

Abstract
HLA-B*4402 and B*4403 are naturally occurring MHC class I alleles that are both found at a high frequency in all human populations, and yet they only differ by one residue on the α2 helix (B*4402 Asp156→B*4403 Leu156). CTLs discriminate between HLA-B*4402 and B*4403, and these allotypes stimulate strong mutual allogeneic responses reflecting their known barrier to hemopoeitic stem cell transplantation. Although HLA-B*4402 and B*4403 share >95% of their peptide repertoire, B*4403 presents more unique peptides than B*4402, consistent with the stronger T cell alloreactivity observed toward B*4403 compared with B*4402. Crystal structures of B*4402 and B*4403 show how the polymorphism at position 156 is completely buried and yet alters both the peptide and the heavy chain conformation, relaxing ligand selection by B*4403 compared with B*4402. Thus, the polymorphism between HLA-B*4402 and B*4403 modifies both peptide repertoire and T cell recognition, and is reflected in the paradoxically powerful alloreactivity that occurs across this “minimal” mismatch. The findings suggest that these closely related class I genes are maintained in diverse human populations through their differential impact on the selection of peptide ligands and the T cell repertoire.
Keywords: class I histocompatibility molecules, antigen presentation, crystallography, X-ray diffraction, graft rejection, polymorphism
Introduction
HLA class I molecules are characterized by a high level of polymorphism presumed to reflect natural selection for protective immunity against microbes (1–3). HLA alleles can differ from each other by only a single amino acid (“micropolymorphism”) or by >30 amino acids (4). It has been suggested that there are nine major HLA class I “supertypes,” or clusters of alleles, that each possess a unique broad specificity for common anchor motifs in antigenic peptides (5). Alleles from each of these supertypic families are distributed in virtually all human populations and account for the majority HLA-A and -B polymorphism, suggesting that their maintenance reflects an adaptable strategy for induction of immunity toward diverse microbial ligands.
The widespread distribution of many low frequency HLA polymorphisms is probably an artifact of human migration and admixture of population groups that originally possessed a limited degree of HLA polymorphism (2). This might explain why most human populations often have only one or two alleles from each of the different HLA class I supertypes (5), such that a complete picture of HLA diversity is only appreciated when multiple racial groups are examined. An exception to this notion is the HLA-B44 family of alleles, in which both B*4402 and B*4403 are present in most human populations at combined phenotypic frequencies of up to 40% (6). HLA-B*4402 and B*4403 are the two most common members of the HLA-B44 supertype and participate in antiviral (7–10), antitumor (11–13), and minor Ag-specific responses (14, 15). HLA-B*4402 (Asp156) and B*4403 (Leu156) differ by only a single amino acid located on the α2 helix, essentially constituting a dimorphism within the HLA-B44 family because the other HLA-B44 alleles are much less common (generally, gene frequency <0.01; reference 6). These two B44 allotypes are also associated with different ancestral MHC haplotypes, suggesting an ancient origin for this di-allelism (16, 17).
Members of the B44 supertype share a preference for peptides with negatively charged residues at P2 (Glu) and hydrophobic residues at the COOH terminus (18, 19). Previous comparisons of ligands eluted from B*4402 and B*4403 molecules have not revealed any difference in their peptide repertoire, raising the question of what functional differences between these alleles support their high prevalence in all human populations.
Despite the similarity of their peptide repertoires, mismatching of B*4402 and B*4403 in hemopoeitic stem cell transplantation has been associated with transplant rejection (19, 20) and acute graft versus host disease (21), indicating that they represent a significant barrier to clinical transplantation. Moreover, CTL can discriminate between B*4402 and B*4403 allotypes, indicating structural differences that are highly relevant to T cells (22–24). The similarity in peptide repertoires of B*4402 and B*4403 suggests that the strong alloresponses between these alleles do not reflect presentation of different peptides (18, 19), but may reflect the accessibility of the α2 domain residue 156 for direct interaction with the TcR (24). Moreover, analyses of HLA-A2 mutants have confirmed the strong influence of position 156 on T cell recognition (25) and suggest a limited influence for this residue on peptide binding (26). On the other hand, residue 156 has also been predicted to influence the D/E pockets (24) that interact with the central core of the bound peptide (P4-P7; reference 27).
To help resolve these issues, we have evaluated the structural and functional properties of HLA-B*4402 and B*4403.
Materials and Methods
Cell Lines and Culture.
The B-lymphoblastoid cell line Hmy2.C1R (28) is class I–deficient, having no detectable HLA-A, very low levels of HLA-B35, normal HLA-Cw4, and intact antigen processing and presentation pathways (29). Transfection of HLA-B*4402 and B*4403 into C1R (C1R.B*4402 and C1R.B*4403, respectively) and cell culture conditions have been described previously (29). LC-13 and DD1 CTL clones were maintained in RPMI 1640 with supplements and restimulated every 10–14 d in the presence of recombinant IL-2 and HLA-matched lymphoblastoid cell lines or PBMCs pulsed with the appropriate peptide antigen. Derivation of CD8+ CTL clone 5101.1999.23 has been described previously (30).
Allogeneic MLR and Intracellular Cytokine Staining.
MLRs were performed using PBMCs from healthy blood donors (n = 13) matched for HLA-A, -B, and sometimes -C, but mismatched for HLA-B*4402 and B*4403 (and HLA class II). The HLA class I typing was as follows: B*4402 group (n = 10: A*02011/9, 03011; B*44021, 5701; and C*0501/02/03, 0602/03/07) and B*4403 group (n = 3: A*02011/9, 3011; B*44031, 5701; and C*0602/03/07, 1601).
In brief, 107 responder and 5 × 106 irradiated (3,000 rad) stimulator cells were cultured in RPMI 1640 plus 10% fetal calf serum, supplemented with 10 U/ml of recombinant IL-2 (Cetus Corporation) for 13 d at 37°C. On day 13, 2 × 105 responder T cells were harvested and restimulated with a panel of APCs (C1R, C1R.B*4402, and C1R.B*4403) at a cell concentration of 105 for 2 h at 37°C, 5% CO2. 10 μg/ml brefeldin was added for an additional 4 h, and responder T cells were stained with anti-CD4 PE (clone SK3; Becton Dickinson) and anti-CD8 CyChrome (BD Biosciences). Cells were fixed with 1% paraformadehyde (ProSciTech), permeabilized with 0.3% saponin (Sigma-Aldrich), and intracellular IFN-γ was detected with an anti–IFN-γ mAb (clone 25723.11; Becton Dickinson). The percentage of CD8+ T cells producing IFN-γ was determined by flow cytometry using FlowJo software (Tree Star Inc.).
Purification of Cell Surface–Associated HLA–B44 Complexes and Peptide Analysis.
Purification of HLA-B*4402 and B*4403 was performed from 5 × 109 C1R.B*4402 and C1R.B*4403 cells grown in roller bottles as described previously (31). Peptides were recovered as described previously (31). Peptides were separated by reverse phase (RP)-HPLC using a SMART system HPLC (Amersham Biosciences) with a μRPC C2 /C18 column (2.1 mm [inside diameter] × 10 cm). Eluted peptides were resolved from contaminating detergent polymers by using a rapid gradient from 0 to 60% acetonitrile in 0.1% aqueous TFA (12% increase in buffer B (organic)/min, 200 μl/min). This material was subjected to pool Edman sequencing and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS).
MS.
MALDI-TOF MS was performed using a Reflex mass spectrometer (Bruker) as described previously (31). Care was taken to ensure reproducibility of MS results on HPLC fractions (Figs. S1–S3 available at http://www.jem.org/cgi/content/full/jem.20030066/DC1). Peptide sequencing by Q-TOF electrospray ionization MS was performed as described previously (31, 32) on a Q-STAR pulsar-i Q-TOF MS (Applied Biosystems). Putative peptide sequences were obtained by database comparison of the fragmentation spectra using the MASCOT algorithm (33) followed by manual assignment of expected fragments from the highest score sequences (Table I).
Table I.
Sequences of Individually Sequenced Peptides
| Peptide sequence | Source | Mass | Frac. | B*4402 | B*4403 |
|---|---|---|---|---|---|
| Shared ligands | Daltons | ||||
| EEFGRAFSF | HLA DPA*0201 | 1,088.5 | 34 | ++++ | ++++ |
| AEDKENYKKF | Hsp90α (428-437) | 1,270.6 | 33 | ++ | ++ |
| AEMGKGSFKY | Translation elongation factor 1α (47-56) |
1,116.5 | 27 | ++ | ++ |
| EEYNGGLVTV | Ig heavy chain (106-115) | 1,079.6 | 31 | +++ | ++ |
| FEQIIERF | Human EST | 1,080.5 | 31 | ++ | ++ |
| EEPTVIKKY | Human EST | 1,105.6 | 28 | ++ | ++ |
| DEVGIVTKY | Unknown | 1,152.6 | 28 | ++ | ++ |
| EEVDLSKDIQHW | Ribonucleoside diphosphate reductase M2 chain |
1,497.7 | 32 | ++ | ++ |
| Ligands preferentially bound to B*4403 |
|||||
| AETPDIKLF | Ribosomal protein s5 | 1,032.5 | 33 | + | +++ |
| KEIKDILIQY | Ribosomal protein s12 | 1,261.7 | 32 | − | +++ |
| AEVDKVTGRF | Ribosomal protein s21 | 1,120.6 | 26 | − | +++ |
| NELNIIHKF | Human EST | 1,126.6 | 32 | + | ++ |
| Ligands preferentially bound to B*4402 |
|||||
| EEVHDLERKY | Human EST | 1,316.6 | 24 | +++ | + |
| YEGLLDYW | Ig heavy chain (86-93) | 1,057.6 | 31 | +++ | + |
Crystallization, Data Collection, and Structure Determination.
Recombinant B44 complexes were prepared from Escherichia coli, refolded, and crystallized as described previously (34). The data were processed and scaled using the HKL package (35) and D*TREK (36), Table II. The HLA-B*4402 structure was solved first using the molecular replacement method, using programs from CCP4 suite (37). The progress of refinement was monitored by the R free value (3% of the data) with neither a sigma, nor a low resolution cut off being applied to the data. The structure was refined using rigid-body fitting followed by the simulated-annealing protocol implemented in CNS (version 1.0; reference 38) and interspersed with rounds of model building using the program “O” (39). Water molecules were included in the model using standard criteria. The subsequent HLA-B*4403 structure was solved using the fully refined HLA-B*4402 structure, minus the bound peptide, water molecules, and site of polymorphism mutated to alanine. The refinement protocol implemented was identical to that of the HLA-B*4402 structure. For the HLA-B*4403 structure, two acetate ions were clearly visible in the electron density map. The quality of the electron density and the stereochemistry for the two fully refined, high resolution HLA-B44 alleles is excellent. The electron density for the bound peptide, as well as the interacting residues, is unambiguous. A summary of the refinement statistics is given in Table II. The coordinates have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb), accession codes 1M60 (B*4402) and 1N2R (B*4403).
Table II.
Data Collection and Refinement Statistics
| Units | B*4402 | B*4403 | |
|---|---|---|---|
| Data collection statistics | |||
| Temperature | K | 100 | 100 |
| Space group | P212121 | P212121 | |
| Cell dimensions (a, b, c) | Å | 50.81, 81.78, 110.07 |
50.72, 82.30, 109.55 |
| Resolution | Å | 1.6 | 1.7 |
| Total No. observations | 282,153 | 176,666 | |
| No. unique observations | 59,302 | 50,615 | |
| Multiplicity | 4.8 | 3.5 | |
| Data completeness | % | 96.8 (92.7) | 96.7 (93.5) |
| No. data >2σ1 | 85.2 | 80.9 | |
| I/σ1 | 9.4 (2.0) | 18.8 (3.2) | |
| Rmerge a | % | 4.9 (27.6) | 5.6 (54.1) |
| Refinement statistics | |||
| Non–hydrogen atom | |||
| Protein | 3,162 | 3,162 | |
| Water | 659 | 481 | |
| Acetate | 2 | ||
| Resolution | Å | 50-1.6 | 50-1.7 |
| Rfactor b | % | 21.4 | 22.9 |
| Rfree | % | 23.4 | 26.2 |
| rms deviations from ideal | |||
| Bond lengths | Å | 0.005 | 0.006 |
| Bond angles | ° | 1.27 | 1.26 |
| Dihedrals | ° | 24.92 | 25.08 |
| Impropers | ° | 0.74 | 0.72 |
| Ramachandran plot | |||
| Most favored | 92.0 | 91.4 | |
| And allowed region | % | 7.4 | 8.0 |
| B-factors | |||
| Average main chain | Å2 | 20.3 | 25.1 |
| Average side chain | Å2 | 22.9 | 28.1 |
| Average water molecule | Å2 | 36.3 | 40.5 |
| rms deviation bonded Bs | 1.6 | 1.8 |
The values in parentheses are for the highest resolution bin (approximate interval 0.1 Å). Two residues (Asp 29, Arg 239) that lie within generously allowed regions of the Ramachandran plot, fit the electron density very well. There are no residues in the disallowed region of the Ramachandran plot.
Rmerge = Σ|Ihkl − <Ihkl>|/ ΣIhkl
Rfactor = ΣIhkl| |Fo| − |Fc| |/ ΣIhkl|Fo| for all data except for 3%, which was used for the Rfree calculation. Two residues (Asp29, Arg239), which lie within generously allowed regions of the Ramachandran plot, fit the electron density very well. There are no residues in the disallowed region of the Ramachandran plot.
Online Supplemental Material.
The general issue of reproducibility and quantitation of the mass spectra are addressed in Figs. S1–S3. Fig. S1 shows the results of an experiment analyzing fractions from two independent elution experiments; Fig. S2 shows how the different peak heights in the mass spectra were compared by using natural internal reference standards; and Fig. S3 shows the results obtained when comparing the same samples but using different types of mass spectroscopy technology (MALDI-TOF versus ESI-QqTOF). Table S1 provides a detailed list of MHC-peptide contacts for the crystal structures of B*4402 and B*4403. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20030066/DC1.
Results
T Cell Discrimination between HLA-B*4402 and B*4403.
Despite only a single amino acid difference in the primary structure of HLA-B*4402 (156Asp) and B*4403 (156Leu), previous papers have shown that CD8+ T cells can readily distinguish between these two molecules (22, 40, 41). For example, allospecific CD8+ CTL clones, LC13 and DD1 (42), kill C1R transfectants expressing HLA-B*4402 and B*4405 but not transfectants expressing HLA-B*4403 (Fig. 1, A and B) . On the other hand, an allospecific, anti-B*4402 CTL clone (30, 43) makes IFN-γ in response to C1R-transfectants expressing HLA-B*4402, B*4403, and B*4407 but not B*4405 (Fig. 1 C). Accordingly, allogeneic T cell reactivity can be used to probe structural differences in MHC allotypes that may influence thymic selection (44) and peripheral diversity of T cells in immunity (3).
Figure 1.

Differential T cell recognition of HLA-B*4402 and HLA-B*4403 by CD8+ T cells. CTL clones, LC13 (A), and DD1 (B) lyse HLA-B*4402 but not HLA-B*4403 targets; untransfected C1R (closed triangles); and C1R-transfected APCs expressing either B*4402 (open squares), B*4403 (closed circles), B*4405 (closed squares), or B*4407 (closed triangles). (C) The CTL clone 5101.1999.23 is alloreactive with HLA-B*4402 and HLA-B*4403. IFN-γ production was assayed after 4 h. APCs: HERLUFF, B*4403 + LCL; PITOUT, B*4402 + LCL; and auto HSV-2, autologous cells infected with HSV-2. (D) PBMCs from donors mismatched for B*4402 and B*4403 were cocultured in a 13-d MLR supplemented with IL-2. The specificity of the responding cells was evaluated by restimulation for 6 h with defined APCs. Vertical axis, IFN-γ staining; horizontal axis, CD8 staining. Gates are shown for IFN-γ–producing CD8+ T cells. The percentage of positive cells is shown in each histogram. (E) The percentage of allospecific CD8+ T cells was assayed as in Fig. 4 D for 28 independent MLR reactions between B*4403 responders and B*4402 stimulators and 13 independent MLR reactions between B*4402 responders and B*4403 stimulators.
Therefore, we evaluated the in vitro alloresponse between individuals mismatched for B*4402 and B*4403, but matched at both HLA-A loci and for the HLA-B trans allele. After reciprocal allogeneic MLRs, CD8+ T lymphocytes were restimulated with defined APCs to determine their specificity, and T cells making IFN-γ were enumerated. Less than 1% of CD8+ T cells responded after the allogeneic MLR when either syngeneic PBMCs or untransfected C1R cells were used as APCs (Fig. 1 D, left). However, ∼20% of CD8+ T cells from B*4402+ responders were activated by C1R.B*4403 APCs (Fig. 1 D, bottom). Similarly, restimulation of B*4403+ responders with C1R.B*4402 APCs provoked IFN-γ production in ∼10% of CD8+ T cells (Fig. 1 D, top).
The median percentage of alloreactive CD8+ T cells in the B*4402 anti-B*4403 vector was 11.3% (n = 28; independent donor pairs) compared with a median of 2% B*4403 anti-B*4402 CD8+ T cells (n = 13; independent donor pairs). Hence, there were nearly sixfold more responding T cells identified in MLRs from B*4402 anti-B*4403 mismatches than vice versa, indicating an asymmetry in the magnitude of the alloresponse stimulated between these two HLA allotypes in vitro (Fig. 1 E). These findings indicate that the single residue that distinguishes B*4402 from B*4403 has a profound effect on T cell recognition of these alloantigens, which is likely to result in differential T cell repertoire selection by these allotypes.
Isolation of HLA-B44–bound Peptides and Analysis of Ligand Specificity.
Differential T cell recognition of B*4402 and B*4403 could result from differences in either peptide repertoire or HLA heavy chain conformation (45, 46). Alternatively, identical peptide repertoires could be presented in an altered manner due to structural changes at the interface between the peptide loaded class I molecule and the TcR (47, 48). Therefore, peptide repertoires of HLA-B*4402 and B*4403 were examined using pool Edman sequencing and high-resolution MALDI-TOF MS. Apart from the previously reported P2Glu and PΩTyr/Phe anchor residues (12, 18, 19), minor differences were noted between subdominant anchor residues with more pronounced yields of valine at P3 for B*4403 and tryptophan at P9 for B*4402 (Fig. 2) .
Figure 2.

Subtle differences in ligand selection by B*4402 and B*4403 despite a shared dominant anchor motif and overlapping peptide repertoires. Pool Edman sequence analysis of peptides eluted from HLA-B*4402 and B*4403. Comparison of B*4402 and B*4403 peptide repertoires by MALDI-TOF MS. Total peptide eluates from HLA-B*4402 (positive polarity spectra) and HLA-B*4403 (negative polarity spectra) after a single-dimension chromatographic separation.
When peptide pools were examined by MALDI-TOF MS, a high degree of overlap was detected in the ligands from HLA-B*4402 and B*4403 (Fig. 2), consistent with previous work (12, 18, 19). Each peptide pool exhibited a complex ionization pattern, with a common prominent signal at (M + H)+ = 1089.6, subsequently shown to be self-peptide derived from HLA DPα46–54 (34). Differences in ligand repertoire were evident between HLA-B*4402 and B*4403, such as the species of m/z 1033.6, 1121.5, and 1145.6 that were all more prevalent in HLA-B*4403 relative to B*4402.
Additional fractionation of the total eluates was performed using a second dimension of RP-HPLC followed by MALDI-TOF MS of each fraction. Fig. 3 A shows the complexity of ligands identified in B*4402 as a function of RP-HPLC retention time (right) and mass spectra (left). The individual species identified in ∼60 RP-HPLC fractions exhibited >95% overlap between HLA-B*4402 and B*4403 (Fig. 3, B–G and unpublished data). Fig. 3 B shows the mass spectra of fraction “B” (Fig. 3 A, right), derived from HLA-B*4402 (MALDI-TOF, positive polarity), and HLA-B*4403 (negative polarity). Here, the species at m/z 1121.3 appears to be specific to the HLA-B*4403 eluate and was not detected in neighboring fractions from either HLA-B*4402 or B*4403. This peptide was identified as amino acids 37–46 of human ribosomal protein S21, a natural ligand of HLA-B*4403 (Table I; reference 18). On the other hand, the species of mass 1180.4 was preferentially recovered from B*4402 eluates. Fractions “C” and “F” (Fig. 3 A, right) revealed greater complexity of peptides bound to HLA-B*4403 versus B*4402 (Fig. 3, C and F), whereas spectra of equal complexity but distinct composition were observed in other fractions (Fig. 3, D and E). Some fractions contained ligands that were mostly identical in B*4402 and B*4403 (Fig. 3 G).
Figure 3.

High resolution peptide mapping of B*4402 and B*4403 ligands reveals ∼5% bound peptides are unique or preferentially bound by one allotype. (A) After an initial RP-HPLC separation of HLA-B*4402 bound peptides from C1R transfectants, a second dimension RP-HPLC separation was performed (right) and mass spectra determined for the selected fractions (left). (B–G) MALDI-TOF mass spectra of ligands from selected fractions after a 2nd round of RP-HPLC (A, right). Peptides from HLA-B*4402 (positive polarity spectra) and HLA-B*4403 (negative polarity spectra).
Approximately 5% of all ligands were either preferentially presented by B*4403 or B*4402, or were uniquely recovered from one or other allotype. A systematic examination of all fractions identified 116 such peptides, of which 80% were “biased” toward presentation by the B*4403 molecule. Only 20% of the biased ligands were preferentially presented by B*4402. This bias was even more marked (>90% B*4403) when the analysis was confined to ligands with the strongest ionization signals. Sequencing of shared or unique peptides associated with HLA-B*4402 and B*4403 verified the observations made by MS (Table I). Tryptophan and valine were identified at the COOH terminus of some eluted peptides. There was no characteristic motif in the peptides preferentially associated with B*4403.
Structural Basis of HLA-B*4402 and B*4403 Binding to a Shared Peptide Ligand.
The crystal structures of B*4402 and B*4403 were determined in complex with a shared natural, high affinity ligand derived from HLA-DPα (DPA*0201, residues 46–54 EEFGRAFSF) (Fig. 2 and Fig. 3 G, (M + H)+ = 1089.6 ± 0.2). The data collection and refinement statistics are shown in Table II. The resolution of the structures was 1.6 and 1.7 Å, respectively. The peptide was bound in an extended conformation in both HLA-B*4402 and B*4403, with a small bulge around Gly 4 (Fig. 4, A–D) . There were 13 water-mediated hydrogen bonds (H-bonds) and 15 direct H-bonds in the HLA–B*4402 complex. Fewer H-bonds were observed in the HLA–B*4403 complex (9 water mediated H-bonds and 15 direct H-bonds). The peptide also participates in three conserved salt bridges in both complexes (Table S1 available at http://www.jem.org/cgi/content/full/jem.20030066/DC1). The dominant solvent-accessible residues of the peptide ligand were Glu1, Gly4, Arg5, and Ser8, whereas Phe3 and Phe7 are only partially solvent accessible (Fig. 4 D). In both structures, Arg5 was the most mobile residue (B factors 29–35, Å2), reflecting the greater solvent accessibility of the polar head group of this residue.
Figure 4.

Structures of bound DPα ligand complexed to two HLA-B44 alleles, highlighting polymorphic amino acids and the electron density of the peptide. The structures of the DPα46–54 (EEFGRAFSF) complexed to (A) HLA-B*4402 and (B) B*4403 highlighting the polymorphism at position 156. (C) Superposition of the two antigen-binding clefts B*4402 (green) and B*4403(yellow) showing the shift in the α1 helix around residues 70–77. (D) Superposition of the two peptide ligands (side view) highlighting the solvent-accessible amino acids.
Structural Basis for Selection of Shared Anchor Residues by the B and F pockets.
The antigen-binding cleft of class I HLA molecules consists of a series of pockets designated A–F (27, 49). In different alleles, these pockets vary in their depth, electrostatic potential, and hydrophobicity, thus determining their ligand specificity. Specificity for P2-Glu occurs in HLA-B44, HLA-A29, and H-2Kk. However, because the structural basis for this specificity has not yet been described, these interactions are described in some detail here and compared with the B pockets of B*2705 (50) and B*3501 (51, 52) that possess alternative specificities (Fig. 5) . The B pocket is formed by residues at positions 9, 24, 45, 66, 67, and 99 of the class I heavy chain; the P2-Glu interacts with all these residues except for Ile66 in our structures and makes an additional interaction (position 70). In B*4402 and B*4403, the peptide anchor Glu2 forms a salt bridge with Lys45, which is situated at the bottom of the B pocket and presumably provides substantial energetic contribution to the specificity of the B pocket (Fig. 5). Furthermore, the charged P2-Glu carboxylate forms direct H-bonds to Tyr9 and Tyr99. Additionally, there is a buried, well-ordered water molecule at the base of the pocket that mediates H-bonds to Ser67 and Thr24 in the HLA-B*4402/03 structures. In B*4403, this conserved water molecule forms a H-bond to Asn70, a residue within the α1-helix that flexes as a result of the polymorphism at position 156. The long aliphatic moiety of Glu2 packs predominantly against the aromatic ring of Tyr7 and is flanked by Ile66, although it doesn't interact with the latter residue.
Figure 5.

Novel structure of the B pocket of HLA-B*4403 and B*4402 determines the selection of dominant anchor residues P2 Asp/Glu. The structure of the B pocket of HLA-B*4403 is compared with previously reported structures for HLA-B*2705 and HLA-B*3501. The upper panels are ball and stick representations of B pocket amino acids 9, 24, 45, 66, 67, 70, and 99 (gray) and the P2 amino acid of the ligand (green); B*4403 (P2 Glu, left), HLA-B*2705 (P2 Arg, middle), and B*3501 (P2 Pro, right). The lower panels represent electrostatic surfaces of the area bounding the B pocket using the program GRASP (70) and showing the bound peptide ligand for the structures in ball and stick format (blue, electropositive; red, electronegative). The B pocket of B*4402 is virtually identical to B*4403.
Some of the structural framework of the B pocket is conserved between B*4403, B*2705, and B*3501 (Fig. 5). Thus, the B pockets of B*2705 (50) and B*4402/B*4403 are deep to accommodate the long aliphatic side chains of arginine or glutamate. However, in contrast to B*4402 and B*4403, the deep electronegative B pocket of B*2705 (50) accommodates a positively charged canonical P2-Arg residue involved in charge complementation with the glutamic acid residue at position 45 of the heavy chain. In the B pocket of HLA-B*3501, the projecting Phe67 creates a hydrophobic, sterically occluded pocket that preferentially accommodates proline residues at P2 of bound peptide ligands (51, 52).
The residues that surround the F pocket are very similar in both the B*4402 and B*4403 structures, and only the structure of B*4402 is shown in Fig. 6 . The P9 anchor residue phenylalanine projects into a hydrophobic F pocket, where it stacks between the aromatic ring of Tyr123 and the aliphatic moiety of Asn77. The main chain of this COOH-terminal residue is tethered by H-bonds to Asn77, Tyr84, and Thr143. In addition, the walls of the F pocket are bound by Trp147, Tyr74, and Ile95. The large cluster of aromatic residues within the F pocket provides a structural basis for the preference of aromatic residues at the P9 position. Asp116 forms the base of this pocket, where the carboxylate group of Asp116 is orientated away from the F pocket, leaving the aliphatic moiety of Asp116 to interact with the hydrophobic Phe side chain (Table S1 and Fig. 6) and to form a direct H-bond with Tyr74 that is lost in B*4403.
Figure 6.

The F pocket of B*4402. The main chain of the peptide carboxy-terminal residue is tethered by H-bonds to Asn 77, Tyr 84, and Thr 143. The P9 anchor residue phenylalanine projects into a hydrophobic F pocket where it stacks between the aromatic ring of Tyr123 and the aliphatic moiety of Asn 77. The walls of the pocket are bounded by Trp 147, Tyr 74, and Ile 95. Peptide residues in green, HLA heavy chain in gray and H-bonds shown as dashed lines.
Impact of α2 Helix Position 156Asp→Leu Polymorphism on HLA-B*4402 and B*4403 Structures.
In the structures of both B*4402 and B*4403, the side chain of amino acid 156 was buried and, therefore, not directly accessible for TcR interaction (Fig. 4, A and B). Superposition of the Cα backbones of peptides bound to B*4402 and B*4403 revealed a root mean square deviation of 0.41 Å with a 1.0-Å shift around Ala6 being the most significant movement between the two structures (Fig. 4 C). In HLA-B*4402, Asp156 forms a salt bridge with Arg97 on the floor of the cleft and participates in a number of H-bond and van der Waals (v.d.w.) interactions. These interactions include water-mediated H-bonds to the Arg97 guanadinium head group and the C5 carbonyl of the peptide ligand backbone, and an unusual direct H-bond to Asp114 (Fig. 7 A). The H-bond between Asp156 and Asp114 orientates the Asp114 side chain, allowing a salt bridge to the guanadinium head group of Arg97 as well as a water-mediated H-bond to Asp116. The pairing of Asp156 and Asp114 has not been seen in any other crystal structures of HLA class I molecules, and is presumably stabilized by salt bridging between both these residues and Arg97, allowing an O–H·····H interaction between carboxylates. However, the potential for H-bond formation between Asp156 and Asp114 exists in several other HLA-B molecules, including other members of the HLA-B44 family. In addition Asp156 forms a v.d.w. interaction with Trp133 and Leu126, as well as with Phe3 of the bound peptide ligand. Here, the peptide Phe3–Asp156 interaction is facilitated by the side-on orientation adopted by the Phe3 side chain.
Figure 7.
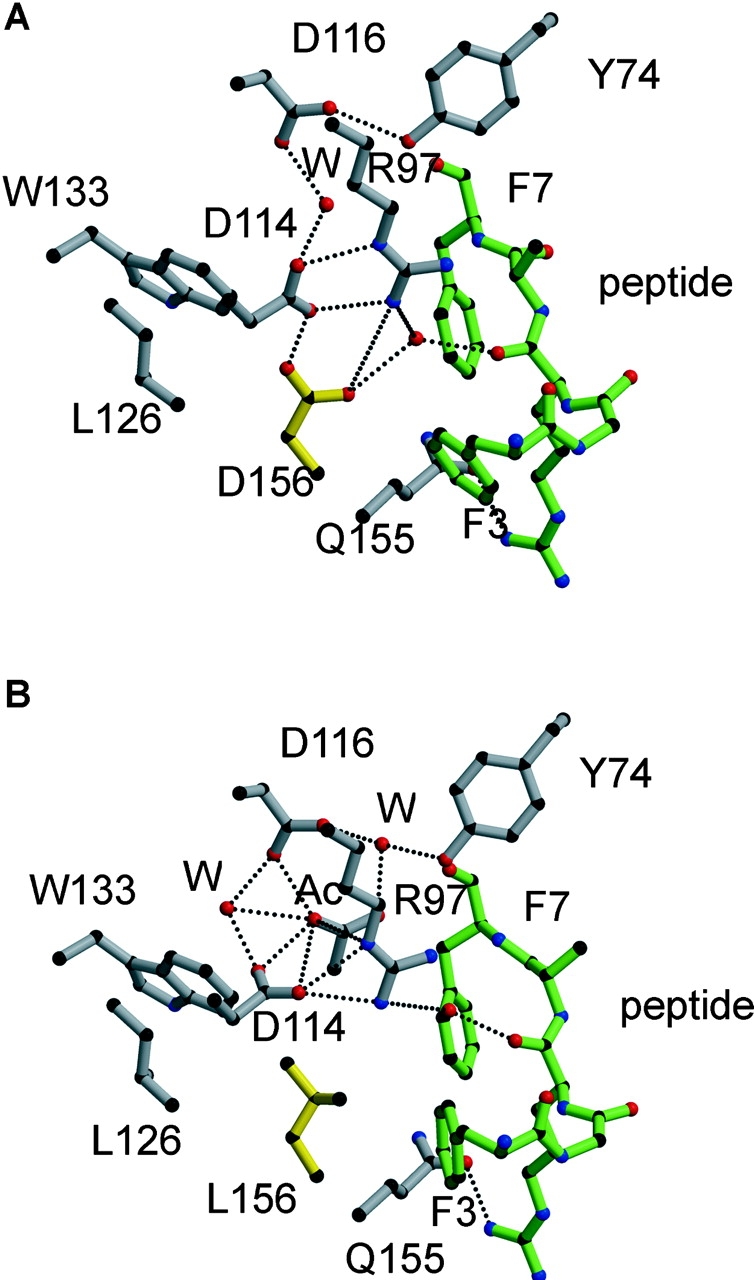
The structural interactions around residue 156 constrain the antigen-binding cleft of HLA-B*4402 (156 Asp) relative to HLA-B*4403 (156 Leu) and alter the orientation of peptide side chains. The side chain of residue 156 is shown in yellow and forms a unique H-bond with Asp 114 in B*4402 (A) that is missing in B*4403 (B), where instead Asp 114 is reoriented to maximize its H-bond network with Arg 97, creating a wider cavity in this region of the cleft. Peptide, green; HLA heavy chain, gray; H-bonds, dotted lines; W, water.
In contrast to B*4402, the nonpolar nature of Leu156 in B*4403 results in loss of a water-mediated H-bond to the peptide backbone, giving rise to a significant repositioning of the peptide between the B*4403 α-helices. On the other hand, the side chain of Leu156 in HLA-B*4403 is rotated by ∼90° (Fig. 7 B), enhancing v.d.w. contacts within the hydrophobic pocket formed by Leu126, Trp133, and Val152. In addition, to avoid unfavorable interactions with Leu156, the orientation of the Asp114 side chain in B*4403 changes by ∼90°, simultaneously maximizing interactions with Arg97 and maintaining a water-mediated H-bond to the C5 carbonyl of the peptide backbone. Hence, despite the loss of some H-bonds in B*4403, including the Asp116–Tyr74 interaction, the stability of the EEFGRAFSF peptide does not seem to be significantly impaired in the B*4403 complex consistent with experiments that show little difference in the thermostability of B*4402 versus B*4403 in complex with the EEFGRAFSF peptide (unpublished data).
However, the altered interactions between residue 156 and neighboring heavy chain residues, and the peptide ligand, significantly change the dimensions of the Ag-binding cleft in B*4402 and B*4403 (Fig. 4, C and D). A “rigid body shift” in the α1 helix, particularly around positions 70–77, results in a 0.6-Å displacement of the α-helices between the antigen-binding cleft of HLA-B*4402 (green) versus B*4403 (Fig. 4 C, yellow). Amino acid residues 70–77 lie adjacent to the central region of the peptide that has undergone a shift of 1.0 Å in the Cα backbone around Ala6. This conformational adjustment is directly associated with the loss of a direct H-bond between Asp116 and Tyr74 of the B*4403 heavy chain (Fig. 7, A and B). Thus, in the HLA-B*4402 structure the direct H-bond between Asp116 and Tyr74 results in a narrower antigen-binding cleft (Figs. 6 and 7 A), whereas in the B*4403 structure loss of this interaction is associated with distortion and relative “breathing” of the binding cleft (Fig. 7 B). Instead of making a direct H-bond with Asp116, Tyr74 participates in a water-mediated H-bond with this residue in HLA-B*4403; this is attributable to changes in the peptide orientation, resulting from the substitution at amino acid 156. Consequently, the slightly larger cavity of the HLA-B*4403 cleft is occupied by an acetate ion in this structure, reflecting potentially greater accommodation of peptide side chains by the B*4403 allotype compared with B*4402 (Fig. 7 B). Any thermodynamic penalty for “breathing” of the B*4403 cleft may not be apparent with some ligands because of compensatory peptide interactions arising from the altered cleft structure, as appears to be the case for B*4403 binding of the EEFGRAFSF peptide.
Discussion
The impact of point mutations and other micropolymorphisms on the structure and function of class I molecules has been studied extensively by in vitro mutagenesis (53, 54) and through exploitation of the experimentally derived Kb mutant system (3, 44, 47, 48). These systems have been enormously important for unraveling key details of structure–function relationships in MHC molecules. However, it is equally important to study the structural and functional consequences of naturally selected microvariation in HLA allotypes found in outbred human populations. To this end, we have determined the structural and functional properties of two closely related, naturally occurring human class I alleles, HLA-B*4402 and B*4403. These were studied because of their evolutionary success as common alleles in diverse human populations, notwithstanding their marked structural similarity and shared peptide binding motif.
Structural differences in B*4402 and B*4403 are distinguishable by many T cells and these allotypes provoke strong, mutual allogeneic T cell responses in vitro. Indeed, up to 25% of CD8+ T cells are specifically alloreactive in extended MLRs between B*4402/B*4403 mismatched individuals. These observations indicate that B*4402 and B*4403 molecules select both shared and unique T cells during thymic development.
The structures of B*4402 and B*4403 show that amino acid 156 is neither solvent accessible, nor directly involved in forming major specificity pockets. Nonetheless, the 156Asp→Leu polymorphism is a nonconservative substitution and markedly alters the characteristics of the P3 specificity (E pocket) as well as the secondary effects of the antigen-binding cleft. Hence, the 156Asp→Leu substitution alters the repertoire of peptide ligands bound by both molecules and directly alters the conformation of the antigen-binding cleft. Subtle differences were observed in preferred residues at minor anchor sites in B*4403 versus HLA-B*4402, however, these differences may be more apparent than real. For instance, a Trp can be modeled at PΩ in B*4402 and B*4403 (unpublished data) and a tryptophan is found at PΩ in the EEVDLSKDIQHW peptide eluted from both B*4402 and 4403 (Table I). Thus, any preference for Trp at PΩ in B*4402 is probably quantitative (statistical) or context-dependent.
The peptide repertoire of HLA-B*4402 and B*4403 showed >95% overlap but most of the unique peptides were associated with B*4403. These differences in peptide selection can be partly understood by an examination of the two structures. Comparison of the antigen-binding cleft of HLA-B*4402 and B*4403 revealed a relative narrowing of the B*4402 cleft between the α-helices, particularly around amino acids 70–77. This distortion of the cleft shape appears to be the result of both indirect and ligand-mediated conformational changes associated with the 156Asp→Leu polymorphism. The Asp114–Asp156 interaction in B*4402 is unusual, and being constrained, appears to limit the type of residue that can sit in the P3, and/or the P7 pocket. A large network of polar interactions at position 156 dissipates this unfavorable interaction and draws the helix 1 closer to the peptide, thereby sterically constraining the makeup of potential bound peptides in B*4402. Thus, the relatively restricted range of ligands recovered from B*4402 compared with B*4403 probably results from this structural constraint that limits the size of side chains that can be accommodated in the narrowed region. In contrast, the 156Asp→Leu substitution in B*4403 results in a loss of this bonding network, allowing the helices to breathe in this region. This breathing of the cleft is further reflected by the presence of an acetate moiety in the extra space around amino acids 70–77 in the B*4403/EEFGRAFSF complex. Breathing, or opening of the binding cleft, is also observed in the structures of HLA-DQ8 (55) and I-Ag7 (56) when these are compared with other class II molecules.
The orientation of side chains in the EEFGRAFSF peptide bound by HLA-B*4402 and B*4403 was also altered by the 156Asp→Leu polymorphism. Structural adjustments within identical ligands bound by related MHC allotypes can be sufficient to induce T cell alloreactivity (24). However, altered heavy chain conformation (57) and binding of unique ligands (58) can also contribute to T cell alloreactivity. Thus, T cell discrimination of B*4402 and B*4403, either during positive selection of the T cell repertoire, or during B*4402 anti-B*4403 alloreactions, could involve any one, or all of the modes of recognition discussed here.
Ironically, the structural similarity between B*4402 and B*4403 is likely to engender strong reciprocal T cell alloreactivity due to thymic selection of a large repertoire of T cells with intrinsic “B44”-reactivity biased toward these allotypes. Hence, the number of T cells that can distinguish subtle structural differences in B*4402 and B*4403 is likely to be very significant. Thus, the magnitude of the alloresponse is paradoxically as great, or even greater, than observed across more disparate HLA allelic differences (19–21)
The subtle differences in peptide repertoire between B*4402 and B*4403 contrast with the differences observed between many HLA class I allotypes, especially where substitutions occur in the dominant anchor residues located within the B and F pockets (59–61). For instance, HLA-B*2705 and B*2709 differ at position 116 (Asp→His) at the base of the F pocket (62, 63), resulting in peptide repertoires that overlap by ∼79 and ∼88%, respectively (64), and producing mutual alloreactivity (62, 65).
However, natural class I polymorphism outside the P2 and PΩ peptide anchor sites appears to modulate peptide repertoire in a much more subtle, often imperceptible manner, as reported for subtypes of HLA-B7, B17 and B15 (66), B5 and B22 (67), and B27 (65–69). Thus, our findings in relation to B*4402 and B*4403 are likely to be of general relevance in understanding the impact of microvariation in HLA class I genes. For instance, among all known HLA class I molecules, there are five different amino acids observed at position 156 (Asp, Leu, Arg, Gln, and Trp). Dimorphism at this residue is found in virtually all the common class I subtypes, suggesting that this position exerts a strong influence on the structure and function of other class I allotypes as well as B44 (25).
The functional basis underlying the retention of the B*4402 and B*4403 alleles in diverse human populations presumably rests with their independent selective advantage in immunity. Selection could be related to differential ligand selection by B*4402 and B*4403 (1); or, alternatively, may reflect differences in either thymic positive selection of T cells (44) or clonal diversification of the peripheral immune response (3). In addition, selection could be influenced by other elements of conserved HLA-B44 haplotypes, although these are not well-preserved in all populations, making this explanation less likely.
We conclude that the relatively small changes in peptide repertoire and MHC–peptide conformation between naturally occurring HLA polymorphisms can be sufficient to maintain selection of these alleles in diverse human populations. These micropolymorphisms are potentially capable of enhancing protective immunity by expanding ligand selection but are also likely to diversify T cell repertoire during thymic positive selection and at the time of the immune response.
Acknowledgments
We would like to thank F. Carbone for critical reading of the manuscript and W. Heath for helpful suggestions. We thank the staff at BioCARS and the Australian Synchrotron Research Program for assistance.
J. Rossjohn is supported by a Wellcome Trust Senior Research Fellowship in Biomedical Science in Australia; A.W. Purcell is a C.R. Roper Fellow of the Faculty of Medicine, Dentistry, and Health Science at The University of Melbourne; A.G. Brooks is supported by a R.D. Wright Fellowship. This work was supported by the National Health Council and the Medical Research Council, the Australian Kidney Foundation, the Roche Organ Transplantation Research Foundation, and the Juvenile Diabetes Research Foundation.
Abbreviations used in this paper: H-bond, hydrogen bond; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; MS, mass spectrometry; RP, reverse phase; v.d.w., van der Waals.
W.A. Macdonald and A.W. Purcell contributed equally to this work.
The online version of this article includes supplemental material.
References
- 1.Hill, A.V., C.E. Allsopp, D. Kwiatkowski, N.M. Anstey, P. Twumasi, P.A. Rowe, S. Bennett, D. Brewster, A.J. McMichael, and B.M. Greenwood. 1991. Common west African HLA antigens are associated with protection from severe malaria. Nature. 352:595–600. [DOI] [PubMed] [Google Scholar]
- 2.Parham, P., and T. Ohta. 1996. Population biology of antigen presentation by MHC class I molecules. Science. 272:67–74. [DOI] [PubMed] [Google Scholar]
- 3.Messaoudi, I., J.A. Patino, R. Dyall, J. LeMaoult, and J. Nikolich-Zugich. 2002. Direct link between MHC polymorphism, T cell avidity, and diversity in immune defense. Science. 298:1797–1800. [DOI] [PubMed] [Google Scholar]
- 4.Marsh, S.G., P. Parham, and L.D. Barber. 2000. The HLA Facts Book. Academic Press, London. 398 pp.
- 5.Sette, A., and J. Sidney. 1999. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics. 50:201–212. [DOI] [PubMed] [Google Scholar]
- 6.Raffoux, C., R.C. Williams, C. Gorodezky, E.D. Albert, E. Gyodi, M.G. Hammond, Z. Layrisse, M. Mariani, G. de la Rosa, J.M. Tiercy, et al. 1997. AHS#7: HLA-B12, B13, B21, B37, B40, B41, B81, B4005. Genetic Diversity of HLA Functional and Medical Implication. D. Charron, editor. EDK Medical and Scientific International Publisher, Paris. 69–72.
- 7.Hiroishi, K., H. Kita, M. Kojima, H. Okamoto, T. Moriyama, T. Kaneko, T. Ishikawa, S. Ohnishi, T. Aikawa, N. Tanaka, et al. 1997. Cytotoxic T lymphocyte response and viral load in hepatitis C virus infection. Hepatology. 25:705–712. [DOI] [PubMed] [Google Scholar]
- 8.Khanna, R., S.R. Burrows, A. Neisig, J. Neefjes, D.J. Moss, and S.L. Silins. 1997. Hierarchy of Epstein-Barr virus-specific cytotoxic T-cell responses in individuals carrying different subtypes of an HLA allele: implications for epitope-based antiviral vaccines. J. Virol. 71:7429–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kita, H., T. Moriyama, T. Kaneko, I. Harase, M. Nomura, H. Miura, I. Nakamura, Y. Yazaki, and M. Imawari. 1993. HLA B44-restricted cytotoxic T lymphocytes recognizing an epitope on hepatitis C virus nucleocapsid protein. Hepatology. 18:1039–1044. [PubMed] [Google Scholar]
- 10.Kita, H., T. Moriyama, T. Kaneko, H. Okamoto, K. Hiroishi, S. Ohnishi, and M. Imawari. 1995. HLA B44-restricted cytotoxic T lymphocyte responses to the peptides of HCV nucleoprotein residues 81-100 in patients with chronic hepatitis C. J. Gastroenterol. 30:809–812. [DOI] [PubMed] [Google Scholar]
- 11.Brichard, V.G., J. Herman, A. Vanpel, C. Wildmann, B. Gaugler, T. Wolfel, T. Boon, and B. Lethe. 1996. A tyrosinase nonapeptide presented by HLA-B44 is recognized on a human melanoma by autologous cytolytic T lymphocytes. Eur. J. Immunol. 26:224–230. [DOI] [PubMed] [Google Scholar]
- 12.Fleischhauer, K., D. Fruci, P. Van Endert, J. Herman, S. Tanzarella, H.J. Wallny, P. Coulie, C. Bordignon, and C. Traversari. 1996. Characterization of antigenic peptides presented by HLA-B44 molecules on tumor cells expressing the gene MAGE-3. Int. J. Cancer. 68:622–628. [DOI] [PubMed] [Google Scholar]
- 13.Herman, J., P. van der Bruggen, I.F. Luescher, S. Mandruzzato, P. Romero, J. Thonnard, K. Fleischhauer, T. Boon, and P.G. Coulie. 1996. A peptide encoded by the human MAGE3 gene and presented by HLA-B44 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE3. Immunogenetics. 43:377–383. [DOI] [PubMed] [Google Scholar]
- 14.Dolstra, H., H. Fredrix, F. Preijers, E. Goulmy, C.G. Figdor, T.M. de Witte, and E. van de Wiel-van Kemenade. 1997. Recognition of a B cell leukemia-associated minor histocompatibility antigen by CTL. J. Immunol. 158:560–565. [PubMed] [Google Scholar]
- 15.Tiercy, J.M., M. Jeannet, and E. Roosnek. 1995. HLA-B44-restricted minor antigen. Transplantation. 60:113–114. [DOI] [PubMed] [Google Scholar]
- 16.Kruskall, M.S., E.E. Eynon, Z. Awdeh, C.A. Alper, and E.J. Yunis. 1987. Identification of HLA-B44 subtypes associated with extended MHC haplotypes. Immunogenetics. 26:216–219. [DOI] [PubMed] [Google Scholar]
- 17.Mattiuz, P.L., E. Di Paolo, V. Fossombroni, A. Menicucci, F. Pradella, B. Porfirio, and G. Rombola. 1997. HLA-B44 subtypes and the chance of finding HLA compatible donor/recipient pairs for bone marrow transplantation: a haplotype study of 303 Italian families. Tissue Antigens. 50:602–609. [DOI] [PubMed] [Google Scholar]
- 18.DiBrino, M., K.C. Parker, D.H. Margulies, J. Shiloach, R.V. Turner, W.E. Biddison, and J.E. Coligan. 1995. Identification of the peptide binding motif for HLA-B44, one of the most common HLA-B alleles in the Caucasian population. Biochemistry. 34:10130–10138. [DOI] [PubMed] [Google Scholar]
- 19.Fleischhauer, K., D. Avila, F. Vilbois, C. Traversari, C. Bordignon, and H.J. Wallny. 1994. Characterization of natural peptide ligands for HLA-B*4402 and -B*4403: implications for peptide involvement in allorecognition of a single amino acid change in the HLA-B44 heavy chain. Tissue Antigens. 44:311–317. [DOI] [PubMed] [Google Scholar]
- 20.Fleischhauer, K., N.A. Kernan, R.J. O'Reilly, B. Dupont, and S.Y. Yang. 1990. Bone marrow-allograft rejection by T lymphocytes recognizing a single amino acid difference in HLA-B44. N. Engl. J. Med. 323:1818–1822. [DOI] [PubMed] [Google Scholar]
- 21.Keever, C.A., N. Leong, I. Cunningham, E.A. Copelan, B.R. Avalos, J. Klein, N. Kapoor, P.W. Adams, C.G. Orosz, P.J. Tutschka, et al. 1994. HLA-B44-directed cytotoxic T cells associated with acute graft-versus-host disease following unrelated bone marrow transplantation. Bone Marrow Transplant. 14:137–145. [PubMed] [Google Scholar]
- 22.Burrows, S.R., R. Khanna, J.M. Burrows, and D.J. Moss. 1994. An alloresponse in humans is dominated by cytotoxic T lymphocytes (CTL) cross-reactive with a single Epstein-Barr virus CTL epitope: implications for graft-versus-host disease. J. Exp. Med. 179:1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burrows, S.R., S.L. Silins, S.M. Cross, C.A. Peh, M. Rischmueller, J.M. Burrows, S.L. Elliott, and J. McCluskey. 1997. Human leukocyte antigen phenotype imposes complex constraints on the antigen-specific cytotoxic T lymphocyte repertoire. Eur. J. Immunol. 27:178–182. [DOI] [PubMed] [Google Scholar]
- 24.Herman, J., V. Jongeneel, D. Kuznetsov, and P.G. Coulie. 1999. Differences in the recognition by CTL of peptides presented by the HLA-B*4402 and the HLA-B*4403 molecules which differ by a single amino acid. Tissue Antigens. 53:111–121. [DOI] [PubMed] [Google Scholar]
- 25.Hogan, K.T., C. Clayberger, E.J. Bernhard, S.F. Walk, J.P. Ridge, P. Parham, A.M. Krensky, and V.H. Engelhard. 1988. Identification by site-directed mutagenesis of amino acid residues contributing to serologic and CTL-defined epitope differences between HLA-A2.1 and HLA-A2.3. J. Immunol. 141:2519–2525. [PubMed] [Google Scholar]
- 26.Tussey, L.G., M. Matsui, S. Rowland-Jones, R. Warburton, J.A. Frelinger, and A. McMichael. 1994. Analysis of mutant HLA-A2 molecules. Differential effects on peptide binding and CTL recognition. J. Immunol. 152:1213–1221. [PubMed] [Google Scholar]
- 27.Saper, M.A., P.J. Bjorkman, and D.C. Wiley. 1991. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 A resolution. J. Mol. Biol. 219:277–319. [DOI] [PubMed] [Google Scholar]
- 28.Zemmour, J., A.M. Little, D.J. Schendel, and P. Parham. 1992. The HLA-A,B “negative” mutant cell line C1R expresses a novel HLA-B35 allele, which also has a point mutation in the translation initiation codon. J. Immunol. 148:1941–1948. [PubMed] [Google Scholar]
- 29.Alexander, J., J.A. Payne, R. Murray, J.A. Frelinger, and P. Cresswell. 1989. Differential transport requirements of HLA and H-2 class I glycoproteins. Immunogenetics. 29:380–388. [DOI] [PubMed] [Google Scholar]
- 30.Koelle, D.M., H.B. Chen, M.A. Gavin, A. Wald, W.W. Kwok, and L. Corey. 2001. CD8 CTL from genital herpes simplex lesions: recognition of viral tegument and immediate early proteins and lysis of infected cutaneous cells. J. Immunol. 166:4049–4058. [DOI] [PubMed] [Google Scholar]
- 31.Purcell, A.W., J.J. Gorman, M. Garcia-Peydro, A. Paradela, S.R. Burrows, G.H. Talbo, N. Laham, C.A. Peh, E.C. Reynolds, J.A. Lopez De Castro, and J. McCluskey. 2001. Quantitative and qualitative influences of tapasin on the class I peptide repertoire. J. Immunol. 166:1016–1027. [DOI] [PubMed] [Google Scholar]
- 32.Purcell, A.W., and J.J. Gorman. 2001. The use of post source decay in matrix assisted laser desorption-ionisation mass spectrometry to delineate T cell determinants. J. Immunol. Methods. 249:17–31. [DOI] [PubMed] [Google Scholar]
- 33.Perkins, D.N., D.J. Pappin, D.M. Creasy, and J.S. Cottrell. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 20:3551–3567. [DOI] [PubMed] [Google Scholar]
- 34.Macdonald, W., D.S. Williams, C.S. Clements, J.J. Gorman, L. Kjer-Nielsen, A.G. Brooks, J. McCluskey, J. Rossjohn, and A.W. Purcell. 2002. Identification of a dominant self-ligand bound to three HLA B44 alleles and the preliminary crystallographic analysis of recombinant forms of each complex. FEBS Letters. 527:27–32. [DOI] [PubMed] [Google Scholar]
- 35.Ottwinowski, Z. 1993. Oscillation data reduction program. Data Collection and Processing, Proceedings of the CCP4 Study Weekend, 29–30, January, 1993. L. Sawyer, N. Issacs, and S.W. Bailey, editors. SERC Daresbury Laboratory, U.K., Warrington, U.K. 56–62.
- 36.Pflugrath, J.W. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55:1718–1725. [DOI] [PubMed] [Google Scholar]
- 37.CCP4. 1994. CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D50:750–763. [DOI] [PubMed] [Google Scholar]
- 38.Brunger, A.T., P.D. Adams, G.M. Clore, W.L. DeLano, P. Gros, R.W. Grosse-Kunstleve, J.S. Jiang, J. Kuszewski, M. Nilges, N.S. Pannu, et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- 39.Jones, T.A., J.-Y. Zou, S.W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr. 47:110–119. [DOI] [PubMed] [Google Scholar]
- 40.Burrows, S.R., S.L. Silins, D.J. Moss, R. Khanna, I.S. Misko, and V.P. Argaet. 1995. T cell receptor repertoire for a viral epitope in humans is diversified by tolerance to a background major histocompatibility complex antigen. J. Exp. Med. 182:1703–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rufer, N., B.S. Breur-Vriesendorp, J.M. Tiercy, A.S. Slavcev, N.M. Lardy, P. Francis, R. Kressig, D.E. Speiser, C. Helg, B. Chapuis, et al. 1993. High-resolution histocompatibility testing of a group of sixteen B44-positive, ABDR serologically matched unrelated donor-recipient pairs. Analysis of serologically undisclosed incompatibilities by cellular techniques, isoelectrofocusing, and HLA oligotyping. Hum. Immunol. 38:235–239. [DOI] [PubMed] [Google Scholar]
- 42.Argaet, V.P., C.W. Schmidt, S.R. Burrows, S.L. Silins, M.G. Kurilla, D.L. Doolan, A. Suhrbier, D.J. Moss, E. Kieff, T.B. Suclley, et al. 1994. Dominant selection of an invariant T cell antigen receptor in response to persistent infection by Epstein-Barr virus. J. Exp. Med. 180:2335–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koelle, D.M., H.B. Chen, C.M. McClurkan, and E.W. Petersdorf. 2002. Herpes simplex virus type 2-specific CD8 cytotoxic T lymphocyte cross-reactivity against prevalent HLA class I alleles. Blood. 99:3844–3847. [DOI] [PubMed] [Google Scholar]
- 44.Dyall, R., I. Messaoudi, S. Janetzki, and J. Nikolic-Zugic. 2000. MHC polymorphism can enrich the T cell repertoire of the species by shifts in intrathymic selection. J. Immunol. 164:1695–1698. [DOI] [PubMed] [Google Scholar]
- 45.Eckels, D.D. 1990. Alloreactivity: allogeneic presentation of endogenous peptide or direct recognition of MHC polymorphism? A review. Tissue Antigens. 35:49–55. [DOI] [PubMed] [Google Scholar]
- 46.Rotzschke, O., K. Falk, S. Faath, and H.G. Rammensee. 1991. On the nature of peptides involved in T cell alloreactivity. J. Exp. Med. 174:1059–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luz, J.G., M. Huang, K.C. Garcia, M.G. Rudolph, V. Apostolopoulos, L. Teyton, and I.A. Wilson. 2002. Structural comparison of allogeneic and syngeneic T cell receptor-peptide-major histocompatibility complex complexes: a buried alloreactive mutation subtly alters peptide presentation substantially increasing Vβ interactions. J. Exp. Med. 195:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rudolph, M.G., J.A. Speir, A. Brunmark, N. Mattsson, M.R. Jackson, P.A. Peterson, L. Teyton, and I.A. Wilson. 2001. The crystal structures of K(bm1) and K(bm8) reveal that subtle changes in the peptide environment impact thermostability and alloreactivity. Immunity. 14:231–242. [DOI] [PubMed] [Google Scholar]
- 49.Garrett, T.P., M.A. Saper, P.J. Bjorkman, J.L. Strominger, and D.C. Wiley. 1989. Specificity pockets for the side chains of peptide antigens in HLA-Aw68. Nature. 342:692–696. [DOI] [PubMed] [Google Scholar]
- 50.Madden, D.R., J.C. Gorga, J.L. Strominger, and D.C. Wiley. 1991. The structure of HLA-B27 reveals nonamer self-peptides bound in an extended conformation. Nature. 353:321–325. [DOI] [PubMed] [Google Scholar]
- 51.Menssen, R., P. Orth, A. Ziegler, and W. Saenger. 1999. Decamer-like conformation of a nona-peptide bound to HLA-B*3501 due to non-standard positioning of the C terminus. J. Mol. Biol. 285:645–653. [DOI] [PubMed] [Google Scholar]
- 52.Smith, K.J., S.W. Reid, K. Harlos, A.J. McMichael, D.I. Stuart, J.I. Bell, and E.Y. Jones. 1996. Bound water structure and polymorphic amino acids act together to allow the binding of different peptides to MHC class I HLA-B53. Immunity. 4:215–228. [DOI] [PubMed] [Google Scholar]
- 53.Murray, R., C.A. Hutchison III, and J.A. Frelinger. 1988. Saturation mutagenesis of a major histocompatibility complex protein domain: identification of a single conserved amino acid important for allorecognition. Proc. Natl. Acad. Sci. USA. 85:3535–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nathenson, S.G., J. Geliebter, G.M. Pfaffenbach, and R.A. Zeff. 1986. Murine major histocompatibility complex class-I mutants: molecular analysis and structure-function implications. Annu. Rev. Immunol. 4:471–502. [DOI] [PubMed] [Google Scholar]
- 55.Latek, R.R., A. Suri, S.J. Petzold, C.A. Nelson, O. Kanagawa, E.R. Unanue, and D.H. Fremont. 2000. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity. 12:699–710. [DOI] [PubMed] [Google Scholar]
- 56.Lee, K.H., K.W. Wucherpfennig, and D.C. Wiley. 2001. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat. Immunol. 2:501–507. [DOI] [PubMed] [Google Scholar]
- 57.Ajitkumar, P., S.S. Geier, K.V. Kesari, F. Borriello, M. Nakagawa, J.A. Bluestone, M.A. Saper, D.C. Wiley, and S.G. Nathenson. 1988. Evidence that multiple residues on both the alpha-helices of the class I MHC molecule are simultaneously recognized by the T cell receptor. Cell. 54:47–56. [DOI] [PubMed] [Google Scholar]
- 58.Heath, W.R., K.P. Kane, M.F. Mescher, and L.A. Sherman. 1991. Alloreactive T cells discriminate among a diverse set of endogenous peptides. Proc. Natl. Acad. Sci. USA. 88:5101–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rojo, S., F. Garcia, J.A. Villadangos, and J.A. Lopez de Castro. 1993. Changes in the repertoire of peptides bound to HLA-B27 subtypes and to site-specific mutants inside and outside pocket B. J. Exp. Med. 177:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barouch, D., T. Friede, S. Stevanovic, L. Tussey, K. Smith, S. Rowland-Jones, V. Braud, A. McMichael, and H.G. Rammensee. 1995. HLA-A2 subtypes are functionally distinct in peptide binding and presentation. J. Exp. Med. 182:1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prilliman, K.R., K.W. Jackson, M. Lindsey, J. Wang, D. Crawford, and W.H. Hildebrand. 1999. HLA-B15 peptide ligands are preferentially anchored at their C termini. J. Immunol. 162:7277–7284. [PubMed] [Google Scholar]
- 62.Ramos, M., and J.A. Lopez de Castro. 2002. HLA-B27 and the pathogenesis of spondyloarthritis. Tissue Antigens. 60:191–205. [DOI] [PubMed] [Google Scholar]
- 63.Hulsmeyer, M., R.C. Hillig, A. Volz, M. Ruhl, W. Schroder, W. Saenger, A. Ziegler, and B. Uchanska-Ziegler. 2002. HLA-B27 subtypes differentially associated with disease exhibit subtle structural alterations. J. Biol. Chem. 277:47844–47853. [DOI] [PubMed] [Google Scholar]
- 64.Ramos, M., A. Paradela, M. Vazquez, A. Marina, J. Vazquez, and J.A. Lopez de Castro. 2002. Differential association of HLA-B*2705 and B*2709 to ankylosing spondylitis correlates with limited peptide subsets but not with altered cell surface stability. J. Biol. Chem. 277:28749–28756. [DOI] [PubMed] [Google Scholar]
- 65.Garcia-Peydro, M., M. Marti, and J. Lopez de Castro. 1999. High T cell epitope sharing between two HLA-B27 subtypes (B*2705 and B*2709) differentially associated to ankylosing spondylitis. J. Immunol. 163:2299–2305. [PubMed] [Google Scholar]
- 66.Barber, L.D., L. Percival, K.L. Arnett, J.E. Gumperz, L. Chen, and P. Parham. 1997. Polymorphism in the alpha 1 helix of the HLA-B heavy chain can have an overriding influence on peptide-binding specificity. J. Immunol. 158:1660–1669. [PubMed] [Google Scholar]
- 67.Barber, L.D., B. Gillece-Castro, L. Percival, X. Li, C. Clayberger, and P. Parham. 1995. Overlap in the repertoires of peptides bound in vivo by a group of related class I HLA-B allotypes. Curr. Biol. 5:179–190. [DOI] [PubMed] [Google Scholar]
- 68.Lopez, D., S. Rojo, V. Calvo, and J.A. Lopez de Castro. 1992. Peptide-presenting similarities among functionally distant HLA-B27 subtypes revealed by alloreactive T lymphocytes of unusual specificity. J. Immunol. 148:996–1002. [PubMed] [Google Scholar]
- 69.Tanigaki, N., D. Fruci, E. Vigneti, G. Starace, P. Rovero, M. Londei, R.H. Butler, and R. Tosi. 1994. The peptide binding specificity of HLA-B27 subtypes. Immunogenetics. 40:192–198. [DOI] [PubMed] [Google Scholar]
- 70.Taylor, N.R., and R. Smith. 1996. The World Wide Web as a graphical user interface to program macros for molecular graphics, molecular modeling, and structure-based drug design. J. Mol. Graph. 14:291–296, 280–282 [DOI] [PubMed] [Google Scholar]