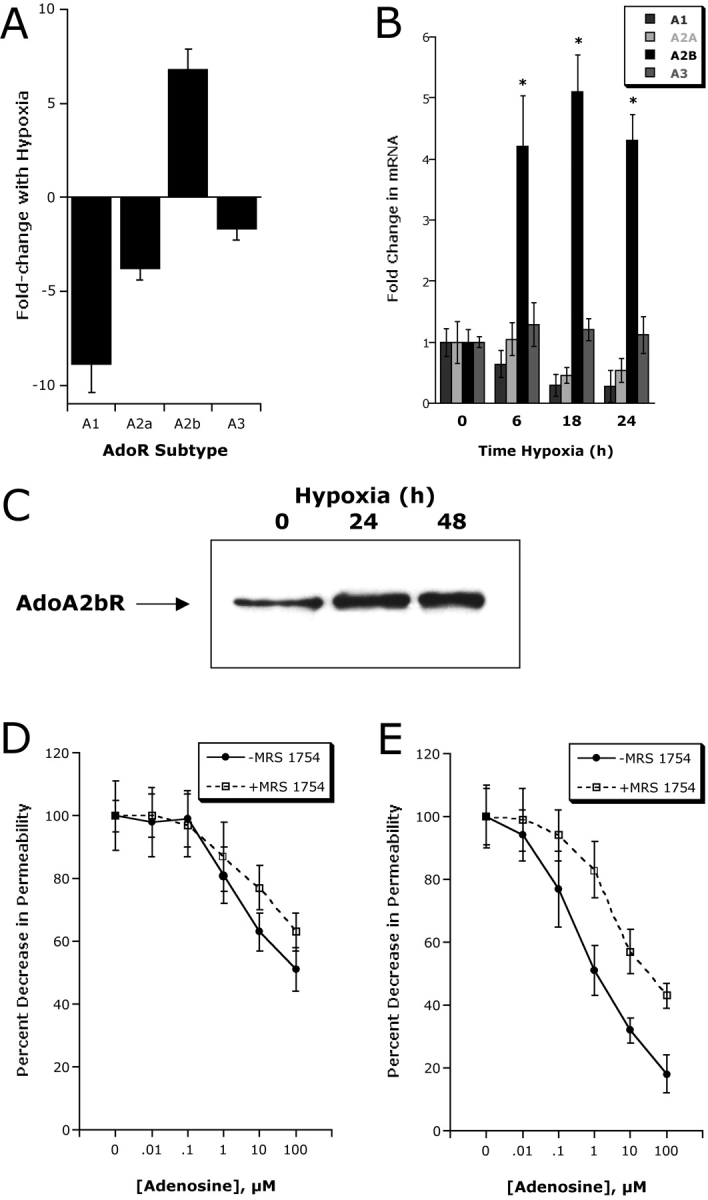
Adenosine A2B receptor (AdoRA2B) induction by hypoxia enhances barrier response. (A) Microarray analysis of individual adenosine receptors in response to hypoxia (AdoRA1, AdoRA2A, AdoRA2B, and AdoRA3). Confluent HMEC-1 were exposed to normoxia or hypoxia (12 h exposure to pO2 20 torr) and the relative expression of individual adenosine receptors was quantified from total RNA by microarray analysis. Data are expressed as fold change ± SD relative to normoxia. (B) Real-time PCR analysis was employed to confirm hypoxia-regulated expression of individual adenosine receptors (AdoRA1, AdoRA2A, AdoRA2B, and AdoRA3) in HMEC-1. Data were calculated relative to internal control (β-actin) and are expressed as fold increase ± SD over normoxia at indicated time points. Results are derived from three experiments in each condition (*, P < 0.01 compared with normoxia). (C) Increased AdoRA2B surface protein with hypoxia. Confluent HMEC-1 monolayers were exposed to indicated periods of hypoxia, monolayers were washed, surface proteins were biotinylated, and cells were lysed. AdoRA2B was immunoprecipitated with mAb to human AdoRA2B and resolved by SDS-PAGE, and resultant Western blots were probed with avidin-peroxidase. A representative experiment of three is shown. (D) Hypoxia induction of AdoRA2B enhances barrier response of HMEC-1 to adenosine. Indicated concentrations of adenosine were added to HMEC-1 monolayers preexposed to normoxia or hypoxia (48 h). Addition of the specific AdoRA2B-antagonist MRS 1754 (100 nM) significantly shifted the adenosine dose response in posthypoxic endothelium (P < 0.01 by ANOVA). Data are derived from 6 monolayers in each condition. Data are expressed as mean ± SD of percent control flux.
To determine whether such hypoxia-induced AdoRA2B expression was functional, endothelial permeability assays were employed using the selective AdoRA2B antagonist MRS1754 (29). As can be seen in Fig. 6 D, MRS 1754 (100 nM) significantly shifted the adenosine dose–response curve to the right, but had little influence on adenosine responses in normoxic endothelia. Such results suggest a relative importance for the expression level of AdoRA2B and that AdoRA2B responses are amplified in posthypoxic endothelia.
Increased Vascular Permeability in cd39-null Animals: Influence of Hypoxia.
As proof of principle for these concepts in vivo, we compared the influence of hypoxia on vascular permeability in wild-type and cd39-null mice using a previously described model (22). After intravenous injection of Evans-blue, we exposed animals to either normoxia or normobaric hypoxia (8% O2 and 92% N2) for 4 h and measured Evans-blue tissue concentrations as a marker of endothelial permeability to albumin. Interestingly, assessment of permeability in nonhypoxic cd39-null animals revealed subtle, but significant increases in all tissues examined, with the exception of skeletal muscle (Fig. 7 A). Consistent with previous studies (3, 30), this model of hypoxia significantly increased permeability in all wild-type control organs examined (Fig. 7 A, P < 0.025 for all). Most importantly, and as shown in Fig. 7 A, this analysis revealed amplified increases in vascular permeability of all organs of cd39-null animals. Gross morphologic changes in overall vascular permeability (Evans blue retention) between hypoxia and normoxia as well as wild-type and cd39-null were evident in open abdominal images taken at necropsy (Fig. 7 B) and were demonstrable in dissected organs such as the colon (Fig. 7 C). These findings in vivo support our hypothesis that CD39 is a critical determinant for permeability changes associated with adenine nucleotide metabolism in the posthypoxic vasculature.
Figure 7.


Role of hypoxia-induced CD39 in vivo: cd39-deficient mice and age, weight, and gender matched controls were administered intravenous Evans blue solution (0.2 ml of 0.5% in PBS) and exposed to normobaric hypoxia (8% O2, 92% N2) or room air for 4 h. Animals were killed and the colon, muscle, kidney, brain, liver, and lungs were harvested. Organ Evans blue concentrations were quantified following formamide extraction (55°C for 2 h) by measuring absorbances at 610 nm with subtraction of reference absorbance at 450 nm. cd39-deficient mice showed higher tissue Evans-blue concentrations in all organs (except muscle) in normoxic and hypoxic conditions as compared with wild-type animals (*, P < 0.05). Hypoxic wild-type and cd39-deficient mice showed higher tissue Evans blue concentrations than normoxic mice, with the exception of hypoxic colon of the cd39-deficient mice (#, P < 0.05). Data are expressed as mean ± SD Evans blue OD/mg wet tissue, and are pooled from four to six animals per condition. Images of abdominal dissections (B) and isolated colons (C) from wild-type and cd39-deficient mice subjected to normoxia and hypoxia. Note increased Evans blue retention with hypoxia and marked differences between wild-type and cd39-deficient animals under both conditions.
Discussion
The vascular endothelium is the predominant interface between the hypoxic insult and surrounding tissues. Simultaneously, the endothelium provides the primary determinant of vascular permeability (31). As such, PMN influx across the protective endothelium, such as occurs during ischemia-reperfusion, creates the potential for endothelial barrier dysfunction, loss of fluid and edema formation (7, 32). In this study we compared the direct influence of activated PMN on paracellular permeability in normoxic and posthypoxic endothelial cells. The results revealed the existence of PMN-derived mediator(s), which selectively promote posthypoxic endothelia. This mediator was identified as ATP based on its chemical characteristics and chromatographic behavior. Further studies demonstrated that hypoxia promotes the induction of CD39, an endothelial surface apyrase responsible for the initiation of ATP phosphohydrolysis to adenosine (33). We show here that increased activity of endothelial CD39, as seen after hypoxic exposure, is necessary for the barrier protective influence of ATP. Similarly, these studies revealed that additional metabolic and signaling molecules (CD73 and AdoRA2B, respectively) are coordinately induced by hypoxia (Fig. 8) . In vivo relevance for this response revealed that cd39-null mice are more susceptible to the development of endothelial barrier dysfunction during hypoxia exposure. Taken together, these studies identify a pathway which results in both elevated tissue levels of adenosine and in amplified endothelial responses to adenosine generated at sites of hypoxia.
Figure 8.

Proposed model of coordinated nucleotide metabolism and nucleoside signaling in posthypoxic endothelial cells: in areas of ongoing inflammation, diminished oxygen supply coordinates the induction of CD39, CD73, and AdoRA2B. At such sites, activated PMN provide a readily available extracellular source of ATP that through two enzymatic steps results in the liberation of extracellular adenosine. Adenosine generated in this fashion is available for activation of surface endothelial adenosine receptors, particularly the AdoRA2B. Postreceptor increases in intracellular cyclic AMP results in enhanced barrier function. As such, this protective mechanism may provide an innate mechanism to preserve vascular integrity and prevent fulminant intravascular fluid loss.
It is well documented that adenosine tissue and plasma levels are increased during hypoxia, however, mechanisms of this response are less clear (34, 35). For example, in human volunteers exposed to ambient hypoxia (SpO2 = 80% over 20 min), plasma adenosine concentrations increased from 21 to 51 nM in the presence of dipyridamole, an inhibitor of adenosine reuptake (36). Similarly, when measuring adenine nucleotide concentrations in the neurally and vascularly isolated, perfused skeletal muscles of anesthetized dogs, normobaric hypoxia is associated with increases of adenosine in the venous blood, but not of AMP, ADP, or ATP (37). In conjunction with these studies, we describe here a metabolic pathway, which results in increased tissue adenosine concentrations. While it is well documented that adenosine levels are increased in hypoxic tissue, the source of adenosine remains unclear. We show here, for the first time, that PMN actively release ATP to the extracellular milieu. At present, we do not know the origin of ATP release. Several mechanisms for ATP release have been proposed, including direct transport through ATP-binding cassette (ABC) proteins, transport through connexin hemichannels, as well as vesicular release (38). As part of the present experiments, some studies were done in an attempt to identify the compartmentalization of ATP (unpublished data). For example, we compared the kinetics of ATP release with activated release of PMN granules (myeloperoxidase, MPO). These studies indicated that while ATP levels were maximal within 1 min, activated degranulation contents such as MPO were maximal at time points >5 min, suggesting that ATP is not granule bound. Moreover, in experiments using isolated granules from unactivated PMN, >95% of MPO activity was associated with granules, <5% of ATP was measurable within this granule pool, suggesting that ATP is unlikely to be granule-bound in PMN. At present, the mechanism by which leukocytes release ATP remains unclear.
As part of these studies, we found that targeted disruption of CD39 in an animal model of ambient hypoxia was associated with barrier dysfunction at baseline and after hypoxic exposure. Such increases in permeability in the cd39-null mice may be related to decreased concentrations of adenosine and activation of endothelial adenosine receptors. In addition, activation of neutrophil A2 adenosine receptors has been shown to play a critical part for the limitation and termination of PMN-mediated systemic inflammatory responses (39, 40). Thus, it is reasonable to propose that the presence of CD39/CD73 is important in modulating inflammatory responses during hypoxia. Coordinated induction of both CD39 and CD73 by hypoxia may provide increased tissue adenosine concentrations, thus leading to increased stimulation of PMN adenosine A2-receptors and decreased leukocyte adherence and transmigration, as has been demonstrated by others (41, 42). Moreover, recent studies have indicated that adenosine A2A-receptors may contribute to attenuated vascular leak associated with leukocyte accumulation (43). As such, direct influences of adenosine via hypoxia-amplified CD39 and CD73 may provide antiinflammatory synergism with the observed protection of endothelial barrier during hypoxia.
As outlined above, leukocyte transmigration associated with hypoxic/ischemic tissues creates the potential for disturbance of the endothelial barrier. We have previously shown that adenosine activation of AdoRA2B leads to a barrier resealing response after PMN transmigration (11). Activation of the AdoRA2B is associated with increases in intracellular cAMP concentration following activation of the adenylate cyclase (44). By inhibition of cAMP formation, the resealing of the endothelial barrier during PMN transmigration can be obviated (11). Such increases in cAMP after activation of the AdoRA2B lead to an activation of protein kinase A (PKA; reference 12). Further studies revealed a central role of PKA-induced phosphorylation of vasodilator-stimulated phosphoprotein (VASP), a protein responsible for controlling the geometry of actin-filaments (45). Adenosine-receptor mediated phosphorylation of VASP is responsible for changes in the geometry and distribution of junctional proteins, thereby affecting the characteristics of the junctional complex and promoting increases in barrier function (12).
We show here that the barrier-protective response to extracellular ATP is most prevalent in posthypoxic endothelial cells. After suppression of CD39 induction with specific siRNA, this barrier-protective influence to ATP is obviated, providing insight that this molecule may efficiently coordinate permeability responses to PMN during inflammation and hypoxia. In parallel, we showed that the ecto-nucleotidase CD73 is similarly induced by hypoxia. These findings in endothelia coincide with recent work in epithelia identifying a predominant role for hypoxia-inducible factor-1 (HIF-1) in CD73 induction by hypoxia (18) and recently in enothelial cells (46). Currently, the mechanism for the hypoxia-inducibility of AdoRA2B and CD39 remains unclear. Relatively little is known about the transcriptional regulation of CD39 and AdoRA2B, and while HIF-1 is currently an area of intense investigation in a variety of disorders (47), including inflammation (5, 17, 18), it remains to be seen whether the coordinated transcription of CD39, CD73, and AdoRA2B have common regulatory elements.
Some precedent exists for coordinated, metabolic responses to hypoxia. For example, in the regulation of oxygen hemostasis, HIF-1 has been shown to regulate multiple metabolic and compartmental steps in several metabolic pathways (47). As such, at least 13 different genes encoding glucose transporters and glycolytic enzymes are coordinately decreased in HIF-1 deficiency (47). Similarly in the present study, transcriptional and metabolic control-points of extracellular nucleotide metabolism and nucleoside signaling are controlled in a coordinated fashion. Interestingly, recent evidence indicates that the AdoRA2A and CD73 are coordinately regulated by mitogenic stimuli in human B cells, and that such a pathway drives increased signal transduction (48). The present results of a coordinated response to decreased oxygen availability of CD39, CD73, and the AdoRA2B suggest a similar evolutionary adaption to hypoxia.
In summary, these results define a previously unappreciated metabolic pathway prevalent at the vascular interface during inflammation/hypoxia. We show here that PMN-derived ATP selectively promotes barrier function of posthypoxic endothelia. The observed differences were attributable to a coordinated transcriptional response initiated by hypoxia, and resulting in amplified metabolism of extracellular ATP to adenosine as well as enhanced signaling through the AdoRA2B. We propose that this feed-forward mechanism may contribute an endogenous, protective pathway for highly vascular tissues during adaptation to hypoxia.
Acknowledgments
The authors wish to acknowledge Dr. Sary Aranki and Barry Shopnick for procurement of human tissue.
This work was supported by NIH grants HL60569, DE13499, and DK50189.
Abbreviations used in this paper: AdoRA2B, adenosine A2B-receptor; HIF-1, hypoxia-inducible factor-1.
References
- 1.Tamura, D.Y., E.E. Moore, D.A. Partrick, J.L. Johnson, P.J. Offner, and C.C. Silliman. 2002. Acute hypoxemia in humans enhances the neutrophil inflammatory response. Shock. 17:269–273. [DOI] [PubMed] [Google Scholar]
- 2.Colgan, S.P., A.L. Dzus, and C.A. Parkos. 1996. Epithelial exposure to hypoxia modulates neutrophil transepithelial migration. J. Exp. Med. 184:1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collard, C.D., K.A. Park, M.C. Montalto, S. Alapati, J.A. Buras, G.L. Stahl, and S.P. Colgan. 2002. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J. Biol. Chem. 277:14801–14811. [DOI] [PubMed] [Google Scholar]
- 4.Rui, T., G. Cepinskas, Q. Feng, Y.S. Ho, and P.R. Kvietys. 2001. Cardiac myocytes exposed to anoxia-reoxygenation promote neutrophil transendothelial migration. Am. J. Physiol. Heart Circ. Physiol. 281:H440–H447. [DOI] [PubMed] [Google Scholar]
- 5.Cramer, T., Y. Yamanishi, B.E. Clausen, I. Forster, R. Pawlinski, N. Mackman, V.H. Haase, R. Jaenisch, M. Corr, V. Nizet, et al. 2003. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 112:645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madara, J.L. 1998. Regulation of the movement of solutes across tight junctions. Annu. Rev. Physiol. 60:143–159. [DOI] [PubMed] [Google Scholar]
- 7.Luscinskas, F.W., S. Ma, A. Nusrat, C.A. Parkos, and S.K. Shaw. 2002. The role of endothelial cell lateral junctions during leukocyte trafficking. Immunol. Rev. 186:57–67. [DOI] [PubMed] [Google Scholar]
- 8.Luscinskas, F.W., S. Ma, A. Nusrat, C.A. Parkos, and S.K. Shaw. 2002. Leukocyte transendothelial migration: a junctional affair. Semin. Immunol. 14:105–113. [DOI] [PubMed] [Google Scholar]
- 9.Carpenter, T.C., and K.R. Stenmark. 2001. Hypoxia decreases lung neprilysin expression and increases pulmonary vascular leak. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L941–L948. [DOI] [PubMed] [Google Scholar]
- 10.Schoch, H.J., S. Fischer, and H.H. Marti. 2002. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 125:2549–2557. [DOI] [PubMed] [Google Scholar]
- 11.Lennon, P.F., C.T. Taylor, G.L. Stahl, and S.P. Colgan. 1998. Neutrophil-derived 5′-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J. Exp. Med. 188:1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comerford, K.M., D.W. Lawrence, K. Synnestvedt, B.P. Levi, and S.P. Colgan. 2002. Role of vasodilator-stimulated phosphoprotein in protein kinase A-induced changes in endothelial junctional permeability. FASEB J. 16:583–585. [DOI] [PubMed] [Google Scholar]
- 13.Collard, C.D., K.A. Park, M.C. Montalto, S. Alipati, J.A. Buras, G.L. Stahl, and S.P. Colgan. 2002. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J. Biol. Chem. 277:14801–14811. [DOI] [PubMed] [Google Scholar]
- 14.Robinson, K.A., F.J. Candal, N.A. Scott, and E.W. Ades. 1995. Seeding of vascular grafts with an immortalized human dermal microvascular endothelial cell line. Angiology. 46:107–113. [DOI] [PubMed] [Google Scholar]
- 15.Mizuno-Yagyu, Y., R. Hashida, C. Mineo, S. Ikegami, S. Ohkuma, and T. Takano. 1987. Effect of PGI2 on transcellular transport of fluorescein dextran through an arterial endothelial monolayer. Biochem. Pharmacol. 36:3809–3813. [DOI] [PubMed] [Google Scholar]
- 16.Sanders, S.E., J.L. Madara, D.K. McGuirk, D.S. Gelman, and S.P. Colgan. 1995. Assessment of inflammatory events in epithelial permeability: a rapid screening method using fluorescein dextrans. Epithelial Cell Biol. 4:25–34. [PubMed] [Google Scholar]
- 17.Furuta, G.T., J.R. Turner, C.T. Taylor, R.M. Hershberg, K. Comerford, S. Narravula, D.K. Podolsky, and S.P. Colgan. 2001. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 193:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Synnestvedt, K., G.T. Furuta, K.M. Comerford, N. Louis, J. Karhausen, H.K. Eltzschig, K.R. Hansen, L.F. Thompson, and S.P. Colgan. 2002. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Invest. 110:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higuchi, R., C. Fockler, G. Dollinger, and R. Watson. 1993. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N.Y.). 11:1026–1030. [DOI] [PubMed] [Google Scholar]
- 20.Pfaffl, M.W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:2002–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enjyoji, K., J. Sevigny, Y. Lin, P.S. Frenette, P.D. Christie, J.S. Esch, II, M. Imai, J.M. Edelberg, H. Rayburn, M. Lech, et al. 1999. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat. Med. 5:1010–1017. [DOI] [PubMed] [Google Scholar]
- 22.Barone, G.W., P.C. Farley, J.M. Conerly, T.L. Flanagan, and I.L. Kron. 1989. Morphological and functional techniques for assessing endothelial integrity: the use of Evans blue dye, silver stains, and endothelial derived relaxing factor. J. Card. Surg. 4:140–148. [DOI] [PubMed] [Google Scholar]
- 23.Narravula, S., P.F. Lennon, B.U. Mueller, and S.P. Colgan. 2000. Regulation of endothelial CD73 by adenosine: paracrine pathway for enhanced endothelial barrier function. J. Immunol. 165:5262–5268. [DOI] [PubMed] [Google Scholar]
- 24.Taylor, C.T., and S.P. Colgan. 1999. Therapeutic targets for hypoxia-elicited pathways. Pharm. Res. 16:1498–1505. [DOI] [PubMed] [Google Scholar]
- 25.Madara, J.L., T.W. Patapoff, B. Gillece-Castro, S.P. Colgan, C.A. Parkos, C. Delp, and R.J. Mrsny. 1993. 5′-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J. Clin. Invest. 91:2320–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaczmarek, E., K. Koziak, J. Sevigny, J.B. Siegel, J. Anrather, A.R. Beaun, F.H. Bach, and S.C. Robson. 1996. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J. Biol. Chem. 271:33116–33122. [DOI] [PubMed] [Google Scholar]
- 27.Wang, T.F., and G. Guidotti. 1996. CD39 is an ecto-(Ca2+,Mg2+)-apyrase. J. Biol. Chem. 271:9898–9901. [PubMed] [Google Scholar]
- 28.Imai, M., E. Kaczmarek, K. Koziak, J. Sevigny, C. Goepfert, O. Guckelberger, E. Csizmadia, J. Schulte Am Esch, II, and S.C. Robson. 1999. Suppression of ATP diphosphohydrolase/CD39 in human vascular endothelial cells. Biochemistry. 38:13473–13479. [DOI] [PubMed] [Google Scholar]
- 29.Ji, X., Y.C. Kim, D.G. Ahern, J. Linden, and K.A. Jacobson. 2001. [3H]MRS 1754, a selective antagonist radioligand for A(2B) adenosine receptors. Biochem. Pharmacol. 61:657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman, G.B., C.T. Taylor, C.A. Parkos, and S.P. Colgan. 1998. Epithelial permeability induced by neutrophil transmigration is potentiated by hypoxia: role of intracellular cAMP. J. Cell. Physiol. 176:76–84. [DOI] [PubMed] [Google Scholar]
- 31.Ramirez, C., C. Colton, K. Smith, M. Stemerman, and R. Lees. 1984. Transport of 125I-albumin across normal and deendothelialized rabbit thoracic aorta in vivo. Arteriosclerosis. 4:283–291. [DOI] [PubMed] [Google Scholar]
- 32.Johnson-Leger, C., M. Aurrand-Lions, and B. Imhof. 2000. The parting of the endothelium: miracle, or simply a junctional affair? J. Cell Sci. 113:921–933. [DOI] [PubMed] [Google Scholar]
- 33.Robson, S.C., K. Enjyoji, C. Goepfert, M. Imai, E. Kaczmarek, Y. Lin, J. Sevigny, and M. Warny. 2001. Modulation of extracellular nucleotide-mediated signaling by CD39/nucleoside triphosphate diphosphohydrolase-1. Drug Dev. Res. 53:193–207. [Google Scholar]
- 34.Gnaiger, E. 2001. Bioenergetics at low oxygen: dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir. Physiol. 128:277–297. [DOI] [PubMed] [Google Scholar]
- 35.O'Farrell, P.H. 2001. Conserved responses to oxygen deprivation. J. Clin. Invest. 107:671–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saito, H., M. Nishimura, H. Shinano, H. Makita, I. Tsujino, E. Shibuya, F. Sato, K. Miyamoto, and Y. Kawakami. 1999. Plasma concentration of adenosine during normoxia and moderate hypoxia in humans. Am. J. Respir. Crit. Care Med. 159:1014–1018. [DOI] [PubMed] [Google Scholar]
- 37.Mo, F.M., and H.J. Ballard. 2001. The effect of systemic hypoxia on interstitial and blood adenosine, AMP, ADP and ATP in dog skeletal muscle. J Physiol. 536:593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novak, I. 2003. ATP as a signaling molecule: the exocrine focus. News Physiol. Sci. 18:12–17. [DOI] [PubMed] [Google Scholar]
- 39.Revan, S., M.C. Montesinos, D. Naime, S. Landau, and B.N. Cronstein. 1996. Adenosine A2 receptor occupancy regulates stimulated neutrophil function via activation of a serine/threonine protein phosphatase. J. Biol. Chem. 271:17114–17118. [DOI] [PubMed] [Google Scholar]
- 40.Ohta, A., and M. Sitkovsky. 2001. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 414:916–920. [DOI] [PubMed] [Google Scholar]
- 41.Wakai, A., J.H. Wang, D.C. Winter, J.T. Street, R.G. O'Sullivan, and H.P. Redmond. 2001. Adenosine inhibits neutrophil vascular endothelial growth factor release and transendothelial migration via A2B receptor activation. Shock. 15:297–301. [DOI] [PubMed] [Google Scholar]
- 42.Zhao, Z.Q., H. Sato, M.W. Williams, A.Z. Fernandez, and J. Vinten-Johansen. 1996. Adenosine A2-receptor activation inhibits neutrophil-mediated injury to coronary endothelium. Am. J. Physiol. 271:H1456–H1464. [DOI] [PubMed] [Google Scholar]
- 43.Montesinos, M.C., A. Desai, J.F. Chen, H. Yee, M.A. Schwarzschild, J.S. Fink, and B.N. Cronstein. 2002. Adenosine promotes wound healing and mediates angiogenesis in response to tissue injury via occupancy of A(2A) receptors. Am. J. Pathol. 160:2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Linden, J. 2001. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 41:775–787. [DOI] [PubMed] [Google Scholar]
- 45.Bear, J.E., T.M. Svitkina, M. Krause, D.A. Schafer, J.J. Loureiro, G.A. Strasser, I.V. Maly, O.Y. Chaga, J.A. Cooper, G.G. Borisy, and F.B. Gertler. 2002. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 109:509–521. [DOI] [PubMed] [Google Scholar]
- 46.Ledoux, S., I. Runembert, K. Koumanov, J.B. Michel, G. Trugnan, and G. Friedlander. 2003. Hypoxia enhances ecto-5′-nucleotidase activity and cell surface expression in endothelial cells: role of membrane lipids. Circ. Res. 92:848–855. [DOI] [PubMed] [Google Scholar]
- 47.Semenza, G.L. 1999. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15:551–578. [DOI] [PubMed] [Google Scholar]
- 48.Napieralski, R., B. Kempkes, and W. Gutensohn. 2003. Evidence for coordinated induction and repression of ecto-5′-nucleotidase (CD73) and the A2a adenosine receptor in a human B cell line. Biol. Chem. 384:483–487. [DOI] [PubMed] [Google Scholar]