

Abstract
Infections are a leading cause of death in stroke patients. In a mouse model of focal cerebral ischemia, we tested the hypothesis that a stroke-induced immunodeficiency increases the susceptibility to bacterial infections. 3 d after ischemia, all animals developed spontaneous septicemia and pneumonia. Stroke induced an extensive apoptotic loss of lymphocytes and a shift from T helper cell (Th)1 to Th2 cytokine production. Adoptive transfer of T and natural killer cells from wild-type mice, but not from interferon (IFN)-γ–deficient mice, or administration of IFN-γ at day 1 after stroke greatly decreased the bacterial burden. Importantly, the defective IFN-γ response and the occurrence of bacterial infections were prevented by blocking the sympathetic nervous system but not the hypothalamo-pituitary-adrenal axis. Furthermore, administration of the β-adrenoreceptor blocker propranolol drastically reduced mortality after stroke. These data suggest that a catecholamine-mediated defect in early lymphocyte activation is the key factor in the impaired antibacterial immune response after stroke.
Keywords: T lymphocytes, natural killer cells, interferon γ, pneumonia, brain ischemia
Introduction
Infectious complications, predominantly chest and urinary tract infections, have been reported to occur in 23–65% of all stroke patients within the first few days after stroke (1–5). Although early mortality is due to direct complications from large strokes, pneumonia is the leading cause of death in the postacute phase of stroke, regardless of hospitalization (6–9). The high frequency of antibiotic treatment on specialized stroke units underlines the clinical problem of infections in stroke patients (10). The high incidence of infections in these patients is likely to be a result of an impaired immune function. Immunodepression has been reported in other potentially life-threatening conditions, such as myocardial infarction, polytrauma, or major surgery, resulting in an increased risk of infectious complications (11–13). In trauma patients, the presence of brain injury was identified as an independent risk factor for infectious complications, possibly due to a central nervous shutdown of the immune defense (14, 15). As early as 1974, an immunosuppressive state was associated with stroke (16). However, the mechanisms leading to an increased susceptibility to infections after stroke are still poorly understood. Here, we demonstrate that focal cerebral ischemia induces a rapid and long-lasting inhibition of cell-mediated immunity resulting in spontaneous systemic bacterial infection, and that early IFN-γ production by T and NK cells is crucial in controlling bacterial infections. Our finding that β-adrenoreceptor blockade enhances cellular immune responses and prevents bacterial infections, provides strong evidence that an activation of the sympathetic nervous system (SNS) is the main immunosuppressive mechanism leading to a high incidence of infections after stroke.
Materials and Methods
Animals.
SV129/J mice (BGVV), B6.129P2-Tcrbtm1Mom (αβ T cell–deficient), B6.129P2-Tcrdtm1Mom (γδ T cell–deficient), B6.129S7-INFγtm1Ts (IFN-γ–deficient), and C57BL/6J wild-type mice (The Jackson Laboratory) were housed in the animal care facility at the Department of Neurology (Charité Hospital, Berlin, Germany) until killed.
Experimental Model of Stroke.
We used gender-mixed 7-wk-old or older SV129/J or, where indicated, C57BL/6J mice weighing ∼18–22 g. The surgical procedure of middle cerebral artery occlusion (MCAO) did not exceed 10 min and was induced as previously described (17). In brief, a monofilament was introduced into the common carotid artery under halothane narcosis, advanced to the origin of the middle cerebral artery (MCA), and left there for 60 min until reperfusion. In sham-operated animals, after inserting the filament to the MCA origin, the filament was immediately withdrawn by 2 mm to avoid ischemia. Occlusion and reperfusion were verified by laser Doppler flowmetry (Peri Flux 4001 Master; Perimed). Mice were kept in heated cages for the next 2 h and body temperature was frequently measured. Animals were then returned to their home cages and allowed free access to food and water. The infarct volumes were measured histologically as previously described (18). All animal experiments were performed according to institutional and state guidelines.
Drug Administration.
All drugs were injected i.p. and the respective diluents were given to the control animals at the same time. RU486 (Sigma-Aldrich) was dissolved in ethanol/sesame oil solution (1:10 vol/vol) at 6 mg/ml and administered (30 mg/kg body weight) 24 h, 5 h, and immediately before MCAO. Propranolol (Sigma-Aldrich) was dissolved in 0.9% sodium chloride at 6 mg/ml, and administered (dose 1–30 mg/kg body weight as indicated) immediately before as well as 4 and 8 h after MCAO. Where indicated, propranolol administration (30 mg/kg body weight) was delayed and given 24, 28, and 32 h after MCAO. 6-hydroxydopamine HBr (6-OHDA; Sigma-Aldrich) was dissolved in sterile 0.01% ascorbic acid/saline and injected (200 mg/kg body weight) 3 d before MCAO. IFN-γ (TEBU) was dissolved in phosphate buffered saline at 20 μg/ml, and administered i.p. (2 μg) at 24 and/or 48 h after MCAO. Control mice received heat-denaturated IFN-γ solution.
Blood Samples, Cell Suspensions, and Cell Count.
Mice were killed and blood was collected into heparinized tubes. Single cell suspensions from thymus and spleen were prepared by forcing the tissues through a fine wire mesh. Cells were washed and resuspended in 2 ml RPMI 1640 medium containing penicillin, streptomycin, 2 mM glutamine, and 10% FCS (Biochrom KG). Residual RBCs were lysed by hypotonic lysis in ice-cold ammonium. Cell counts were performed in triplicates.
Flow Cytometry.
For flow cytometric analysis the following fluorescently labeled anti–mouse monoclonal antibodies (BD Biosciences) were used: CD3 (145-2C11; T cells), CD4 (L3T4), CD8 (Ly-2), CD45R (RA3-6B2; B cells), anti–pan-NK cell marker (DX5), CD11b (M1/70; monocytes/macrophages, granulocytes), and CD11c (HL3; dendritic cells). In blood and spleen samples, RBCs were lysed with BD Lysis Solution (Becton Dickinson) before analysis. The degree of apoptotic cell death in thymus and spleen single cell suspensions was quantified using fluorescein-labeled annexin V (Qbiogene). Cell phenotyping and identification of apoptotic cells was performed by four-color flow cytometry on a FACSCalibur™ using CELLQuest™ Software (BD Biosciences).
Analysis of Ex Vivo Cytokine Production.
Whole blood was diluted 1:5 in heparinized RPMI 1640 and incubated at 37°C and 5% CO2. For analysis of TNF-α synthesis, samples were stimulated with 100 ng/ml LPS (endotoxin) from Escherichia coli 0127:B8 (Sigma-Aldrich) for 4 h. For analysis of IFN-γ and IL-4 production, blood samples were stimulated with 100 μg/ml Con A (Sigma-Aldrich) for 24 h. For analysis of IFN-γ production in spleen cells, 106 cells/ml were stimulated with 10 μg/ml Con A for 24 h. Concentrations of cytokines in culture supernatants were determined using commercially available kits and according to the manufacturer's instructions (BD Biosciences). Each sample was assayed in duplicate.
Adoptive Transfer of Spleen Cells.
Single cell suspensions were prepared from spleens of wild-type or knockout mice as described above. Mice that underwent MCAO were injected intraperitoneally with 5 × 106 splenocytes 24 h after MCAO. Control mice were injected with medium. In some experiments, splenic T, B, and NK cell subsets were depleted using commercially available magnetic bead kits (CD90, CD45R, DX5 microbeads; Miltenyi Biotec). Depletion efficacy was verified as >95% by flow cytometry.
Histopathological Analysis of Lung Tissue.
After transcardial perfusion with ethanol-formalin-acetic acid, lungs were removed and embedded in paraffin wax. 12-μm thick sections were obtained by microtome dissection and stained with hematoxylin/eosin (HE). 20 representative sections of lungs were chosen per animal (n = 4 in each group) and evaluated by two investigators, who were blinded to the treatment groups. The sections were graded according to the following criteria: no definite damage represented no histological changes or minor changes, including unequal distension of alveolar units, mild thickening of the alveolar septa, and perivascular and peribronchiolar edema. Definite damage was observed as lung consolidation, thickened alveolar septae, and the presence of intra-alveolar inflammatory infiltrates.
Histochemistry and In Situ Terminal Deoxynucleotide Transferase-mediated dUTP Nick-end Labeling (TUNEL) Assay.
After transcardial perfusion with ethanol-formalin-acetic acid, thymi were removed and embedded in paraffin wax. Sections were obtained by microtome dissection and stained with HE. Based on HE staining, two representative 12-μm sections of thymus were chosen per animal (n = 6) and processed for TUNEL. The Apoptag Kit (Intergen) was used according to the manufacturer's instructions. Visualization was achieved using the Vectorstain ABC elite kit (Vector Laboratories) in conjunction with 3,3′-diaminobenzidine/H2O2 (Sigma-Aldrich).
Bacteriological Analysis.
The anesthetized mice were washed with 70% ethanol under sterile conditions. Blood was collected by decapitation. The lungs were removed after thoracotomy and homogenized. For determination of CFU, 100 μl tissue homogenate or blood was serially diluted, plated onto blood agar plates (Merck), incubated at 37°C for 18 h, and bacterial colonies were counted.
Statistical Analysis.
Bacteriological data are presented as box plots and other data are given as mean ± SD. Data were analyzed by Kruskal-Wallis analysis of variance (ANOVA) after pairwise comparison with Dunn's method or Mann-Whitney U test using SPSS software (SPSS Inc.). Differences in the group survival were determined using Fisher's exact test. Values of P < 0.05 were considered significant.
Results
Ischemia was induced in mice by temporary occlusion of the MCAO. Sham controls received surgery identical to that of MCAO but with regional cerebral blood flow remaining unaffected. After 60 min of MCAO, animals developed large infarcts (90 ± 10 mm3) of cortex, striatum, and hippocampus.
Spontaneous Bacteremia and Pneumonia after Focal Cerebral Ischemia.
3 d after ischemia, all MCAO animals suffered from spontaneous bacterial infections. Bacterial cultures from peripheral blood and lungs invariably demonstrated >95% E. coli (Fig. 1 a). Significant bacterial loads were observed in lungs and blood 24 and 48 h after ischemia, respectively. Histological examination of lungs revealed typical signs of bacterial pneumonia (19, 20) in all analyzed MCAO-treated animals 72 h after experimental stroke (n = 4, Fig. 1 b). In contrast, blood and lung cultures from sham-operated animals remained sterile at 6, 12, 24, 48 (not depicted), and 72 h (Fig. 1 a). In addition, lung sections of sham controls revealed no signs of pneumonia (n = 4, Fig. 1 b). Therefore, susceptibility to infection resulted from stroke and not from surgical stress.
Figure 1.
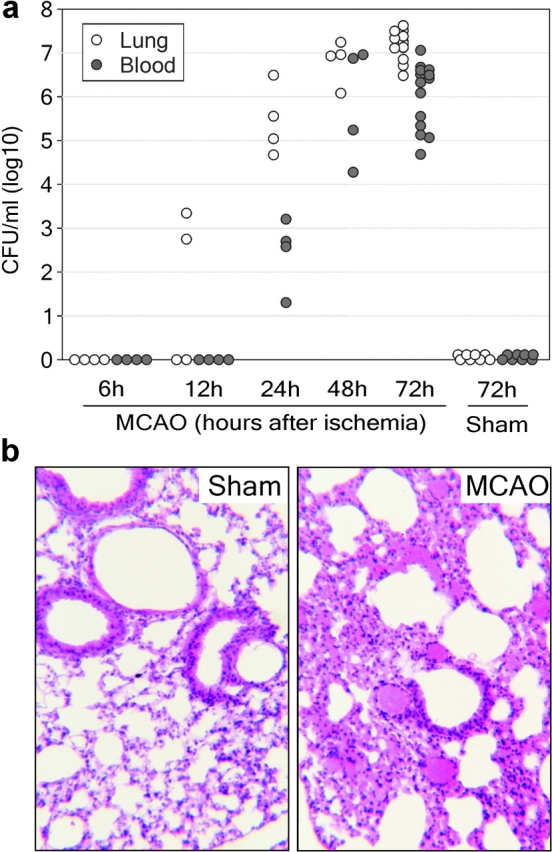
Stroke induces bacteremia and pneumonia. (a) Lungs and blood samples from sham (n = 10) and MCAO-treated mice (n = 4–14 per group) were collected for bacteriological analysis at the indicated time points after surgery. Data are given in CFU/ml (log 10) blood or lung tissue homogenate. (b) In a different set of experiments, lungs from sham-operated (n = 4) and MCAO mice (n = 4) were collected after 72 h for histological examination. A representative 12-μm section of HE-stained lung from MCAO but not from sham animals revealed signs (thickening of alveolar walls and neutrophilic infiltrates) of E. coli pneumonia. ×160.
Long-lasting Lymphopenia and Impaired Cellular Immune Functions after Cerebral Ischemia.
To characterize the underlying mechanisms of spontaneous infections after stroke, we analyzed the cellularity and leukocyte populations in lymphoid organs and peripheral blood from MCAO, sham, and control mice using four-color flow cytometry. We found a striking reduction of lymphocyte counts in blood, spleen, and thymus after cerebral ischemia (Fig. 2) . The absolute numbers of T, B, and NK lymphocytes were decreased 3–10-fold in blood and 2–3-fold in spleen as early as 12 h after MCAO (Fig. 2, a–f). There was only a marginal decrease in the number of lymphocytes in sham animals, indicating that lymphopenia was mainly caused by ischemia and only partly due to surgical stress. The postischemic total lymphocyte count in the blood was significantly lower even after 14 d (MCAO: 2.7 ± 0.6 vs. control: 4.8 ± 0.5 × 106/ml; P < 0.05), whereas the number of splenic lymphocytes remained reduced for 42 d (MCAO: 26.9 ± 0.8 vs. control: 46.5 ± 4.7 × 106/ml; P < 0.05; Fig. 2, a–f). Cellularity in the thymus was also severely reduced (Fig. 2 g) and characterized by a rapid loss of immature CD4+ CD8+ thymocytes and subsequent reduction in the number of mature single positive T cells (not depicted). Interestingly, splenic T and B cell counts increased 2 d after stroke, but dropped again at day 5 (Fig. 2, d and e) coinciding with the occurrence of bacteremia.
Figure 2.

Stroke induces long-lasting lymphopenia and impaired cytokine expression. Spleen, thymus, and peripheral blood samples of untreated SV129/J mice (control) and sham or MCAO mice were collected at different time points (sham, 12 h; MCAO, as indicated) after surgery. Leukocyte counts in blood (a–c), spleen (d–f), and thymus (g) single cell suspensions were determined as described in Materials and Methods. The lymphocyte subsets were determined by flow cytometry and their absolute numbers were calculated. (h and i) Aliquots of blood samples were stimulated ex vivo with either LPS for analysis of monocytic TNF-α expression or Con A for analysis of IFN-γ and IL-4 synthesis, as described in Materials and Methods. Cytokines were determined in supernatants by ELISA. The Con A–induced lymphokine expression is given as the ratio of IFN-γ and IL-4 production calculated for each individual. Data are shown as mean ± SD. *, results differed from the control group; *, P < 0.05; **, P < 0.01; ***, P < 0.001; #, results differed from the sham-operated group; #, P < 0.05; ##, P < 0.01. Mann-Whitney U test, n = 3–11 per group.
Recently, we have reported that patients with brain surgery showed diminished monocytic HLA-DR expression and endotoxin-induced TNF-α secretion as signs of systemic immunodepression (13, 21). To test whether cerebral ischemia is also associated with alterations in cellular immune functions, we examined the endotoxin-induced TNF-α secretion as well as Con A–induced IFN-γ and IL-4 production ex vivo in whole blood cultures, as parameters of monocyte and T lymphocyte functions, respectively. Secretion of endotoxin-induced TNF-α was significantly decreased after 12 h and 2 d after MCAO but returned to control levels on day 5 (Fig. 2 h). The decrease in the monocytic TNF-α release was not due to reduced monocyte cell numbers, as blood monocyte counts in MCAO animals did not differ from those in control mice (not depicted). Furthermore, we found reduced IFN-γ but increased IL-4 production in Con A–stimulated blood cultures resulting in a significant decrease of the IFN-γ/IL-4 ratio for at least 14 d after MCAO (Fig. 2 i). In contrast, sham-operated animals did not show significant alterations in any of the measured cytokine secretion parameters (Fig. 2, h and i). Importantly, the described alterations of immune parameters after experimental stroke preceded bacterial infections (compare Figs. 1 a and 2) and, therefore, are more likely to be the cause than the result of bacterial infections.
Brain Ischemia Causes Rapid and Extensive Apoptosis in Lymphatic Organs.
To test whether lymphopenia was a result of increased apoptosis, we analyzed spleen and thymus cell suspensions by flow cytometry and found a marked increase in apoptotic lymphocytes 12 h after MCAO. In splenocytes, increased apoptosis affected all lymphocyte subsets, as determined by annexin V and cell-type–specific surface marker staining (Fig. 3 a). Similarly, enhanced apoptosis was found in all thymocyte subsets, mostly affecting the immature CD3dim CD4+ CD8+ double positive thymocytes (Fig. 3 b). In contrast, there was no significant difference in the degree of apoptosis in splenocytes from sham-operated mice, and only a very modest increase in apoptosis in double positive immature thymocytes was noted (Fig. 3, a and b). In accordance with these results, light microscopic examination of thymi from MCAO mice, but not sham mice, showed many apoptotic cells with features of pyknosis or karyorrhexis (Fig. 3 c, top). Moreover, TUNEL staining demonstrated that the apoptosis occurred predominantly in the thymic cortex, which is mainly populated by immature double positive thymocytes, and relatively spared the thymic medulla (Fig. 3 c, bottom). In addition, thymocyte DNA from MCAO animals showed extensive laddering (not depicted).
Figure 3.

Stroke induces increased apoptosis in lymphoid organs. Spleen and thymus from untreated SV129/J mice (control) and sham or MCAO mice were collected 12 h after surgery. Splenocytes (a) and thymocytes (b) were isolated and investigated for apoptosis by annexin V labeling and flow cytometry. Lymphocyte subpopulations were determined by staining with mAbs against cell-type–specific surface markers. Data are shown as mean ± SD. *, results differed from the control group; *, P < 0.05; **, P < 0.01; ***, P < 0.001; #, results differed from the sham-operated group; #, P < 0.05; ##, P < 0.01; ###, P < 0.001. Mann-Whitney U test, n = 8 per group. (c) Thymic tissue sections from sham- and MCAO-treated mice were stained with HE (top) or examined by the fluorescent TUNEL method (bottom). Thymi from MCAO mice showed many apoptotic cells with typical features of nuclear condensation and fragmentation (HE; ×400). Note also the high number of TUNEL+ cells (bright red nuclei) in the thymus cortex from MCAO animals as compared with sham mice. ×100.
Role of Stress Mediators in Postischemic Immunodepression.
The activation of the hypothalamic-pituitary-adrenal axis (HPA) and the SNS, resulting in a systemic release of adrenal steroid hormones and catecholamines, is an essential component of the response to major surgery, brain injury, or trauma. As high levels of these stress mediators are known to be immunosuppressive (22), we investigated the effects of the β-adrenoreceptor antagonist propranolol and the glucocorticoid receptor inhibitor RU486 on the immunoinhibitory alterations induced by stroke. Treatment of MCAO animals with either propranolol or RU486 decreased the percentage of apoptotic splenocytes to levels observed in sham-operated mice and prevented the decrease in peripheral blood lymphocyte counts. The protective effects of both compounds extended to all lymphocyte subsets in spleen and blood (Fig. 4 , a and b). Both compounds also significantly increased the ex vivo endotoxin-induced TNF-α release, but only propranolol normalized the IFN-γ/IL-4 ratio after Con A stimulation of whole blood cultures (Fig. 4 c).
Figure 4.

Effects of β-adrenoreceptor and glucocorticoid receptor blockade on stroke-induced immunodepression and bacterial infection. SV129/J mice underwent sham or MCAO surgery (n = 9–12 per group). MCAO mice received the β-adrenoreceptor antagonist propranolol, the glucocorticoid receptor inhibitor RU486, or only diluent before or after surgery, as described in Materials and Methods. Spleens and blood samples were collected 12 h after surgery. (a) Splenocytes were isolated and investigated for apoptosis by annexin V labeling, staining with mAbs against cell-type–specific surface markers, and flow cytometry. (b) Total leukocyte counts in blood samples were determined as described in Materials and Methods. The percentages of different lymphocyte classes were determined by flow cytometry using mAbs against cell-type–specific surface markers and their absolute numbers calculated. (c) For determination of cytokine production, blood samples were processed and cytokine expression was analyzed as described in Fig. 2. Data are shown as mean ± SD. #, results differed from the sham-operated group; #, P < 0.05; ##, P < 0.01; §, results differed from the diluent-treated MCAO group; §, P < 0.05; §§, P < 0.01; §§§, P < 0.001. Mann-Whitney U test, n = 9–12 per group. (d) In a different set of experiments, lungs and blood samples from sham and inhibitor- or diluent-treated MCAO mice were collected for bacteriological analysis 72 h after surgery. Data are shown as box plots in CFU/ml (log 10) blood or lung tissue homogenate.
Inhibition of the SNS but Not Steroid Receptor Blockade Prevents Systemic Infection.
Treatment with propranolol (3 × 30 mg/kg BW) completely prevented both bacteremia and pneumonia in 8 out of 10 animals (Fig. 4 d). In the remaining two mice, bacterial counts in blood and lung were reduced by the order of 104 and 106, respectively. In contrast, the glucocorticoid receptor antagonist RU486 had no effect on blood or pulmonary bacterial burden. The protective effect of propranolol was dose and time dependent. Propranolol doses (3 × 10 and 3 × 30 mg/kg BW starting immediately after MCAO) sufficient to reduce or prevent bacterial dissemination at 72 h after MCAO (Fig. 5 a) also restored the defective IFN-γ response determined 12 h after experimental stroke (Fig. 5 b), whereas lower doses had no effect on either bacterial load or IFN-γ production. Importantly, delayed administration of propranolol (3 × 30 mg/kg BW starting 24 h after cerebral ischemia) neither increased IFN-γ production nor decreased bacterial load (Fig. 5), demonstrating that blockade of sympathetic nerves activation early after MCAO is crucial to prevent lymphocyte dysfunction and, subsequently, bacterial infections. To confirm that the protective effect of propranolol was due to inhibition of sympathetic nerve signaling, MCAO mice were pretreated with 6-OHDA, which selectively enters and destroys sympathetic noradrenergic terminals, thereby depleting tissue catecholamines (23). Like propranolol, chemical sympathectomy by 6-OHDA up-regulated IFN-γ production and significantly reduced bacterial load in lungs and blood (Fig. 5).
Figure 5.
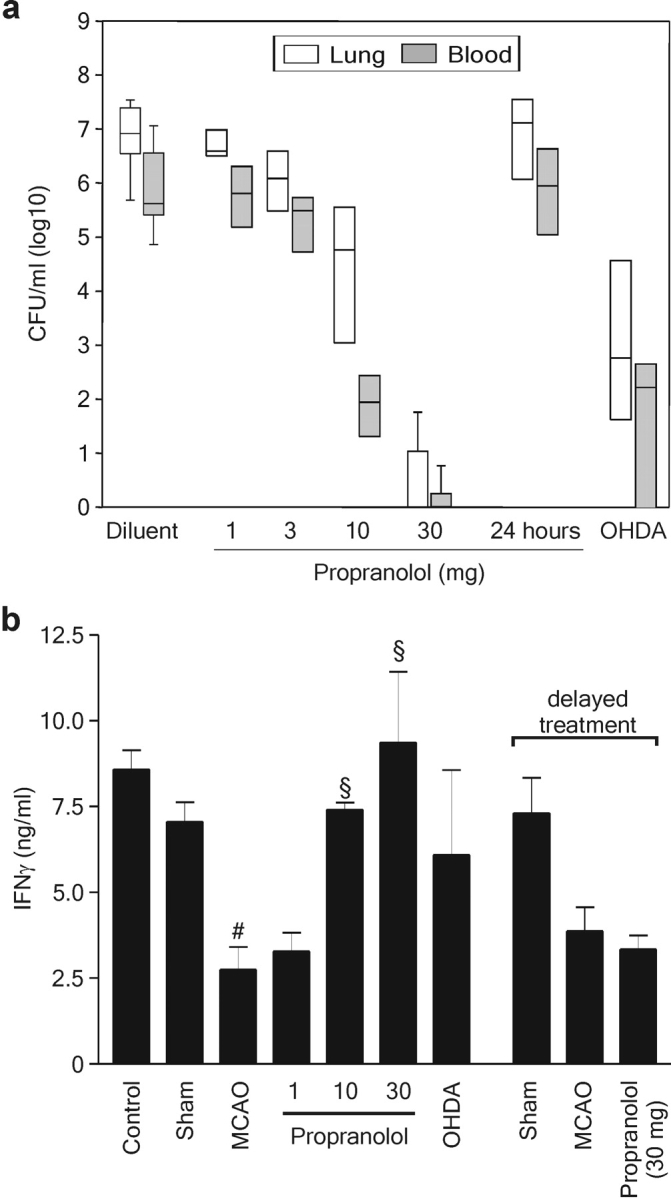
Prevention of bacterial infections and restoration of defective IFN-γ response by SNS inhibitors is dose and time dependent. (a) MCAO mice were treated with diluent or different doses of propranolol (3 × 1–3 × 30 mg/kg BW) commencing immediately after MCAO or 24 h (3 × 30 mg/kg BW) after MCAO, or with 6-OHDA before MCAO as described in Materials and Methods and as indicated (n = 4 in each group). Lungs and blood samples were collected for bacteriological analysis 72 h after MCAO. Data are shown as box plots in CFU/ml (log 10) blood or lung tissue homogenate. (b) In a different experiment, spleens of untreated mice (control), sham-, or MCAO-operated mice, which received diluent, propranolol, or 6-OHDA as described above, were collected 12 or 36 h (delayed propranolol treatment) after surgery. 106/ml spleen cells were stimulated ex vivo with Con A for 24 h and IFN-γ production was analyzed by ELISA, as described in Materials and Methods. Data are shown as mean ± SD. #, results differed from the sham-operated group; #, P < 0.05; §, results differed from the MCAO animals; §, P < 0.05. Mann-Whitney U test, n = 3–6 per group.
Inhibition of the SNS Improves Survival after Experimental Stroke.
Mortality in MCAO animals sharply increased from 13% on day 3 to 60% on day 7 after ischemia (n = 15). Because animals developed severe hypothermia between days 3 and 5 (<33°C measured rectally, not depicted) accompanied by bacteremia, sepsis was the likely cause of death. The early mortality in propranolol-treated (3 × 10 mg/kg BW) and diluent-treated animals equally amounted to ∼7-13% until day 3 after stroke. However, only an additional 7% of the propranolol-treated animals died, whereas 60% of mice treated with diluent were dead by day 7 (P = 0.02, diluent vs. propranolol; Fig. 6) .
Figure 6.

Blocking the SNS improves survival after experimental stroke. Mice underwent MCAO and received either diluent or 10 mg/kg BW immediately before, 4, and 8 h after MCAO (n = 15 per group). Differences in survival between propranolol-treated versus diluent-treated mice were tested for significance on day 11 after MCAO and are indicated by asterisks. *, P < 0.05. Fisher's exact test.
Adoptive Lymphocyte Transfer Prevents Bacterial Infections after Stroke.
Both RU486 and propranolol prevented most of the stroke-induced immunodepressive effects except the changes in the lymphocyte cytokine production, which was only normalized by β-adrenoreceptor blockade. To test whether bacterial infection in MCAO animals was caused by the (functional) loss of lymphocytes, we conducted a series of adoptive cell transfer experiments. 24 h after MCAO, mice received either unseparated splenocytes (5 × 106) or subset-depleted spleen cells from normal littermates. Bacterial counts in blood and lungs were determined 48 h later. Transfer of nonseparated spleen cells virtually abolished bacteremia and reduced pulmonary bacterial load by the order of 105 (Fig. 7 a). This effect was absent upon transfer of spleen cell suspensions depleted of all lymphocyte subsets (thus containing mostly monocytes/macrophages). Depletion of B cells only marginally affected the ability to reduce the bacterial load. In contrast, T cell–depleted splenocytes reduced bacteremia only by two orders of magnitude and failed to reduce pulmonary bacterial counts. In addition, NK cell–depleted splenocytes lowered the bacterial load significantly in the lung but not in peripheral blood (Fig. 7 a). To further investigate the role of specific T cell populations in the antibacterial defense, we performed adoptive transfer studies with spleen cells (5 × 106) from αβ T cell– or γδ T cell–deficient mice. Spleen cells from γδ T cell knockout mice were unable to prevent pulmonary infection, whereas they substantially reduced blood bacterial dissemination (Fig. 7 b). In contrast, spleen cells from αβ T cell knockout mice reduced the bacterial burden in the lung but hardly had any effect on bacteremia. These data indicate that T and NK lymphocytes are critical components of the acute response to bacterial infections after stroke. In particular, γδ T cells and αβ T cells are important for an effective host response to bacterial pneumonia and bacteremia, respectively.
Figure 7.
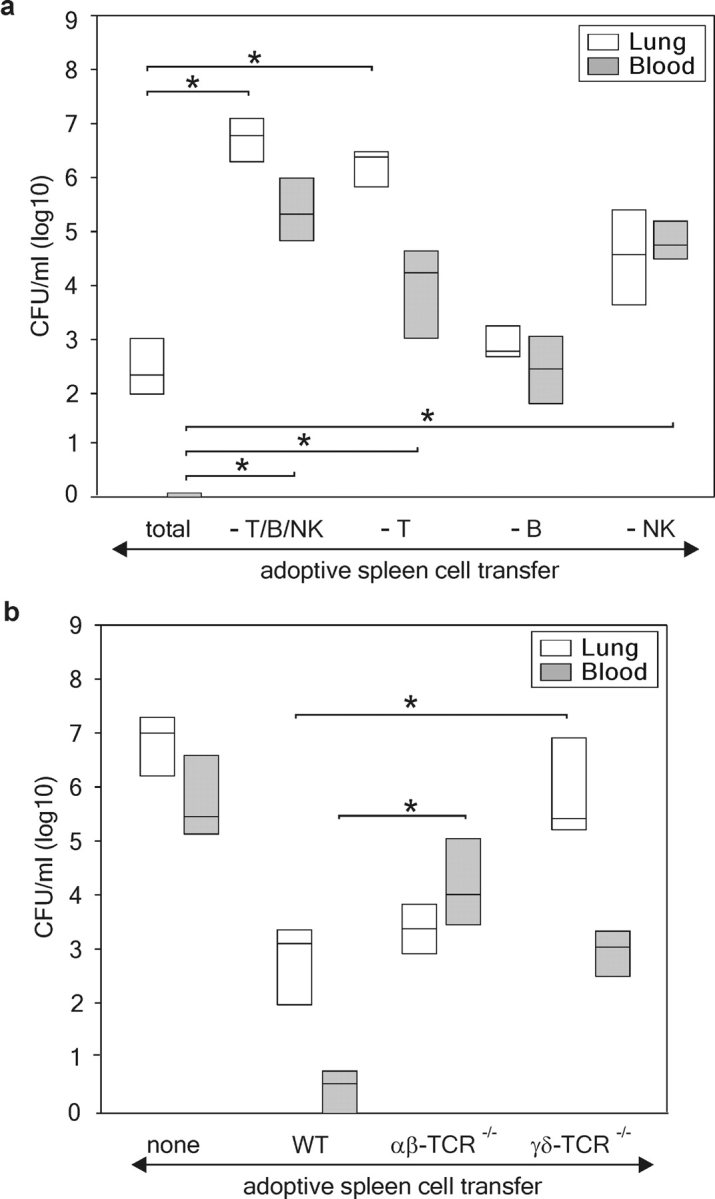
Adoptive lymphocyte transfer after experimental stroke prevents infection. (a) Adoptive lymphocyte transfer was performed 24 h after MCAO using either unseparated (total) or T, B, and NK cell, or T cell–, B cell–, or NK cell–depleted spleen cell suspensions from wild-type SV129/J mice. *, results differed from unseparated splenocyte transfer group. Kruskal-Wallis analysis of variance (ANOVA) after pairwise comparison with Dunn's method test, n = 4 per group. (b) In a different set of experiments, splenocytes from αβ T cell–deficient (αβ-TCR−/−), γδ T cell–deficient (γδ-TCR−/−), or wild-type B6 mice (WT) were transferred into B6 mice 24 h after MCAO. 48 h later, blood and lung samples were analyzed for bacterial counts. Data are shown as box plots in CFU/ml (log 10) blood or lung tissue homogenate. *, results differed from WT splenocyte transfer group. Kruskal-Wallis analysis of variance (ANOVA) after comparison of pairs with Dunn's method test, n = 8 per group.
IFN-γ Is Crucial in Controlling Bacterial Infections after Stroke.
IFN-γ is one of the key effector cytokines produced by NK and T cells that enhances microbicidal activity of macrophages and neutrophils. As shown above, blockade of catecholamines restored both IFN-γ production and bacterial clearance. Therefore, we examined whether the beneficial effect of lymphocyte transfer in stroke-induced infections is dependent on their ability to produce IFN-γ. Adoptive transfer of splenocytes from IFN-γ−/− mice failed to reduce pulmonary and blood bacterial counts in comparison to spleen cells from wild-type B6 mice (Fig. 8 a). Moreover, recombinant IFN-γ reduced bacterial dissemination in peripheral blood and lungs when administered 24 h, but not 48 h, after cerebral ischemia (Fig. 8 b).
Figure 8.

IFN-γ is essential in preventing the bacterial infections. (a) Adoptive lymphocyte transfer experiments 24 h after MCAO revealed an impaired ability of splenocytes from IFN-γ–deficient mice (IFN-γ−/−) to prevent bacterial infection in blood and lung when compared with splenocytes from wild-type B6 mice (n = 8 per group). (b) Administration of 2 μg recombinant IFN-γ 24 h, but not 48 h, after cerebral ischemia reduced the bacterial burden in peripheral blood and lungs as measured 72 h after MCAO (n = 4 per group). For data representation see Fig. 5.
Discussion
In this study, we provide for the first time experimental proof that a neuroendocrine-mediated systemic immunosuppression after acute central nervous system lesion results in the development of spontaneous systemic bacterial infections. Our study provides the long-sought explanation for a stroke-induced immunosuppressive state. Our findings may have general implications for the understanding of how severe stress and injury may lead to an increased susceptibility to bacterial infections. We demonstrate, in an experimental model, that cerebral ischemia induces long-lasting depression of the cell-mediated immunity (monocyte deactivation, lymphopenia, Th1/Th2 shift) associated with spontaneous bacteremia and pneumonia. We provide strong evidence that an impaired early NK and T cell response, in particular a reduced IFN-γ production, is the critical stroke-induced defect in the antibacterial defense. Thus, either adoptive transfer of IFN-γ–producing lymphocytes (i.e., NK and T, but not B cells) or early treatment with recombinant IFN-γ inhibited bacteremia and pneumonia. Importantly, the occurrence of life-threatening spontaneous systemic bacterial infections after experimental stroke and the defective IFN-γ response were only prevented by blocking the SNS, but not the HPA, suggesting that catecholamine-mediated lymphocyte dysfunction is the key factor in the impaired antibacterial immune response after stroke. We call this phenomenon stroke-induced immunodeficiency syndrome.
It has been long known that activation of the SNS and the HPA by proinflammatory cytokines in systemic inflammation results in the release of glucocorticoids and catecholamines, which in turn inhibit further production of proinflammatory mediators. More recently, another physiological feedback mechanism termed the “cholinergic antiinflammatory pathway” has been uncovered. Activation of the vagus nerve by inflammatory cytokines during endotoxemia was found to inhibit macrophage cytokine production through release of acetylcholine (24, 25). The rapid activation of these pathways in inflammatory conditions protects the organism against any adverse effects of an overwhelming immune response. However, an excessive activation of inhibitory neuroendocrine pathways without systemic inflammation can inappropriately suppress the immune system and increase the risk of infections. We have recently reported that the intrathecal release of proinflammatory cytokines is associated with signs of systemic immunodepression and a high incidence of infections in neurosurgical patients (21). Increased intrathecal levels of proinflammatory cytokines like IL-1β, IL-6, and TNF-α have been found in various brain disorders including trauma, subarachnoidal hemorrhage, and ischemia, which are commonly associated with HPA and SNS activation (22, 26–32). Interestingly, diminished ex vivo endotoxin-induced TNF-α production in rats, after intracerebroventricular TNF-α or IL-1β infusion, was prevented by hypophysectomy or β-adrenoreceptor blockade (33), suggesting that a local increase of proinflammatory cytokines in damaged brain tissue can trigger a systemic immunoinhibitory response via sympathoadrenal activation (13, 21).
In this study, we demonstrated that pharmacological inhibition of the activation of either SNS or HPA prevented the stroke-induced lymphocyte apoptosis, lymphopenia, and monocytic deactivation. However, poststroke lymphocyte dysfunction (i.e., decrease of IFN-γ and increase of IL-4 production) and bacterial infections were only prevented by SNS inhibitors (propranolol and 6-OHDA) and not by the glucocorticoid receptor blocker RU486. Catecholamines have been shown to suppress Th1 activities and cellular immune responses either directly, inhibiting the IFN-γ synthesis by Th1 cells, or indirectly, by inhibition of Th1-polarizing cytokines like IL-12 and TNF-α produced by antigen-presenting cells (34). Instead, catecholamines appear to promote Th2 cell differentiation and cytokine production (34), and trigger the secretion of IL-10 (a potent inhibitor of Th1 and monocyte functions) by monocytes (13). Because IFN-γ stimulates the microbicidal activity of phagocytes, whereas IL-4 inhibits phagocytosis, a decreased ratio of IFN-γ/IL-4 production indicates a compromised antibacterial defense (35). A decreased Th1/Th2 ratio has been found after major surgery or trauma and associated with lower survival and decreased resistance to infections after major burn injury (36–39). Our data on stress mediator blockade underline the importance of functional defects in IFN-γ production in the control of infectious complications after stroke. This conclusion is further supported by the adoptive transfer experiments: prevention of bacterial infections by spleen cell transfer depends not only on T and NK cells, but also on their ability to secret IFN-γ. Moreover, we show that early (24 h) but not late (48 h) administration of recombinant IFN-γ greatly reduced blood and pulmonary bacterial burden, suggesting that a rapid production of IFN-γ by lymphocytes is crucial in controlling bacterial infections. Our data support and extend recent findings in models of sepsis and pneumonia suggesting an important role for early lymphocyte responses in mounting an efficient antibacterial defense (40–42). In murine models of Gram-negative (Klebsiella pneumoniae) and Gram-positive (Nocardia asteroides) pneumonia, γδ T cells are critical for survival (43, 44). In stroke-induced infections, γδ T cells are essential for pulmonary bacterial clearance, whereas αβ T cells are more critical in the peripheral blood.
The results of our study may also have important implications for models of stroke as well as sepsis. The high mortality that we have observed in our study is well known in rodent models of stroke (45–47) and seems to be largely dependent on the duration of the MCAO as well as the strain of animals used (48–51). Previously, the high mortality was directly attributed to the central nervous system lesion (45, 46). Interestingly, the time course of the survival rate shows two phases. In the first phase (up to day 3) ∼13% of the mice die. The mortality in this phase is predominantly caused by the brain lesion directly. After day 3, mortality increased dramatically and reached ∼60% by day 7. The results of this study strongly suggest that this second phase is a consequence of stroke-induced immunodeficiency syndrome–associated severe infections and not directly due to the brain lesion because prevention of infection by antibiotic therapy (unpublished data) or by blockade of SNS (Fig. 6) improves survival dramatically without affecting infarct size (unpublished data). Although we have not found a systematic analysis of mortality in MCAO models in the literature, historic data also suggest a two phase time course of survival (45, 47, 51–54). Although infectious complications (in particular, bacterial pneumonia) and their relevance for mortality are well known in acute stroke (55, 56), these complications have not been reported previously in experimental stroke. To our knowledge, this is the first report of a model with stress-mediated immunodepression promoting spontaneous systemic bacterial infections. This model will be useful to further investigate the neuroimmunological mechanisms contributing to immunodepression and development of infections after trauma and stress, and to evaluate new therapeutical approaches to prevent or reverse immunodepression and its infectious complications.
In stroke patients, the occurrence of pneumonia is of particular clinical importance. Importantly, the 30-d mortality in patients who developed pneumonia is increased up to sixfold in comparison with stroke patients not suffering from pneumonia (55). Because, conservatively estimated, at least 10% of deaths occurring in stroke patients are attributable to pneumonia, further research to identify patients at increased risk for pneumonia and alternative approaches for prevention are needed (55). Additionally, the extent of infectious complications is proportional to the severity of the initial ischemic deficit (2–4, 55, 56). With respect to stroke size, our murine model is comparable with large infarctions in the human MCA region, which may explain the high incidence of infections in this model. Although pneumonia is thought to develop in stroke patients most often as a result of aspiration secondary to dysphagia, not all patients who aspirate develop infections (57, 58). In addition, up to a half of patients who develop pneumonia do not aspirate (59). Thus, although after severe strokes aspiration due to dysphagia is a known risk factor for pneumonia, other factors have been postulated that might predispose stroke patients to pneumonia, e.g., an impaired immune responsiveness (57, 58, 60). Our data strongly support this hypothesis and provide conclusive experimental evidence that aspiration alone is not sufficient for the development of bacteremia and pneumonia, for the following reasons. First, we demonstrated that bacterial infections can be prevented by propranolol, administration of INF-γ, as well as by adoptive lymphocyte transfer. These interventions, specifically the latter two, restore the immune function and are unlikely to prevent only aspiration. Second, the described changes in immune parameters preceded infections. Third, antibiotic treatment prevents infection but does not restore the altered immune profile (unpublished data). Rather, a defective antibacterial defense after cerebral ischemia is the main factor that determines the occurrence of systemic bacterial infections. However, we cannot exclude that aspiration is a prerequisite for pneumonia in stroke.
In conclusion, we have characterized for the first time a stroke-induced immunodeficiency that leads to severe bacterial infections, which could have important implications in improving therapy. Prevention of infections in patients with cerebral infarctions could reduce morbidity and mortality, as chest infections significantly affect the outcome in stroke patients (61, 62) and preventive antibiotic treatment reduces neurological deficit and mortality in this model of stroke by >60% (unpublished data). In addition to antimicrobial approaches, immunotherapeutic approaches, such as inhibition of lymphocyte apoptosis (e.g., by caspase inhibitors) or use of immunostimulatory drugs (e.g., IFN-γ, GM-CSF), may help to prevent or even reverse inappropriate immunodepression and decrease the risk of infectious complications after stroke. Considering the currently limited arsenal of stroke therapies, these new strands of treatment may significantly improve the clinical outcome.
Acknowledgments
We thank Claudia Muselmann and Rolf Eckert for excellent technical assistance and Anita Milicic for critically reviewing the manuscript.
This work was supported by the Hermann and Lilly Schilling Stiftung and the Deutsche Forschungsgemeinschaft.
Abbreviations used in this paper: 6-OHDA, 6-hydroxydopamine HBr; HE, hematoxylin/eosin; HPA, hypothalamic-pituitary-adrenal axis; MCA, middle cerebral artery; MCAO, middle cerebral artery occlusion; SNS, sympathetic nervous system; TUNEL, in situ terminal deoxynucleotide transferase-mediated dUTP nick-end labeling.
K. Prass and C. Meisel contributed equally to this work.
C. Meisel's present address is Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, OX3 7BN Oxford, United Kingdom.
References
- 1.Davenport, R.J., M.S. Dennis, I. Wellwood, and C.P. Warlow. 1996. Complications after acute stroke. Stroke. 27:415–420. [DOI] [PubMed] [Google Scholar]
- 2.Johnston, K.C., J.Y. Li, P.D. Lyden, S.K. Hanson, T.E. Feasby, R.J. Adams, R.E. Faught, Jr., and E.C. Haley, Jr. 1998. Medical and neurological complications of ischemic stroke: experience from the RANTTAS trial. RANTTAS investigators. Stroke. 29:447–453. [DOI] [PubMed] [Google Scholar]
- 3.Georgilis, K., A. Plomaritoglou, U. Dafni, Y. Bassiakos, and K. Vemmos. 1999. Aetiology of fever in patients with acute stroke. J. Intern. Med. 246:203–209. [DOI] [PubMed] [Google Scholar]
- 4.Grau, A.J., F. Buggle, P. Schnitzler, M. Spiel, C. Lichy, and W. Hacke. 1999. Fever and infection early after ischemic stroke. J. Neurol. Sci. 171:115–120. [DOI] [PubMed] [Google Scholar]
- 5.Langhorne, P., D.J. Stott, L. Robertson, J. MacDonald, L. Jones, C. McAlpine, F. Dick, G.S. Taylor, and G. Murray. 2000. Medical complications after stroke: a multicenter study. Stroke. 31:1223–1229. [DOI] [PubMed] [Google Scholar]
- 6.Kalra, L., A. Evans, I. Perez, M. Knapp, N. Donaldson, and C.G. Swift. 2000. Alternative strategies for stroke care: a prospective randomised controlled study of stroke unit, stroke team, and domiciliary management of stroke. Lancet. 356:894–899. [DOI] [PubMed] [Google Scholar]
- 7.Henon, H., O. Godefroy, D. Leys, F. Mounier-Vehier, C. Lucas, P. Rondepierre, A. Duhamel, and J.P. Pruvo. 1995. Early predictors of death and disability after acute cerebral ischemic event. Stroke. 26:392–398. [DOI] [PubMed] [Google Scholar]
- 8.Kalra, L., G. Yu, K. Wilson, and P. Roots. 1995. Medical complications during stroke rehabilitation. Stroke. 26:990–994. [DOI] [PubMed] [Google Scholar]
- 9.Viitanen, M., B. Winblad, and K. Asplund. 1987. Autopsy-verified causes of death after stroke. Acta Med. Scand. 222:401–408. [DOI] [PubMed] [Google Scholar]
- 10.Ronning, O.M., and B. Guldvog. 1998. Stroke unit versus general medical wards, II: neurological deficits and activities of daily living: a quasi-randomized controlled trial. Stroke. 29:586–590. [DOI] [PubMed] [Google Scholar]
- 11.Livingston, D.H., S.H. Appel, S.R. Wellhausen, G. Sonnenfeld, and H.C. Polk, Jr. 1988. Depressed interferon gamma production and monocyte HLA-DR expression after severe injury. Arch. Surg. 123:1309–1312. [DOI] [PubMed] [Google Scholar]
- 12.Docke, W.D., F. Randow, U. Syrbe, D. Krausch, K. Asadullah, P. Reinke, H.D. Volk, and W. Kox. 1997. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat. Med. 3:678–681. [DOI] [PubMed] [Google Scholar]
- 13.Woiciechowsky, C., K. Asadullah, D. Nestler, B. Eberhardt, C. Platzer, B. Schoning, F. Glockner, W.R. Lanksch, H.D. Volk, and W.D. Docke. 1998. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nat. Med. 4: 808–813. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez, J.L., K.J. Gibbons, L.G. Bitzer, R.E. Dechert, S.M. Steinberg, and L.M. Flint. 1991. Pneumonia: incidence, risk factors, and outcome in injured patients. J. Trauma. 31:907–912. [PubMed] [Google Scholar]
- 15.Hsieh, A.H., M.J. Bishop, P.S. Kubilis, D.W. Newell, and D.J. Pierson. 1992. Pneumonia following closed head injury. Am. Rev. Respir. Dis. 146:290–294. [DOI] [PubMed] [Google Scholar]
- 16.Howard, R.J., and R.L. Simmons. 1974. Acquired immunologic deficiencies after trauma and surgical procedures. Surg. Gynecol. Obstet. 139:771–782. [PubMed] [Google Scholar]
- 17.Hara, H., P.L. Huang, N. Panahian, M.C. Fishman, and M.A. Moskowitz. 1996. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J. Cereb. Blood Flow Metab. 16:605–611. [DOI] [PubMed] [Google Scholar]
- 18.Prass, K., K. Ruscher, M. Karsch, N. Isaev, D. Megow, J. Priller, A. Scharff, U. Dirnagl, and A. Meisel. 2002. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J. Cereb. Blood Flow Metab. 22:520–525. [DOI] [PubMed] [Google Scholar]
- 19.Wang, Q., P. Teder, N.P. Judd, P.W. Noble, and C.M. Doerschuk. 2002. CD44 deficiency leads to enhanced neutrophil migration and lung injury in Escherichia coli pneumonia in mice. Am. J. Pathol. 161:2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matute-Bello, G., C.W. Frevert, W.C. Liles, M. Nakamura, J.T. Ruzinski, K. Ballman, V.A. Wong, C. Vathanaprida, and T.R. Martin. 2001. Fas/Fas ligand system mediates epithelial injury, but not pulmonary host defenses, in response to inhaled bacteria. Infect. Immun. 69:5768–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asadullah, K., C. Woiciechowsky, W.D. Docke, C. Liebenthal, H. Wauer, W. Kox, H.D. Volk, S. Vogel, and R. von Baehr. 1995. Immunodepression following neurosurgical procedures. Crit. Care Med. 23:1976–1983. [DOI] [PubMed] [Google Scholar]
- 22.Woiciechowsky, C., B. Schoning, W.R. Lanksch, H.D. Volk, and W.D. Docke. 1999. Mechanisms of brain-mediated systemic anti-inflammatory syndrome causing immunodepression. J. Mol. Med. 77:769–780. [DOI] [PubMed] [Google Scholar]
- 23.Cao, L., N.M. Filipov, and D.A. Lawrence. 2002. Sympathetic nervous system plays a major role in acute cold/restraint stress inhibition of host resistance to Listeria monocytogenes. J. Neuroimmunol. 125:94–102. [DOI] [PubMed] [Google Scholar]
- 24.Bernik, T.R., S.G. Friedman, M. Ochani, R. DiRaimo, L. Ulloa, H. Yang, S. Sudan, C.J. Czura, S.M. Ivanova, and K.J. Tracey. 2002. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J. Exp. Med. 195:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tracey, K.J. 2002. The inflammatory reflex. Nature. 420:853–859. [DOI] [PubMed] [Google Scholar]
- 26.Feuerstein, G.Z., T. Liu, and F.C. Barone. 1994. Cytokines, inflammation, and brain injury: role of tumor necrosis factor-alpha. Cerebrovasc. Brain Metab. Rev. 6:341–360. [PubMed] [Google Scholar]
- 27.Tarkowski, E., L. Rosengren, C. Blomstrand, C. Wikkelso, C. Jensen, S. Ekholm, and A. Tarkowski. 1997. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin. Exp. Immunol. 110:492–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.del Zoppo, G., I. Ginis, J.M. Hallenbeck, C. Iadecola, X. Wang, and G.Z. Feuerstein. 2000. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 10:95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fassbender, K., R. Schmidt, R. Mossner, M. Daffertshofer, and M. Hennerici. 1994. Pattern of activation of the hypothalamic-pituitary-adrenal axis in acute stroke. Relation to acute confusional state, extent of brain damage, and clinical outcome. Stroke. 25:1105–1108. [DOI] [PubMed] [Google Scholar]
- 30.Cruse, J.M., R.E. Lewis, Jr., G.R. Bishop, W.F. Kliesch, E. Gaitan, and R. Britt. 1993. Decreased immune reactivity and neuroendocrine alterations related to chronic stress in spinal cord injury and stroke patients. Pathobiology. 61:183–192. [DOI] [PubMed] [Google Scholar]
- 31.Sander, D., K. Winbeck, J. Klingelhofer, T. Etgen, and B. Conrad. 2001. Prognostic relevance of pathological sympathetic activation after acute thromboembolic stroke. Neurology. 57:833–838. [DOI] [PubMed] [Google Scholar]
- 32.Myers, M.G., J.W. Norris, V.C. Hachniski, and M.J. Sole. 1981. Plasma norepinephrine in stroke. Stroke. 12:200–204. [DOI] [PubMed] [Google Scholar]
- 33.Woiciechowsky, C., B. Schoning, N. Daberkow, K. Asche, G. Stoltenburg, W.R. Lanksch, and H.D. Volk. 1999. Brain-IL-1beta induces local inflammation but systemic anti-inflammatory response through stimulation of both hypothalamic-pituitary-adrenal axis and sympathetic nervous system. Brain Res. 816:563–571. [DOI] [PubMed] [Google Scholar]
- 34.Elenkov, I.J., R.L. Wilder, G.P. Chrousos, and E.S. Vizi. 2000. The sympathetic nerve-an integrative interface between two supersystems: the brain and the immune system. Pharmacol. Rev. 52:595–638. [PubMed] [Google Scholar]
- 35.Abbas, A.K., A.H. Lichtman, and J.S. Pober. 2000. Cellular and Molecular Immunology. Fourth edition. W.B. Saunders, Philadelphia. 235 pp.
- 36.O'Sullivan, S.T., J.A. Lederer, A.F. Horgan, D.H. Chin, J.A. Mannick, and M.L. Rodrick. 1995. Major injury leads to predominance of the T helper-2 lymphocyte phenotype and diminished interleukin-12 production associated with decreased resistance to infection. Ann. Surg. 222:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Decker, D., M. Schondorf, F. Bidlingmaier, A. Hirner, and A.A. von Ruecker. 1996. Surgical stress induces a shift in the type-1/type-2 T-helper cell balance, suggesting down-regulation of cell-mediated and up-regulation of antibody-mediated immunity commensurate to the trauma. Surgery. 119:316–325. [DOI] [PubMed] [Google Scholar]
- 38.Mack, V.E., M.D. McCarter, H.A. Naama, S.E. Calvano, and J.M. Daly. 1996. Dominance of T-helper 2-type cytokines after severe injury. Arch. Surg. 131:1303–1308. [DOI] [PubMed] [Google Scholar]
- 39.Zedler, S., R.C. Bone, A.E. Baue, G.H. von Donnersmarck, and E. Faist. 1999. T-cell reactivity and its predictive role in immunosuppression after burns. Crit. Care Med. 27:66–72. [DOI] [PubMed] [Google Scholar]
- 40.Hotchkiss, R.S., K.W. Tinsley, P.E. Swanson, K.C. Chang, J.P. Cobb, T.G. Buchman, S.J. Korsmeyer, and I.E. Karl. 1999. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc. Natl. Acad. Sci. USA. 96:14541–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hotchkiss, R.S., K.C. Chang, P.E. Swanson, K.W. Tinsley, J.J. Hui, P. Klender, S. Xanthoudakis, S. Roy, C. Black, E. Grimm, et al. 2000. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat. Immunol. 1:496–501. [DOI] [PubMed] [Google Scholar]
- 42.Rubins, J.B., and C. Pomeroy. 1997. Role of gamma interferon in the pathogenesis of bacteremic pneumococcal pneumonia. Infect. Immun. 65:2975–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King, D.P., D.M. Hyde, K.A. Jackson, D.M. Novosad, T.N. Ellis, L. Putney, M.Y. Stovall, L.S. Van Winkle, B.L. Beaman, and D.A. Ferrick. 1999. Cutting edge: protective response to pulmonary injury requires gamma delta T lymphocytes. J. Immunol. 162:5033–5036. [PubMed] [Google Scholar]
- 44.Moore, T.A., B.B. Moore, M.W. Newstead, and T.J. Standiford. 2000. Gamma delta-T cells are critical for survival and early proinflammatory cytokine gene expression during murine Klebsiella pneumonia. J. Immunol. 165:2643–2650. [DOI] [PubMed] [Google Scholar]
- 45.Gendron, A., J. Teitelbaum, C. Cossette, S. Nuara, M. Dumont, D. Geadah, P. du Souich, and E. Kouassi. 2002. Temporal effects of left versus right middle cerebral artery occlusion on spleen lymphocyte subsets and mitogenic response in Wistar rats. Brain Res. 955:85–97. [DOI] [PubMed] [Google Scholar]
- 46.Hatcher, J.P., D. Virley, S.J. Hadingham, J. Roberts, A.J. Hunter, and A.A. Parsons. 2002. The behavioural effect of middle cerebral artery occlusion on apolipoprotein-E deficient mice. Behav. Brain Res. 131:139–149. [DOI] [PubMed] [Google Scholar]
- 47.Wang, R.Y., P.S. Wang, and Y.R. Yang. 2003. Effect of age in rats following middle cerebral artery occlusion. Gerontology. 49:27–32. [DOI] [PubMed] [Google Scholar]
- 48.Neumann-Haefelin, T., A. Kastrup, A. de Crespigny, M.A. Yenari, T. Ringer, G.H. Sun, and M.E. Moseley. 2000. Serial MRI after transient focal cerebral ischemia in rats: dynamics of tissue injury, blood-brain barrier damage, and edema formation. Stroke. 31:1965–1972. [DOI] [PubMed] [Google Scholar]
- 49.Aspey, B.S., F.L. Taylor, M. Terruli, and M.J. Harrison. 2000. Temporary middle cerebral artery occlusion in the rat: consistent protocol for a model of stroke and reperfusion. Neuropathol. Appl. Neurobiol. 26:232–242. [DOI] [PubMed] [Google Scholar]
- 50.Huang, J., D.B. Agus, C.J. Winfree, S. Kiss, W.J. Mack, R.A. McTaggart, T.F. Choudhri, L.J. Kim, J. Mocco, D.J. Pinsky, et al. 2001. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc. Natl. Acad. Sci. USA. 98:11720–11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maier, C.M., G.H. Sun, D. Kunis, M.A. Yenari, and G.K. Steinberg. 2001. Delayed induction and long-term effects of mild hypothermia in a focal model of transient cerebral ischemia: neurological outcome and infarct size. J. Neurosurg. 94:90–96. [DOI] [PubMed] [Google Scholar]
- 52.Kondo, T., A.G. Reaume, T.T. Huang, E. Carlson, K. Murakami, S.F. Chen, E.K. Hoffman, R.W. Scott, C.J. Epstein, and P.H. Chan. 1997. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J. Neurosci. 17:4180–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schabitz, W.R., F. Li, K. Irie, B.W. Sandage, Jr., K.W. Locke, and M. Fisher. 1999. Synergistic effects of a combination of low-dose basic fibroblast growth factor and citicoline after temporary experimental focal ischemia. Stroke. 30:427–431. [DOI] [PubMed] [Google Scholar]
- 54.Virley, D., J.S. Beech, S.C. Smart, S.C. Williams, H. Hodges, and A.J. Hunter. 2000. A temporal MRI assessment of neuropathology after transient middle cerebral artery occlusion in the rat: correlations with behavior. J. Cereb. Blood Flow Metab. 20:563–582. [DOI] [PubMed] [Google Scholar]
- 55.Katzan, I.L., R.D. Cebul, S.H. Husak, N.V. Dawson, and D.W. Baker. 2003. The effect of pneumonia on mortality among patients hospitalized for acute stroke. Neurology. 60:620–625. [DOI] [PubMed] [Google Scholar]
- 56.Hilker, R., C. Poetter, N. Findeisen, J. Sobesky, A. Jacobs, M. Neveling, and W.D. Heiss. 2003. Nosocomial pneumonia after acute stroke: implications for neurological intensive care medicine. Stroke. 34:975–981. [DOI] [PubMed] [Google Scholar]
- 57.Perry, L., and C.P. Love. 2001. Screening for dysphagia and aspiration in acute stroke: a systematic review. Dysphagia. 16:7–18. [DOI] [PubMed] [Google Scholar]
- 58.Finegold, S.M. 1991. Aspiration pneumonia. Rev. Infect. Dis. 13:S737–S742. [DOI] [PubMed] [Google Scholar]
- 59.Riquelme, R., A. Torres, M. El-Ebiary, J.P. de la Bellacasa, R. Estruch, J. Mensa, J. Fernandez-Sola, C. Hernandez, and R. Rodriguez-Roisin. 1996. Community-acquired pneumonia in the elderly: a multivariate analysis of risk and prognostic factors. Am. J. Respir. Crit. Care Med. 154:1450–1455. [DOI] [PubMed] [Google Scholar]
- 60.Marik, P.E. 2001. Aspiration pneumonitis and aspiration pneumonia. N. Engl. J. Med. 344:665–671. [DOI] [PubMed] [Google Scholar]
- 61.Evans, A., I. Perez, F. Harraf, A. Melbourn, J. Steadman, N. Donaldson, and L. Kalra. 2001. Can differences in management processes explain different outcomes between stroke unit and stroke-team care? Lancet. 358:1586–1592. [DOI] [PubMed] [Google Scholar]
- 62.Hajat, C., S. Hajat, and P. Sharma. 2000. Effects of poststroke pyrexia on stroke outcome: a meta-analysis of studies in patients. Stroke. 31:410–414. [DOI] [PubMed] [Google Scholar]