

Abstract
T cell–specific adaptor protein (TSAd) is a T lineage–restricted signaling adaptor molecule that is thought to participate in the assembly of intracellular signaling complexes in T cells. Previous studies of TSAd-deficient mice have revealed a role for TSAd in the induction of T cell interleukin 2 secretion and proliferation. We now show that TSAd-deficient mice are susceptible to lupus-like autoimmune disease. On the nonautoimmune-prone C57BL/6 genetic background, TSAd deficiency results in hypergammaglobulinemia that affects all immunoglobulin (Ig)G subclasses. Older C57BL/6 TSAd-deficient mice (1 yr of age) accumulate large numbers of activated T and B cells in spleen, produce autoantibodies against a variety of self-targets including single stranded (ss) and double stranded (ds) DNA, and, in addition, develop glomerulonephritis. We further show that immunization of younger C57BL/6 TSAd-deficient mice (at age 2 mo) with pristane, a recognized nonspecific inflammatory trigger of lupus, results in more severe glomerulonephritis compared with C57BL/6 controls and the production of high titer ss and ds DNA antibodies of the IgG subclass that are not normally produced by C57BL/6 mice in this model. The development of autoimmunity in TSAd-deficient mice is associated with defective T cell death in vivo. These findings illustrate the role of TSAd as a critical regulator of T cell death whose absence promotes systemic autoimmunity.
Keywords: apoptosis, autoimmunity, T lymphocyte, signal transduction, glomerulonephritis
Introduction
T cell–specific adapter protein (TSAd) is an SH2 domain–containing intracellular adaptor-like molecule that is expressed in thymocytes and activated mature T cells (1, 2). The exact role played by TSAd in T cell signal transduction is unknown. TSAd has been described to interact physically with the Src family tyrosine kinase, Lck, the Tec family tyrosine kinases, Rlk and Itk, and the MAP3 kinase, MEKK2 (2–4). However, although these interactions can be readily detected in yeast cells, it has been more difficult to demonstrate binding in mammalian cells, particularly between cell-endogenous proteins. Therefore, the significance of interaction with these kinases is unclear. In fact, recent studies have indicated that TSAd may function predominantly in the nucleus where it acts to positively regulate the transcription of different T cell target genes, including the gene for the T cell autocrine growth factor, IL-2 (5). This would be consistent with the finding that splenic T cells from TSAd-deficient mice secrete less IL-2 and proliferate less upon TCR engagement in vitro (3).
In this study, we have further examined the immunological function of TSAd-deficient mice. We found that TSAd-deficient mice are susceptible to spontaneous and induced lupus-like autoimmune disease. In addition, we found that T cells from TSAd-deficient mice are resistant to antigen-induced cell death in vivo, a process that is thought to prevent the development of autoimmunity in healthy animals. These findings highlight the role of TSAd as a novel regulator of T cell death and lupus-like autoimmunity.
Materials and Methods
Mice.
C57BL/6 TSAd-deficient mice were obtained from J. Bluestone (University of California at San Francisco, San Francisco, CA) and were derived by backcrossing 129/Sv × C57BL/6 TSAd-deficient mice (3) with C57BL/6 mice for eight generations. In these studies, we produced ninth generation C57BL/6 TSAd heterozygotes that were then intercrossed to generate littermate C57BL/6 wild-type, heterozygote, and TSAd-deficient mice for use in experiments. Most experiments were performed with these littermate mice. For some experiments, C57BL/6 wild-type and TSAd-deficient mice were generated from separate matings in our facilities. Additional C57BL/6 mice and control MRL/lpr mice were purchased from Taconic Farms and The Jackson Laboratory, respectively. Mice were housed and bred under standard conditions. Serial serum samples taken from sentinel mice in the colony as well as from the test mice directly were consistently negative for common murine viral pathogens. All experiments were performed in compliance with Hospital for Special Surgery and University of Michigan guidelines and were approved by the respective institutional animal care and use committees.
Pristane Administration.
C57BL/6 wild-type and TSAd-deficient mice were given a single i.p. injection of 0.5 ml Pristane (Sigma-Aldrich) at 2 mo of age. After 6 mo, the presence of autoantibodies in sera was determined and the mice were killed for histopathological analysis.
Serology.
Serum total IgG, IgG subclass, and IgM concentrations were determined by ELISA using an SBA Clonotyping™ System (Southern Biotechnology Associates, Inc.). Anti-cardiolipin antibodies were detected with the use of an anti-cardiolipin ELISA kit (Sigma-Aldrich). Anti–double stranded (ds) DNA antibodies and rheumatoid factor were detected using mouse anti–ds DNA and rheumatoid factor ELISA kits (Alpha Diagnostic International). Antibodies against single stranded (ss) DNA were detected by ELISA as previously described (6). The secondary detection reagents used in autoantibody ELISAs were goat anti–mouse IgG (GAMIgG) or IgM coupled to alkaline phosphatase (Sigma-Aldrich). To determine levels of nonspecific binding associated with known IgG or IgM concentrations in test samples, serial dilutions of normal mouse IgG or IgM were also assayed in autoantibody ELISA experiments. Specific autoantibody binding was thus determined by subtracting from the test sample OD value, the OD value obtained with the same concentration of normal IgG or IgM calculated from a standard curve. In these experiments, test samples were scored positive if their specific OD value was greater than an arbitrary 0.25 units.
To detect anti-nuclear antibodies and confirm the presence of anti-ds DNA specificities, serum samples (1:100 dilution) were incubated with fixed and permeabilized HEp-2 cells (Bion Enterprises, Ltd.) or Crithidia luciliae organisms (Wampole Laboratories), respectively. Slides were washed and then incubated with GAMIgG coupled to Alexa 488 (Molecular Probes). After further washing and mounting, cells were viewed on a Nikon Eclipse E600 fluorescent microscope.
Western Blotting.
Western blotting experiments were performed as previously described using nuclear lysates prepared from Jurkat T leukemia cells (5). Mouse sera were used at a dilution of 1:1,000 and the secondary detection reagent was GAMIgG coupled to horseradish peroxidase (Santa Cruz Biotechnology, Inc.).
Pathology.
Different organs from C57BL/6 wild-type, TSAd heterozygote, and TSAd-deficient mice were fixed in formalin and embedded in paraffin for histological studies. Sections were stained with hematoxylin and eosin (H&E) and additionally periodic acid-Schiff and hematoxylin (for kidneys only). For immunohistochemistry, kidneys were quick frozen in OCT compound. Kidney sections were first blocked by incubation in 10% goat serum followed by incubation with GAMIgG Alexa 488 to detect IgG deposited in glomeruli. Sections were viewed on a Nikon Eclipse E600 microscope.
Staphylococcal enterotoxin B (SEB) Immunization.
Mice were immunized i.p. with 50 μg SEB (Sigma-Aldrich). For T cell death experiments, immediately before and at d 4 and 11 after SEB immunization, 300–400 μl venous blood was collected from mice by orbital bleeding and the percentage of TCR Vβ6+ or TCR Vβ8+ T cells was determined (see below). To examine T cell activation marker and cytokine expression, flow cytometric analyses were performed upon splenic T cells 24 h after SEB administration (see below).
Flow Cytometry.
The following labeled monoclonal antibodies were used for flow cytometry: H57-597-CyChrome (TCRβ chain), RA3-6B2-PE (CD45R/B220), H1.2F3-PE (CD69), Jo2-FITC (CD95/Fas), MEL-14-FITC (CD62L), IM7-PE (CD44), RR4-7-PE and FITC (TCR Vβ6), MR5-2-PE and FITC (TCR Vβ8), GK1.5-FITC (CD4), JES6-5H4-PE (IL-2), XMG1.2-PE (IFN-γ), A95-1-PE (rat IgG2b isotype control for IL-2), and R3-34-PE (rat IgG1 isotype control for IFN-γ). All antibodies were from BD Biosciences with the exception of XMG1.2-PE, which was from Caltag. To enumerate percentages of TCRαβ1 T cells and B cells in splenocyte populations, samples were double stained with TCRβ and B220 antibodies. Expression of activation markers (CD69, CD44, CD95, and CD62L) upon splenic TCRαβ1 T cells from older mice was determined by double staining of splenocytes with a TCRβ antibody together with the appropriate activation marker antibodies. The percentage of TCR Vβ6+ or TCR Vβ8+ cells among CD4+ T cells in peripheral blood of SEB-immunized mice was assessed by double staining with PE-labeled TCR antibodies and CD4-FITC. Likewise, the percentage of TCR Vβ6+ or TCR Vβ8+ T cells that express CD69, IL-2, and IFN-γ in spleens of SEB-immunized mice was determined by double staining using FITC-labeled TCR antibodies. For IL-2 and IFN-γ analyses, harvested splenocytes were first restimulated with 50 ng/ml PMA and 500 ng/ml ionomycin for 4 h and GolgiStop (BD Biosciences) was added for the last 2 h of culture. Cells were then fixed and permeabilized with Cytofix/CytoPerm reagent (BD Biosciences) before staining.
Gene Profiling.
CD4+ T cells were purified from eight pooled C57BL/6 wild-type and eight pooled C57BL/6 TSAd-deficient spleens (from mice aged 2 mo) using mouse CD4 Dynabeads and DETACHaBEAD mouse CD4 reagent (Dynal) according to the manufacturer's instructions. As determined by flow cytometry, CD4+ T cells were >98% pure and were not activated as a result of the isolation procedure. CD4+ T cells were not stimulated or were stimulated by culture for 20 h in the wells of 12-well tissue culture plates that had been precoated with 2 μg/ml of the 145-2C11 anti-CD3 antibody (BD Biosciences). Cells were plated at a density of 2.5 × 106/ml and the total well volume was 2 ml. Soluble 37.51 anti-CD28 antibody was included in the culture medium at 2 μg/ml. Total RNA was prepared from unstimulated and stimulated CD4+ T cells using RNeasy Mini Kits (QIAGEN) and T7-dT primed ds cDNA was synthesized using a SuperScript Choice System (Invitrogen). Biotinylated cRNA was prepared with the use of T7 BioArray High Yield RNA Transcript Labeling Kit (Enzo Diagnostics). 15 μg fragmented cRNA was incorporated into cocktails that were hybridized with U74Av2 arrays (Affymetrix, Inc.) containing probe sets representing ∼12,000 distinct murine genes. All hybridization, washing, and staining procedures were performed according to the manufacturer's instructions.
Data were analyzed with the use of Affymetrix Suite (Affymetrix, Inc.) and GeneSpring (Silicon Genetics) software. To assist in the identification of genes that were positively or negatively regulated by TSAd, we applied several restrictions to data. First, because TSAd is up-regulated upon T cell activation, we selected genes that were increased (positive regulation) or decreased (negative regulation) at least threefold in wild-type T cells in response to CD3/CD28. Second, because TSAd would be expected to exert a greater influence upon gene expression in stimulated versus unstimulated T cells (based on increased expression) we selected genes in which the ratio of gene expression between stimulated wild-type and knockout T cells was greater than (positive regulation) or less than (negative regulation) the ratio of gene expression between unstimulated wild-type and knockout T cells by at least 1.5-fold. Third, because TSAd would be predicted to influence gene expression at least in stimulated T cells, we only considered genes that were decreased (positive regulation) or increased (negative regulation) by a factor of 1.5-fold in stimulated knockout T cells relative to stimulated wild-type T cells. Fourth, so as to increase the reliability of results, we considered only those genes that were assigned as present (P) by Affymetrix in stimulated wild-type T cells (for positively regulated genes) or in stimulated knockout T cells (for negatively regulated genes).
Online Supplemental Material.
Fig. S1, which shows total serum Ig levels by mouse gender, and Tables S1–S8, which depict raw data values of TSAd positively regulated genes in microarray experiments, are available at http://www.jem.org/cgi/content/full/jem.20021358/DC1.
Results
Hypergammaglobulinemia in TSAd-deficient Mice.
As part of an effort to examine in more detail the immunological characteristics of TSAd-deficient mice, we assayed serum Ig concentrations. Altered serum Ig concentrations could indicate a role for TSAd in the generation of appropriate T cell help for B cell antibody responses. Serum IgG and IgM levels in C57BL/6 TSAd-deficient and littermate control wild-type and TSAd heterozygote mice were determined by ELISA (Fig. 1) . In comparison with wild-type mice, TSAd-deficient mice showed elevated levels of serum IgG (up to threefold) at all ages examined starting from 2 mo. In addition, IgM levels in TSAd-deficient mice were elevated (up to twofold) starting at 3–4 mo of age. In contrast, heterozygote TSAd mice, showed only small increases in serum IgG and essentially no increases in IgM compared with wild-type mice. Hypergammaglobulinemia in TSAd-deficient mice was independent of mouse gender (see Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20021358/DC1). All subclasses of IgG were elevated in TSAd-deficient mice suggesting a lack of bias toward either Th1 or Th2 differentiation programs in these animals (Fig. 1).
Figure 1.

Serum Ig levels in TSAd-deficient mice. Concentrations of total IgG, IgM, and different IgG subclasses in sera from TSAd-deficient (−/−) and littermate control wild-type (+/+) and TSAd heterozygote (+/−) mice at the indicated ages were determined by ELISA (IgG subclass analyses were performed upon 3–4-mo-old mice). Data are represented as mean plus 1 SE. Except for IgM at 2 mo, all increases in Ig concentration in TSAd-deficient mice (relative to wild-type mice) are statistically significant (all P < 0.05 using Student's two-sample t test). Increases in Ig concentrations seen in TSAd heterozygotes (relative to wild-type) are statistically significant only for IgG at 2 and 3–4 mo. Within each genotype and age group category mixed numbers of males and females were analyzed (total numbers are shown in parentheses above bars). Increases in Ig concentrations seen in TSAd-deficient mice were independent of gender (see Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20021358/DC1).
TSAd-deficient Mice Develop Antinuclear Antibodies (ANA).
Hypergammaglobulinemia is encountered commonly in mouse models of lupus-like systemic autoimmune disease (7, 8). One such classic model of lupus-like autoimmunity is the MRL/lpr mouse (8). MRL/lpr mice develop a severe lupus-like disease with 50% mortality by 6 mo of age. Hypergammaglobulinemia in TSAd-deficient mice was not as pronounced as in MRL/lpr mice (of 12 examined MRL/lpr mice aged 3–6 mo, mean IgG levels were 4.62 mg/ml with a SE of 0.71 mg/ml) but was comparable to that seen in other mouse lupus models such as NZB/W and BXSB mice (7). Therefore, to determine if TSAd-deficient mice also succumb to systemic autoimmunity, we first assayed sera for the presence of ANA. ANA were detected by immunofluorescent staining of permeabilized HEp-2 epithelial cells (Fig. 2, A and B) . At all ages examined, from 2 up to 12–14 mo, the percentage of sera that were ANA+ was significantly higher in TSAd-deficient mice than in littermate control wild-type and TSAd heterozygote mice (Fig. 2 A). At 12–14 mo, 11 out of 16 (69%) TSAd-deficient mice showed ANA reactivity compared with 11 out of 12 (92%) MRL/lpr mice at 3–6 mo of age. There were no obvious sex differences in the propensity of TSAd-deficient mice to develop ANA. A variety of different patterns of antinuclear reactivity were observed in TSAd-deficient mice, each of which has been noted previously in human SLE (9). These included homogenous nuclear reactivity with or without cytoplasmic dots, nuclear rim staining, and nucleolar staining, again, with or without cytoplasmic dots (Fig. 2 B). These findings are consistent with reactivity against nuclear and ribosomal proteins and/or nucleic acids. To confirm that sera from TSAd-deficient mice contained antinuclear protein specificities, we examined sera for their reactivity with nuclear lysates prepared from T leukemia cells in Western blotting experiments (Fig. 2 C). When compared with sera from control wild-type mice, sera from older ANA+ TSAd-deficient mice were seen to react strongly and heterogeneously with a number of different nuclear proteins.
Figure 2.

ANA in sera from TSAd-deficient mice. (A) The presence of ANA antibodies in sera from TSAd-deficient and littermate control wild-type and TSAd heterozygote mice was determined by immunofluorescent staining of HEp-2 epithelial cells. Shown is the percentage of ANA+ sera for each genotype at the indicated ages (total numbers of mice analyzed are shown in parentheses). Differences between TSAd-deficient and wild-type control mice are statistically significant in all age groups (all P < 0.05 calculated using the chi-squared test of independence). nd, not determined. (B) Different patterns of nuclear staining of HEp-2 epithelial cells observed with TSAd-deficient sera. Clockwise from top left: homogenous nuclear, rim, homogenous nuclear with cytoplasmic dots, and nucleolar with cytoplasmic dots. (C) Reactivity of sera from four 7–9-mo-old ANA+ TSAd-deficient mice and age-matched control wild-type (wt) mice with proteins in nuclear lysates of T leukemia cells as detected by Western blotting. The wild-type serum was pooled from three different wild-type mice.
Autoantibody Specificities in TSAd-deficient Mice.
Antibodies against ss DNA, ds DNA, cardiolipin, and IgG (rheumatoid factor) are found commonly in sera of lupus-prone mice and human SLE patients (7, 8, 10–12). Therefore, we examined TSAd-deficient sera for the presence of these autoantibodies by ELISA. Up to 9 mo of age, antibodies against each of these antigens were rarely detected. After 9 mo, however, all four specificities were detected in the sera of TSAd-deficient mice, although rarely in the sera of control wild-type mice (Fig. 3 A). We compared TSAd-deficient sera with MRL/lpr sera for frequency and titer of the different autoantibody specificities (Fig. 3, A and C). Notably, the frequency of anti-ss DNA antibodies was lower in TSAd-deficient mice compared with MRL/lpr mice (37 vs. 75%, respectively). However, the frequency of anti-ds DNA, anticardiolipin, and rheumatoid factor antibodies was only slightly decreased or not at all decreased in TSAd-deficient mice compared with MRL/lpr mice (26 vs. 42%, 58 vs. 42%, and 42 vs. 42%, respectively; Fig. 3 A). Furthermore, whereas titers of anti-ss DNA antibodies were lower in TSAd-deficient mice, titers of anti-ds DNA and anticardiolipin antibodies were comparable between TSAd-deficient and MRL/lpr mice and titers of rheumatoid factor antibodies were higher in the TSAd-deficient mice (Fig. 3 C). For individual TSAd-deficient mice (and MRL/lpr mice), the presence or titer of antibodies against one target was not necessarily predictive of the presence or titer of antibodies against another target, thus emphasizing the heterogeneity of the autoantibody response (Fig. 3 C).
Figure 3.

Reactivity of TSAd-deficient sera with self-antigens. (A) The presence of IgG antibodies against ss DNA, ds DNA, cardiolipin, and IgG in sera from 1-yr-old TSAd-deficient (n = 19), 1-yr-old control C57BL/6 wild-type (n = 18, except for anti-ss DNA where n = 20), and 3–6-mo-old MRL/lpr (n = 12) mice was determined by ELISA. Sera were tested at a dilution of 1:50 and were scored positive based on criteria outlined in Materials and Methods. Shown is the percentage of positive sera for each autoantigen. Differences between TSAd-deficient and C57BL/6 mice are statistically significant for all autoantigens (all P < 0.05 determined using the chi-squared test). (B) Representative staining of Crithidia luciliae by TSAd-deficient sera that scored positive for anti-ds DNA antibodies by ELISA. Shown is intense staining of the ds DNA-containing kinetochore and the dimmer staining nucleus. Reactivity with the kinetochore (which is free of ss DNA and not complexed to protein) confirms the presence of anti-ds DNA antibodies. (C) Titers of autoantibodies in TSAd-deficient and MRL/lpr mice. Reactivity of diluted sera from seven 1-yr-old TSAd-deficient mice and six 4–6-mo-old MRL/lpr mice with the indicated autoantigens was determined by ELISA as indicated in A. Data from individual mice are represented by different symbols. For each mouse the same symbol is used for each antigen.
Kidney Disease and Lung Leukocytic Infiltration in TSAd-deficient Mice.
In lupus-like syndromes, immune complexes become deposited in kidney glomeruli leading to glomerulonephritis (7, 8, 10, 11). To determine if TSAd-deficient mice develop glomerulonephritis, older mice were killed and kidneys were subject to histopathological and immunohistochemical analyses. Kidneys from control wild-type mice revealed glomeruli with normal cellularity, mesangium, and glomerular basement membranes (Fig. 4 A and Table I). In contrast, kidneys of TSAd-deficient mice were frequently abnormal and showed signs of glomerulonephritis (Fig. 4 A and Table I). These included a marked increase in glomerular size and cellularity (accounted for by an increase in the number of mesangial cells and by the presence of inflammatory cells, including polymorphonuclear leukocytes), an increase in the amount of mesangial matrix, a thickening of glomerular basement membranes, and the presence of intratubular proteinaceous hyaline casts indicative of glomerular dysfunction (Fig. 4 B). Furthermore, IgG immune complex deposits were detected in glomeruli (Fig. 4 C). These features were qualitatively and quantitatively similar to those observed in kidneys of 5–6-mo-old MRL/lpr mice with the exception that glomeruli in TSAd-deficient kidneys showed little evidence of sclerosis (not depicted and references 6–8). None of the changes observed in kidneys of TSAd-deficient mice were apparent in kidneys of TSAd heterozygote mice (unpublished data). Consistent with the kidney pathology, the majority of older TSAd-deficient mice showed evidence of proteinuria. Of 19 examined 1-yr-old TSAd-deficient mice, 12 had urine protein levels in the range of 30 mg/dL and 4 had urine protein levels in the range of 100 mg/dL. Only three mice showed the trace urine protein levels found in wild-type mice (P < 5 × 10−7 using the chi-squared test).
Figure 4.
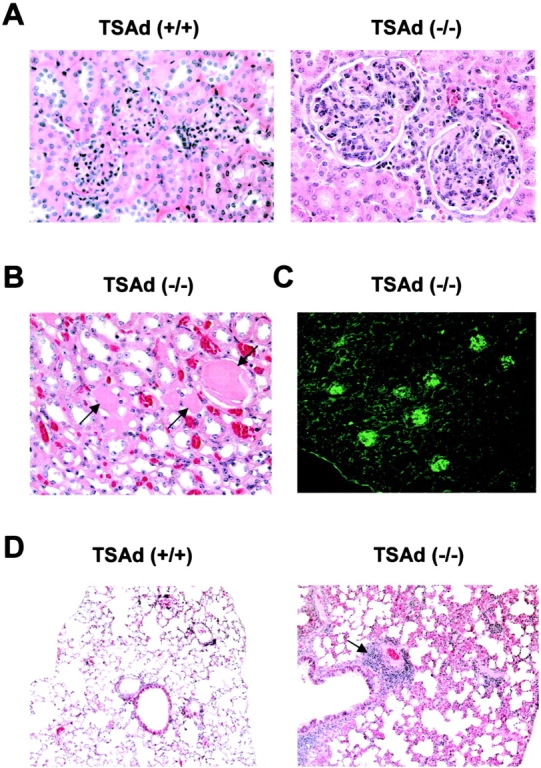
Kidney and lung pathology of TSAd-deficient mice. (A and B) H&E stained kidney sections from 1-yr-old control wild-type and TSAd-deficient mice. Representative glomeruli are shown in A. Note gross increase in mesangial cellularity in TSAd-deficient glomeruli. (B) Intratubular hyaline casts (indicated by arrows) seen in TSAd-deficient kidneys. (C) IgG immune complex deposits in glomeruli of a 9-mo-old TSAd-deficient mouse detected by immunofluorescence. (D) H&E stained lung sections from 1-yr-old control wild-type and TSAd-deficient mice. Note perivascular inflammatory aggregate (indicated by arrow) and thickening of alveolar interstitial spaces in the TSAd-deficient lung.
Table I.
Glomerular Pathology of TSAd (+/+) and TSAd (−/−) Micea
| Mouse | Genotype | Pristaneb | Age (mo) | Sexc | Mesangialhypercellularity | Mesangialmatrix increase | Basementmembrane thickness |
|---|---|---|---|---|---|---|---|
| WT1 | +/+ | No | 12 | F | 0 | 0 | 0 |
| WT2 | +/+ | No | 12 | F | 0 | 0 | 0 |
| WT3 | +/+ | No | 13 | F | 0 | 0 | 0 |
| WT4 | +/+ | No | 12 | M | 0 | 0 | 0 |
| WT5 | +/+ | No | 13 | M | 0 | 0 | 0 |
| K1 | −/− | No | 11 | F | 4 | 2 | 1 |
| K2 | −/− | No | 14 | F | 1 | 1 | 1 |
| K3 | −/− | No | 13 | F | 1 | 0 | 1 |
| K4 | −/− | No | 12 | F | 4 | 2 | 3 |
| K5 | −/− | No | 13 | F | 2 | 2 | 1 |
| K6 | −/− | No | 13 | F | 1 | 2 | 1 |
| K7 | −/− | No | 11 | M | 2 | 1 | 0 |
| K8 | −/− | No | 13 | M | 1 | 1 | 0 |
| K9 | −/− | No | 13 | M | 0 | 1 | 0 |
| K10 | −/− | No | 13 | M | 1 | 1 | 1 |
| K11 | −/− | No | 13 | M | 2 | 2 | 1 |
| K12 | −/− | No | 13 | M | 1 | 1 | 0 |
| WTP1 | +/+ | Yes | 8 | F | 0 | 0 | 0 |
| WTP2 | +/+ | Yes | 8 | F | 1 | 1 | 1 |
| WTP3 | +/+ | Yes | 8 | F | 1 | 1 | 0 |
| WTP4 | +/+ | Yes | 8 | F | 1 | 1 | 0 |
| WTP5 | +/+ | Yes | 8 | F | 1 | 1 | 1 |
| WTP6 | +/+ | Yes | 8 | F | 1 | 1 | 0 |
| WTP7 | +/+ | Yes | 8 | M | 0 | 0 | 0 |
| WTP8 | +/+ | Yes | 8 | M | 0 | 0 | 0 |
| WTP9 | +/+ | Yes | 8 | M | 1 | 2 | 1 |
| WTP10 | +/+ | Yes | 8 | M | 1 | 1 | 1 |
| WTP11 | +/+ | Yes | 8 | M | 0 | 1 | 0 |
| WTP12 | +/+ | Yes | 8 | M | 1 | 1 | 1 |
| WTP13 | +/+ | Yes | 8 | M | 1 | 1 | 0 |
| KP1 | −/− | Yes | 8 | F | 2 | 2 | 1 |
| KP2 | −/− | Yes | 8 | F | 4 | 4 | 3 |
| KP3 | −/− | Yes | 8 | F | 1 | 2 | 3 |
| KP4 | −/− | Yes | 8 | F | 3 | 3 | 3 |
| KP5 | −/− | Yes | 8 | M | 1 | 1 | 0 |
| KP6 | −/− | Yes | 8 | M | 3 | 3 | 3 |
| KP7 | −/− | Yes | 8 | M | 3 | 3 | 3 |
| KP8 | −/− | Yes | 8 | M | 2 | 3 | 2 |
Glomeruli were scored in a blinded fashion on a 0–4 scale for the indicated parameters. 0, normal; 4, pronounced change. Scores are based upon analysis of 30–40 glomeruli in each kidney section.
Pristane was administered to mice at 2 mo of age. Analysis was performed at 8 mo.
M, male; F, female.
Another common feature of lupus-like syndromes is the invasion of nonlymphoid organs by leukocytes (7, 8). Therefore, we examined a number of different nonlymphoid tissues from older TSAd-deficient mice for evidence of leukocytic infiltration. Particularly in lung, there were large perivascular accumulations of leukocytes that were not observed in control mice (Fig. 4 D). These leukocytic infiltrates extended deep into the pulmonary parenchyma. In addition, there was a thickening of interstitial alveolar spaces and the fine alveolar architecture observed in control mice was lost in the TSAd-deficient mice. Leukocytic infiltrates were less frequently observed in other nonlymphoid organs.
Susceptibility of TSAd-deficient Mice to Experimentally Induced Lupus-like Disease.
Because several of the key lupus-like disease features of TSAd-deficient mice are only observed in older mice, we next asked if younger TSAd-deficient mice are more susceptible to experimentally induced lupus. To examine this, we immunized 2-mo-old mice with the isoprenoid alkane, pristane, which has been shown previously to act as a nonspecific inflammatory trigger of lupus (13–15). We took advantage of the fact that compared with other strains, C57BL/6 mice mount a weaker autoimmune response to pristane that is not, for example, accompanied by the production of IgG isotype anti-ss DNA or anti-ds DNA antibodies (15–17). Confirming these previous reports, 6 mo after immunization with pristane, C57BL/6 wild-type mice produced low titers of IgM anti-ss DNA antibodies (11 out of 13 mice) but essentially failed to produce IgG anti-ss DNA antibodies and IgM and IgG anti-ds DNA antibodies (Fig. 5 ; low titers of these antibodies were detected in 1 or 2 out of 13 wild-type mice). In contrast, C57BL/6 TSAd-deficient mice produced higher titer IgM anti-ss DNA antibodies and, in addition, produced IgG anti-ss DNA antibodies (10 out of 10 mice) and IgM and IgG anti-ds DNA antibodies (9 and 4 out of 10 mice, respectively) after immunization with pristane (Fig. 5).
Figure 5.

Pristane induction of anti-DNA antibodies in TSAd-deficient mice. Control wild-type and TSAd-deficient mice were immunized with pristane at 2 mo of age. 6 mo after immunization, the presence of the indicated anti-DNA antibodies in sera was determined by ELISA. All sera were tested at a dilution of 1:100. Data points represent different individual mice of the indicated genotypes. The difference in mean OD value for IgM anti-ss DNA antibodies between wild-type and TSAd-deficient mice is statistically significant (P < 3.6 × 10−4 determined using the Student's two-sample t test). Differences between wild-type and TSAd-deficient mice with respect to the percentage of mice that develop the other types of anti-DNA antibodies (see text and Materials and Methods) are also statistically significant (all P < 5 × 10−5 determined using the chi-squared test).
We also examined kidneys of pristane-immunized mice for evidence of glomerulonephritis (Table I). 6 mo after pristane immunization, kidneys of wild-type pristane-immunized mice showed histological evidence of only mild glomerulonephritis. In contrast, kidneys of pristane-immunized TSAd-deficient mice showed clear evidence of more severe glomerulonephritis that was manifest as increased mesangial hypercellularity, mesangial matrix deposition, and glomerular basement membrane thickness. In accordance with these findings, TSAd-deficient mice showed evidence of increased proteinuria. Thus, of 10 examined TSAd-deficient mice, 6 mo after pristane immunization, 6 had urine protein concentrations in the 30 mg/dL range and 4 had urine protein concentrations in the 100 mg/dL range. In comparison, of 13 examined wild-type mice at the same time point, all had urine protein concentrations in the 30 mg/ml range (P < 5 × 10−5).
Lymphoid Hyperplasia in TSAd-deficient Mice.
Next, we examined whether lymphoid organs of TSAd-deficient mice contain increased numbers of T cells that could point to deregulated T cell function as a cause of autoimmunity. However, it was previously reported that primary and secondary lymphoid organs from TSAd-deficient mice contained normal numbers and ratios of T cells as well as B cells (3). Therefore, during our analysis of TSAd-deficient mice we reexamined this issue in case subtle differences had been overlooked in the original studies (Table II). Indeed, comparison of spleen weights between 29 control wild-type mice and 30 TSAd-deficient mice, aged 2–3 mo, revealed mild splenomegaly in the TSAd-deficient mice (an ∼1.6-fold increase in spleen weight). Similarly, thymus weight was notably increased in TSAd-deficient mice. These increased spleen and thymus weights were associated with increased cellularity. For spleen, the hypercellularity affected both T and B compartments. For thymus, as reported previously (3), no alterations in the ratios of CD4/CD8 double negative, double positive, or single positive thymocytes were noted (not depicted).
Table II.
Lymphoid Organ Weights and Lymphocyte Numbers in TSAd (+/+) and TSAd (−/−) Mice
| TSAd (+/+) | TSAd (−/−) | Pa | |
|---|---|---|---|
| Spleen weight (mg)b | 61.2 ± 1.9 | 98.2 ± 6.1 | <4.8 × 10−7 |
| Total spleen cell number | 73.3 ± 4.9 × 106 | 120.1 ± 10.9 × 106 | <2.0 × 10−3 |
| Percentage T cells | 32.7 ± 1.4 | 26.3 ± 1.2 | <5.3 × 10−3 |
| Total T cell number | 23.7 ± 1.2 × 106 | 31.0 ± 2.0 | <4.8 × 10−3 |
| Percentage B cells | 55.9 ± 1.2 | 60.0 ± 2.0 | <1.0 × 10−1 |
| Total B cell number | 41.2 ± 3.5 × 106 | 71.8 ± 6.2 × 106 | <9.8 × 10−4 |
| Thymus weight (mg) | 57.0 ± 4.0 | 79.9 ± 4.3 | <2.0 × 10−3 |
Shown are mean values ± 1 SE.
Probabilities were calculated using the Student's two-sample t test.
Spleen figures are derived from 29 TSAd (+/+) and 30 TSAd (−/−) mice, aged 2–3 mo. All other figures are based upon data from 7 TSAd (+/+) and 7 TSAd (−/−) mice, aged exactly 8 wk.
T Cells from Older TSAd-deficient Mice Display an Activated Phenotype.
By 9 mo of age, splenomegaly in TSAd-deficient mice was more pronounced (Fig. 6 A). Furthermore, histological examination of spleens and lymph nodes from older TSAd-deficient mice revealed hyperreactive features. There was an effacement of architecture seen as a lack of clear distinction between red and white pulp areas in spleen or between cortical and medullary areas in lymph nodes (Fig. 6 B). To determine if T cells in older TSAd-deficient spleens were in an activated state, splenocytes were double stained with TCRβ antibodies and different activation marker antibodies (Fig. 6 C). In comparison with T cells from spleens of control mice, T cells from TSAd-deficient mice showed increased expression of CD69, CD44, and CD95/Fas activation markers and decreased expression of CD62L/l-selectin (whose expression correlates inversely with activation status). The same results were obtained with all 11 tested TSAd-deficient mice age 9–13 mo. In contrast, T cells from older TSAd heterozygotes did not show increased expression of CD69, CD44, or CD95, nor decreased expression of CD62L (not depicted). Similar to T cells, B cells from older TSAd-deficient spleens showed increased expression of CD69, CD44, and CD95 although up-regulation of these activation markers on B cells was less dramatic (not depicted). In summary, these findings indicate that older TSAd-deficient spleens contain large numbers of activated T and B cells.
Figure 6.

Splenic pathology of TSAd-deficient mice. (A) Top: spleens from 9-mo-old wild-type (left) and TSAd-deficient (right) mice. Middle: spleen weights of six 1-yr-old wild-type and TSAd-deficient mice. Bottom: the total number of T cells and B cells in spleens from three 1-yr-old wild-type and TSAd-deficient mice were determined. (B) H&E stained spleen and inguinal lymph node sections from 1-yr-old wild-type and TSAd-deficient mice. Note the loss of normal architecture (clearly defined areas of red and white pulp in spleen and cortical and medullary regions in lymph node) in TSAd-deficient mice. (C) Expression of the indicated activation markers on splenic T cells from 9-mo-old wild-type (blue or green) and TSAd-deficient (red) mice was determined by flow cytometry.
Defective T Cell Death in TSAd-deficient Mice.
We considered the possibility that the development of autoimmunity in TSAd-deficient mice was consequent to a failure of antigen-induced T cell death that would be consistent with the spleen findings. To examine this, control wild-type and TSAd-deficient mice were immunized with the superantigen, SEB. In healthy animals, immunization with SEB leads to a reduction in the number of responding TCR Vβ8+ T cells from the peripheral T cell pool as they die by apoptosis (18). To examine whether TCR Vβ8+ T cells are as efficiently eliminated in TSAd-deficient mice, we determined the percentage of Vβ8+ T cells amongst total CD4+ peripheral blood T cells at different time points after SEB immunization (Fig. 7 A). In control mice, immunization with SEB led to a time-dependent decrease in the percentage of Vβ8+ T cells that was specific to Vβ8+ T cells because the percentage of Vβ6+ T cells was not significantly altered in these experiments. Likewise, in TSAd-deficient mice, immunization with SEB led to a reduction in the percentage of Vβ8+ T cells and not Vβ6+ T cells. However, elimination of Vβ8+ T cells in TSAd-deficient mice was clearly less efficient than in the controls. The same results were obtained in two identical repeat experiments. Importantly, the slower kinetics of T cell elimination in TSAd-deficient mice could not be explained on the basis that the initial activation of Vβ8+ T cells in response to SEB is impaired in these animals. Thus, Vβ8+ T cells from TSAd-deficient and control wild-type mice were induced to express comparable levels of the CD69 activation marker when examined 24 h after immunization with SEB (Fig. 7 B). In addition, up to 3 d after SEB immunization, Vβ8+ T cells from TSAd-deficient and wild-type mice were seen to undergo the same number of cell divisions in vivo (unpublished data).
Figure 7.

Defective T cell death in TSAd-deficient mice. (A) Percentages of TCR Vβ6+ or TCR Vβ8+ T cells amongst total CD4+ T cells in venous blood from wild-type and TSAd-deficient mice (age 2 mo) at the indicated times after immunization with SEB were determined by flow cytometry. Data are from eight mice in each group and are represented as means ± 1 SE. At days 4 and 11 differences between wild-type and TSAd-deficient mice with respect to the percentage of Vβ8+ T cells are statistically significant (both P < 0.005 calculated using the Student's two sample t test). (B) CD69 expression on TCR Vβ6+ and Vβ8+ T cells from wild-type and TSAd-deficient mice 24 h after immunization with SEB was determined by flow cytometry. The percentages of Vβ6+ and Vβ8+ T cells that express CD69 are indicated in parentheses.
Microarray Analysis of TSAd-regulated Gene Transcription.
The Fas death receptor pathway has been studied extensively as a major pathway involved in the control of T cell death (19–21). In in vitro studies, TSAd-deficient T cells showed partial resistance to Fas-mediated apoptosis that was not associated with reduced expression of either Fas or Fas ligand (unpublished data). However, the significance of these findings to the impaired T cell death seen in TSAd-deficient mice in vivo is uncertain because recent studies have indicated that superantigen-induced T cell death is Fas independent (22–24). Therefore, to shed light upon possible mechanisms by which TSAd might regulate T cell death, we undertook gene profiling experiments to determine in total which genes are regulated by TSAd in activated T cells. In these experiments we compared gene expression between purified wild-type and TSAd-deficient CD4+ T cells that were either not stimulated or stimulated for 20 h with a CD3 antibody (directed against the TCR complex) plus an antibody against the CD28 T cell costimulatory receptor. To uncover genes that were positively or negatively regulated by TSAd, several filters were placed upon the data derived from these experiments (refer to Materials and Methods). With the use of these filters, we identified a total of 107 genes (out of ∼12,000) that were positively regulated by TSAd in CD4+ T cells (Fig. 8) . In contrast, using the same filters, we did not identify any genes that were negatively regulated by TSAd. Within the group of positively regulated genes, several have been previously ascribed a proapoptotic function in different cell types. These include the transmembrane protein, BAP29 (25), the intracellular chloride channel, CLIC4 (26), the proto-oncogene, c-myc (27), the molecular chaperone, Hsp60 (28), and IFN-γ (29). Furthermore, confirming earlier findings, IL-2 was identified as a major positively regulated TSAd target gene in these experiments. Although IL-2 is known to function as a T cell growth factor in vitro, numerous studies have shown that IL-2 acts only to promote T cell death in vivo (30–34). Thus, impaired induction of IL-2 in vivo could be an important factor accounting for the resistance of TSAd-deficient T cells to apoptotic death. Given these findings, therefore, we sought to verify that TSAd-deficient T cells produce less IL-2 protein in vivo. For this, we examined IL-2 protein levels in Vβ8+ splenic T cells 24 h after immunization of mice with SEB using an intracellular staining technique. As shown in Fig. 9 , TSAd-deficient Vβ8+ T cells synthesized considerably less IL-2 protein (and IFN-γ) in response to SEB as determined in three repeat experiments. In contrast, for the other apoptosis-related genes, we were unable to detect any differences in protein expression despite the changes in mRNA levels (unpublished data). Thus, TSAd-deficient T cells produce less IL-2 in vivo as well as in vitro, which may account for the development of systemic autoimmunity in these animals.
Figure 8.
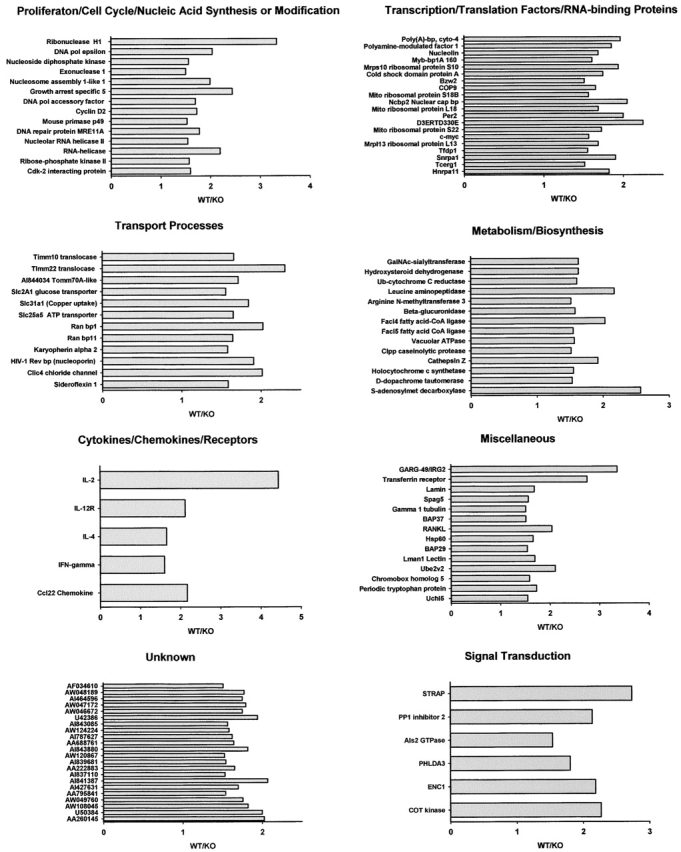
Genes positively regulated by TSAd in CD4+ T cells. Purified CD4+ splenic T cells from 2-mo-old wild-type (WT) and TSAd-deficient (KO) mice were not stimulated (0) or stimulated with CD3 plus CD28 antibodies for 20 h (20) in vitro. Relative gene expression in unstimulated and stimulated T cells was determined in gene profiling experiments using Affymetrix U74Av2 microchips. Shown are those genes (107 total out of ∼12,000) where WT (20) > WT (0) by a factor of at least threefold, WT (20)/KO (20) > WT (0)/KO (0) by at least 1.5-fold, and WT (20) > KO (20) by at least 1.5-fold. Genes are categorized on the basis of known or inferred (based on amino acid sequence homology) function. For each gene the WT/KO ratio at 20 h is depicted. For all data values for this gene set see Tables S1–S8, available at http://www.jem.org/cgi/content/full/jem.20021358/DC1.
Figure 9.

Impaired cytokine production by TSAd-deficient T cells in vivo. Control wild-type and TSAd-deficient mice were immunized with SEB. After 24 h, IL-2 and IFN-γ expression in Vβ8+ splenic T cells was determined by intracellular staining and flow cytometry. Filled and open histograms represent isotype control and anticytokine-stained Vβ8+ T cells, respectively. The percentages of cytokine+ Vβ8+ T cells are indicated in parentheses.
Discussion
The main finding of these studies is that TSAd-deficient mice develop spontaneously with age a systemic lupus-like autoimmune disease that involves hypergammaglobulinemia, production of autoantibodies against different targets including ss and ds DNA, lymphoid hyperplasia, accumulation of large numbers of activated lymphocytes, leukocytic infiltration of nonlymphoid organs, and glomerulonephritis. These features are similar to those seen in the NZB/W mouse and the Fas-deficient MRL/lpr mouse, although the disease in TSAd-deficient mice is less severe than in these classical models of mouse lupus. Disease in MRL/lpr and NZB/W mice is characterized by early production of autoantibodies, acute renal failure, and early death that affects females more than males (7, 8). In contrast, disease in TSAd-deficient mice is later in onset, does not result in significantly increased mortality up to 15 mo (unpublished data), and does not affect females preferentially. In this regard, the disease in TSAd-deficient mice is more akin to the lupus-like disease that occurs in several other genetically engineered mouse models such as cbl-b, stra13, and bim-deficient mice, CD45E613R knock-in mice, and bcl-2 transgenic mice where significant early mortality has not been reported and/or sex differences are not apparent (35–39).
Several factors could account for the prolonged lifespan of TSAd-deficient mice despite the development of marked glomerulonephritis. Most notable of these is the absence of a vasculitic response in TSAd-deficient mice. Vasculitis occurs in both MRL/lpr and NZB/W mice and in the former at least contributes in a major way to early mortality (6–8). It should also be noted that the spontaneous autoimmune disease that results from TSAd deficiency described herein is observed on the C57BL/6 genetic background. In comparison with other strains, C57BL/6 mice are less prone to develop lupus-like autoimmunity (16, 40, 41). For example, young wild-type C57BL/6 mice are relatively resistant to pristane-induced lupus compared with other strains. However, further illustrating the fact that impaired expression of TSAd promotes systemic autoimmunity is the finding here that young C57BL/6 TSAd-deficient mice are highly susceptible to pristane-induced lupus.
T cells from TSAd-deficient mice were shown to be resistant to superantigen-induced death in vivo. This finding is highly significant, pointing to the mechanism by which TSAd-deficient mice develop autoimmunity. Superantigen-induced T cell death is known to involve mitochondrial dysfunction rather than death receptors (22–24). This implies that the mitochondrial death pathway is impaired in TSAd-deficient T cells. In turn, it has been shown previously that impaired mitochondrial death in T cells results in systemic autoimmunity (23, 24, 42). It is also possible that an observed partial resistance of T cells to Fas-mediated death in vitro is relevant to the development of autoimmunity in TSAd-deficient mice. Although Fas plays little role in superantigen-induced T cell death, Fas is known to be involved in the death of T cells that are reactive with persistent self-antigens, thus accounting for the development of autoimmunity in Fas-deficient MRL/lpr mice (7, 8, 42). However, there are several counter-arguments to the notion that Fas-mediated apoptosis is significantly impaired in C57BL/6 TSAd-deficient mice in vivo or that impaired Fas-mediated apoptosis contributes to autoimmune disease. First, C57BL/6/lpr mice, in contrast to MRL/lpr mice, do not develop significant lupus-like disease characterized by the production of anti-DNA antibodies and the development of glomerulonephritis (40, 41). Second, Fas-deficient mice, regardless of background strain, acquire a large population of CD4− CD8− (double negative) B220+ peripheral T cells that are not seen in TSAd-deficient mice (7, 8). Third, C57BL/6 Fas-deficient and Fas ligand–deficient mice are more resistant to pristane-induced lupus than C57BL/6 controls, which is in contrast to C57BL/6 TSAd-deficient mice, which are more susceptible (see above; 17).
Previous studies have indicated that TSAd can function as a transcriptional regulator in T cells (5). Therefore, we performed microarray experiments to identify any proapoptotic or antiapoptotic genes that might be down-regulated or up-regulated, respectively, in TSAd-deficient T cells. Several genes that encode proapoptotic proteins were found to be down-regulated in TSAd-deficient T cells. Prominent amongst these was the gene for IL-2, thus confirming that TSAd is a major regulator of this cytokine. Indeed, because IL-2 is known to function as a major T cell growth factor in vitro, reduced expression of many of the other genes identified in these microarray experiments, such as those encoding DNA polymerases, cell cycle proteins, and metabolic enzymes, might be consequent to deficient expression of IL-2. However, despite the fact that IL-2 can act as a T cell growth factor in vitro, IL-2 acts mainly as an inducer of T cell death in vivo. Consequently, T cells from IL-2 and IL-2R–deficient mice are resistant to superantigen-induced death in vivo and, similar to TSAd-deficient mice, IL-2 and IL-2R–deficient mice develop generalized autoimmune disease (30–33). The demonstration, therefore, that TSAd-deficient T cells produce less IL-2 in vivo suggests a molecular mechanism that links TSAd deficiency to impaired T cell death and the development of autoimmunity. Significantly, however, there are differences in the nature of the autoimmune disease in IL-2 and IL-2R–deficient mice versus that seen in TSAd-deficient mice. IL-2 and IL-2R–deficient mice do not develop glomerulonephritis but instead develop inflammatory bowel disease and severe autoimmune hemolytic anemias that are not seen in TSAd-deficient mice. It is possible that expression of inflammatory bowel disease and hemolytic anemia requires total or near total absence of IL-2 signaling, whereas in TSAd-deficient mice significant IL-2 signal transduction would presumably still occur. Further, because IL-2 and IL-2R–deficient mice die at an early age, it could be argued that there is not sufficient time for the development of glomerulonephritis in these mice. Another possible explanation for the observed differences is that in TSAd-deficient mice reduced expression of other identified TSAd-regulated genes, together with impaired expression of IL-2, contributes to phenotype.
Aside from mechanism, these studies highlight how deficient expression of TSAd can lead to the development of systemic autoimmunity. In this regard, TSAd is set further apart from other adaptor proteins expressed in T cells whose deficient expression does not similarly result in autoimmune disease (43). These studies, therefore, also raise the possibility that deficient expression of TSAd might be a causative factor in human autoimmunity. Thus far, loss of function TSAd polymorphisms associated with human SLE have not been identified (nor looked for). Interestingly, however, a promoter polymorphism of TSAd has recently been characterized that results in reduced levels of TSAd expression and shows a statistically significant association with human multiple sclerosis (44). Taken together with the findings presented here, this supports the idea that TSAd may represent a general susceptibility factor for human autoimmunity.
Acknowledgments
We thank Dr. Hai Nguyen (Weill Medical College of Cornell University) for assistance with pathology. Dr. Jeff Bluestone is thanked for providing TSAd-deficient mice.
This study was supported by Public Health Service grants AI044926 and AI050699 to P.D. King.
Abbreviations used in this paper: ANA, antinuclear antibodies; ds, double stranded; GAMIgG, goat anti–mouse IgG; H&E, hematoxylin and eosin; SEB, staphylococcal enterotoxin B; ss, single stranded; TSAd, T cell–specific adapter protein.
J. Drappa and L.A. Kamen contributed equally to this work.
The online version of this article contains supplemental material.
References
- 1.Spurkland, A., J.E. Brinchmann, G. Markussen, F. Pedeutour, E. Munthe, T. Lea, F. Vartdal, and H.C. Aasheim. 1998. Molecular cloning of a T cell-specific adapter protein (TSAd) containing an Src homology (SH) 2 domain and putative SH3 and phosphotyrosine binding sites. J. Biol. Chem. 273:4539–4546. [DOI] [PubMed] [Google Scholar]
- 2.Choi, Y.B., C.K. Kim, and Y. Yun. 1999. Lad, an adapter protein interacting with the SH2 domain of p56lck, is required for T cell activation. J. Immunol. 163:5242–5249. [PubMed] [Google Scholar]
- 3.Rajagopal, K., C.L. Sommers, D.C. Decker, E.O. Mitchell, U. Korthauer, A.I. Sperling, C.A. Kozak, P.E. Love, and J.A. Bluestone. 1999. RIBP, a novel Rlk/Txk- and itk-binding adaptor protein that regulates T cell activation. J. Exp. Med. 190:1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun, W., K. Kesavan, B.C. Schaefer, T.P. Garrington, M. Ware, N.L. Johnson, E.W. Gelfand, and G.L. Johnson. 2001. MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J. Biol. Chem. 276:5093–5100. [DOI] [PubMed] [Google Scholar]
- 5.Marti, F., N.H. Post, E. Chan, and P.D. King. 2001. A transcription function for the T cell–specific adapter (TSAd) protein in T cells: critical role of the TSAd Src homology 2 domain. J. Exp. Med. 193:1425–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bullard, D.C., P.D. King, M.J. Hicks, B. Dupont, A.L. Beaudet, and K.B. Elkon. 1997. Intercellular adhesion molecule-1 deficiency protects MRL/MpJ-Fas(lpr) mice from early lethality. J. Immunol. 159:2058–2067. [PubMed] [Google Scholar]
- 7.Theofilopoulos, A.N., and F.J. Dixon. 1985. Murine models of systemic lupus erythematosus. Adv. Immunol. 37:269–390. [DOI] [PubMed] [Google Scholar]
- 8.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 9.Tan, E.M., E.K. Chan, K.F. Sullivan, and R.L. Rubin. 1988. Antinuclear antibodies (ANAs): diagnostically specific immune markers and clues toward the understanding of systemic autoimmunity. Clin. Immunol. Immunopathol. 47:121–141. [DOI] [PubMed] [Google Scholar]
- 10.Dubois, E.L. 1975. Serologic abnormalities in spontaneous and drug-induced systemic lupus erythematosus. J. Rheumatol. 2:204–214. [PubMed] [Google Scholar]
- 11.Gershwin, M.E., and A.D. Steinberg. 1974. Qualitative characteristics of anti-DNA antibodies in lupus nephritis. Arthritis Rheum. 17:947–954. [DOI] [PubMed] [Google Scholar]
- 12.Harris, E.N., A.E. Gharavi, M.L. Boey, B.M. Patel, C.G. Mackworth-Young, S. Loizou, and G.R. Hughes. 1983. Anticardiolipin antibodies: detection by radioimmunoassay and association with thrombosis in systemic lupus erythematosus. Lancet. 2:1211–1214. [DOI] [PubMed] [Google Scholar]
- 13.Satoh, M., and W.H. Reeves. 1994. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J. Exp. Med. 180:2341–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Satoh, M., A. Kumar, Y.S. Kanwar, and W.H. Reeves. 1995. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proc. Natl. Acad. Sci. USA. 92:10934–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards, H.B., M. Satoh, M. Shaw, C. Libert, V. Poli, and W.H. Reeves. 1998. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J. Exp. Med. 188:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satoh, M., H.B. Richards, V.M. Shaheen, H. Yoshida, M. Shaw, J.O. Naim, P.H. Wooley, and W.H. Reeves. 2000. Widespread susceptibility among inbred mouse strains to the induction of lupus autoantibodies by pristane. Clin. Exp. Immunol. 121:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satoh, M., J.P. Weintraub, H. Yoshida, V.M. Shaheen, H.B. Richards, M. Shaw, and W.H. Reeves. 2000. Fas and Fas ligand mutations inhibit autoantibody production in pristane-induced lupus. J. Immunol. 165:1036–1043. [DOI] [PubMed] [Google Scholar]
- 18.Kawabe, Y., and A. Ochi. 1991. Programmed cell death and extrathymic reduction of Vbeta8+ CD4+ T cells in mice tolerant to Staphylococcus aureus enterotoxin B. Nature. 349:245–248. [DOI] [PubMed] [Google Scholar]
- 19.Brunner, T., R.J. Mogil, D. LaFace, N.J. Yoo, A. Mahboubi, F. Echeverri, S.J. Martin, W.R. Force, and D.H. Lynch. 1995. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 373:441–444. [DOI] [PubMed] [Google Scholar]
- 20.Dhein, J., H. Walczak, C. Baumler, K.M. Debatin, and P.H. Krammer. 1995. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature. 373:438–441. [DOI] [PubMed] [Google Scholar]
- 21.Ju, S.T., D.J. Panka, H. Cui, R. Ettinger, M. el-Khatib, D.H. Sherr, B.Z. Stanger, and A. Marshak-Rothstein. 1995. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 373:444–448. [DOI] [PubMed] [Google Scholar]
- 22.Hildeman, D.A., Y. Zhu, T.C. Mitchell, P. Bouillet, A. Strasser, J. Kappler, and P. Marrack. 2002. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 16:759–767. [DOI] [PubMed] [Google Scholar]
- 23.Hildeman, D.A., Y. Zhu, T.C. Mitchell, J. Kappler, and P. Marrack. 2002. Molecular mechanisms of activated T cell death in vivo. Curr. Opin. Immunol. 14:354–359. [DOI] [PubMed] [Google Scholar]
- 24.Rathmell, J.C., and C.B. Thompson. 2002. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 109:S97–S107. [DOI] [PubMed] [Google Scholar]
- 25.Breckenridge, D.G., M. Nguyen, S. Kuppig, M. Reth, and G.C. Shore. 2002. The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 99:4331–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandez-Salas, E., K.S. Suh, V.V. Speransky, W.L. Bowers, J.M. Levy, T. Adams, K.R. Pathak, L.E. Edwards, D.D. Hayes, C. Cheng, et al. 2002. mtCLIC/CLIC4, an organellular chloride channel protein, is increased by DNA damage and participates in the apoptotic response to p53. Mol. Cell. Biol. 22:3610–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juin, P., A.O. Hueber, T. Littlewood, and G. Evan. 1999. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 13:1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xanthoudakis, S., S. Roy, D. Rasper, T. Hennessey, Y. Aubin, R. Cassady, P. Tawa, R. Ruel, A. Rosen, and D.W. Nicholson. 1999. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 18:2049–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Refaeli, Y., L. Van Parijs, S.I. Alexander, and A.K. Abbas. 2002. Interferon γ is required for activation-induced death of T lymphocytes. J. Exp. Med. 196:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadlack, B., H. Merz, H. Schorle, A. Schimpl, A.C. Feller, and I. Horak. 1993. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 75:253–261. [DOI] [PubMed] [Google Scholar]
- 31.Sadlack, B., J. Lohler, H. Schorle, G. Klebb, H. Haber, E. Sickel, R.J. Noelle, and I. Horak. 1995. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur. J. Immunol. 25:3053–3059. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki, H., T.M. Kundig, C. Furlonger, A. Wakeham, E. Timms, T. Matsuyama, R. Schmits, J.J. Simard, P.S. Ohashi, and H. Griesser. 1995. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 268:1472–1476. [DOI] [PubMed] [Google Scholar]
- 33.Willerford, D.M., J. Chen, J.A. Ferry, L. Davidson, A. Ma, and F.W. Alt. 1995. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 3:521–530. [DOI] [PubMed] [Google Scholar]
- 34.Li, X.C., G. Demirci, S. Ferrari-Lacraz, C. Groves, A. Coyle, T.R. Malek, and T.B. Strom. 2001. IL-15 and IL-2: a matter of life and death for T cells in vivo. Nat. Med. 7:114–118. [DOI] [PubMed] [Google Scholar]
- 35.Bachmaier, K., C. Krawczyk, I. Kozieradzki., Y.Y. Kong, T. Sasaki, A. Oliveira-dos-Santos, S. Mariathasan, D. Bouchard, A. Wakeham, A. Itie, et al. 2000. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 403:211–216. [DOI] [PubMed] [Google Scholar]
- 36.Sun, H., B. Lu, R.Q. Li, R.A. Flavell, and R. Taneja. 2001. Defective T cell activation and autoimmune disorder in Stra13-deficient mice. Nat. Immunol. 2:1040–1047. [DOI] [PubMed] [Google Scholar]
- 37.Bouillet, P., D. Metcalf, D.C. Huang, D.M. Tarlinton, T.W. Kay, F. Kontgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- 38.Majeti, R., Z. Xu, T.G. Parslow, J.L. Olson, D.I. Daikh, N. Killeen, and A. Weiss. 2000. An inactivating point mutation in the inhibitory wedge of CD45 causes lymphoproliferation and autoimmunity. Cell. 103:1059–1070. [DOI] [PubMed] [Google Scholar]
- 39.Strasser, A., S. Whittingham, D.L. Vaux, M.L. Bath, J.M. Adams, S. Cory, and A.W. Harris. 1991. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA. 88:8661–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelley, V.E., and J.B. Roths. 1985. Interaction of mutant lpr gene with background strain influences renal disease. Clin. Immunol. Immunopathol. 37:220–229. [DOI] [PubMed] [Google Scholar]
- 41.Izui, S., V.E. Kelley, K. Masuda, H. Yoshida, J.B. Roths, and E.D. Murphy. 1984. Induction of various autoantibodies by mutant gene lpr in several strains of mice. J. Immunol. 133:227–233. [PubMed] [Google Scholar]
- 42.Van Parijs, L., D.A. Peterson, and A.K. Abbas. 1998. The Fas/Fas ligand pathway and Bcl-2 regulate T cell responses to model self and foreign antigens. Immunity. 8:265–274. [DOI] [PubMed] [Google Scholar]
- 43.Ohashi, P.S. 2002. T-cell signalling and autoimmunity: molecular mechanisms of disease. Nat. Rev. Immunol. 2:427–438. [DOI] [PubMed] [Google Scholar]
- 44.Dai, K.Z., H.F. Harbo, E.G. Celius, A. Oturai, P.S. Sorensen, L.P. Ryder, P. Datta, A. Svejgaard, J. Hillert, S. Fredrikson, et al. 2001. The T cell regulator gene SH2D2A contributes to the genetic susceptibility of multiple sclerosis. Genes Immun. 2:263–268. [DOI] [PubMed] [Google Scholar]