

Abstract
In the current study, we address the underlying mechanism for the selective generation of gut-homing T cells in the gut-associated lymphoid tissues (GALT). We demonstrate that DCs in the GALT are unique in their capacity to establish T cell gut tropism but in vivo only confer this property to T cells in the presence of DC maturational stimuli, including toll-like receptor-dependent and -independent adjuvants. Thus, DCs from mesenteric LNs (MLNs), but not from spleen, supported expression of the chemokine receptor CCR9 and integrin α4β7 by activated CD8+ T cells. While DCs were also required for an efficient down-regulation of CD62L, this function was not restricted to MLN DCs. In an adoptive CD8+ T cell transfer model, antigen-specific T cells entering the small intestinal epithelium were homogeneously CCR9+α4β7 +CD62Llow, and this phenotype was only generated in GALT and in the presence of adjuvant. Consistent with the CCR9+ phenotype of the gut-homing T cells, CCR9 was found to play a critical role in the localization of T cells to the small intestinal epithelium. Together, these results demonstrate that GALT DCs and T cell expression of CCR9 play critical and integrated roles during T cell homing to the gut.
Keywords: lymphocytes, antigen-presenting cell, inflammation, chemokines, intestinal mucosa
Introduction
Whereas naive T cell migration is restricted to secondary lymphoid organs, effector T cells have the ability to localize to peripheral tissues such as the intestinal mucosa and inflamed skin. Effector T cell subsets display preferential homing potential for different peripheral tissues, a process that is mediated by the selective expression of cell adhesion molecules and chemokine receptors (1). Effector T cells homing to the intestine express high levels of the β7 integrin α4β7, whose ligand MAdCAM-1 is expressed on post capillary venules in the intestinal lamina propria (2), and the chemokine receptor CCR9 (3), whose ligand CCL25 is selectively expressed by small intestinal epithelial cells (4, 5). β7 integrins and CCL25 are important for T cell localization to intestinal effector sites, since β7-deficient T cells are severely impaired in their ability to localize to the intestinal mucosa, and neutralizing antibodies to CCL25 partially block T cell localization to the small intestinal epithelium (6, 7).
Recent data indicate that peripheral tissue-homing receptors are induced on T cells during their activation in secondary lymphoid organs and that distinct secondary lymphoid organ microenvironments underlie the generation of effector T cells with differential homing capacity (7, 8). Thus, T cells activated in mesenteric LNs (MLNs) express α4β7 and CCR9, whereas those undergoing activation in peripheral LNs (PLNs) are induced to express P-selectin ligands. In addition, during the preparation of this article, Mora et. al. (9) reported that Peyer's patch (PP) DCs, but not splenic or PLN DCs, induce CD8+ T cells to express α4β7 and to respond to the CCR9 ligand CCL25. In the current study, we have examined the underlying mechanism by which antigen driven T cell activation in MLN leads to the selective generation of intestinal homing effector T cell populations.
Materials and Methods
Mice.
OT-1 (provided by A. Mowat, University of Glasgow, Glasgow, UK), CCR9−/− (10) and C57BL/6J-Ly5.1 mice were bred and maintained at the Biomedical Center Animal Facility at Lund University. CCR9−/− OT-I mice were obtained by crossing OT-I × C57BL/6J (F7) CCR9−/− litters with C57BL/6J (F7) CCR9−/− mice and screening the offspring by flow cytometry and PCR. Ly5.2+Ly5.1+OT-1 mice were obtained from OT-1 × C57BL/6J-Ly5.1 matings.
Reagents.
OVA (grade VI; Sigma-Aldrich) was purified from endotoxins by Detoxi-Gel™ (Pierce Chemical Co.) chromatography. OVA257–264 peptide SIINFEKL was from Innovagen. LPS (Escherichia coli, serotype 055:B5) and polyinosine polycytidylic acid (pI:C) were from Sigma-Aldrich. SNARF®-1 carboxylic acid acetate succinimidyl ester (SNARF-1) and 5- and 6-carboxy-fluorescein diacetate succinimidyl ester (CFSE) were from Molecular Probes. The chemokines CCL25 and CXCL10 were from R&D Systems.
Purification of DCs, DC-depleted APCs, and OT-1 T Cells.
CD11c+ DCs were isolated using anti–CD11c-conjugated MACS beads and LS columns (Miltenyi Biotech) according to the manufacturer's protocol. DC-depleted cells were obtained by passing the CD11c− fraction obtained after isolation of DCs through a high magnetic field LD column (Miltenyi Biotech). DC preparations were >90% CD11c+ MHCII+, whereas DC-depleted preparations contained <0.2% CD11c+MHC II+ cells. MLN DCs were also labeled with FITC-conjugated anti-CD11c and PE-conjugated anti-CD8α mAbs, and sorted into CD11c+ CD8α+ and CD11c+ CD8α− subsets using a FACS® Vantage cell sorter (BD Biosciences). Splenic CD8β+ T cells were obtained (>98% pure) from OT-1 mice using biotinylated anti-CD8β mAb followed by streptavidin-conjugated magnetic beads according to standard MACS procedures (Miltenyi Biotech).
In Vitro Cultures.
Purified APCs were pulsed at ∼5 × 106 cells/ml with 1 nM SIINFEKL peptide for 2 h at 37°C. Peptide-loaded APCs were extensively washed and used to stimulate CFSE-labeled OT-1 cells (7) in flat bottom 96-well plates. Unless stated, 105 DCs/well or 5 × 105 DC-depleted APCs/well were used to stimulate 2 × 105 OT-1 cells. OT-1 cells were also stimulated with 10 μg/ml plate-adsorbed anti-CD3 mAb (145–2C11; American Type Culture Collection) plus soluble anti-CD28 (1 μg/ml; BD, PharMingen). Cells were cultured in complete medium for 4 d and thereafter analyzed by flow cytometry.
Flow Cytometry Analysis.
Flow cytometry analysis was performed as described previously (7). Specificity of the CCR9 staining was confirmed by preincubating the polyclonal rabbit anti-CCR9 Ab (11) with 10 μg/ml of the corresponding antigenic peptide. Anti–mouse CXCR3 mAb (4C4; Millennium Pharmaceuticals) was revealed by Cy5-conjugated goat anti–rat IgM (μ-chain specific; Jackson ImmunoResearch Laboratories). All other mAbs were used as FITC, PE, or allophycocyanin conjugates (BD PharMingen).
Chemotaxis Assay.
OT-1 cells activated by spleen and MLN DCs were labeled with 1 μM SNARF-1 and CFSE, respectively, as previously described for CFSE labeling (7), mixed at a 1:1 ratio, and their ability to migrate to optimal concentrations of CCL25 (250 nM) or CXCL10 (100 nM) was determined in chemotaxis assays (7). The number of SNARF-1+ (red fluorescence) and CFSE+ (green fluorescence) cells in the starting population (cellsstart) and in the population migrating to chemokine (cellschemokine) or medium alone (cellsmedium) was determined by flow cytometry analysis. Specific migration is expressed for SNARF-1+ cells (primed by spleen DCs) and CFSE+ cells (primed by MLN DCs) as 100 × [number of cellschemokine − number of cellsmedium] / number of cellsstart.
Adoptive Transfer Experiments.
CD8β+ OT-1 cells (3–5 × 106) were injected i.v. into C57BL/6J-Ly5.1 mice, and 1–2 d later recipient mice received an i.p. injection of 200 μl PBS containing 5 mg OVA with or without 100 μg pI:C or 100 μg LPS, or 2.5 mg alum-precipitated OVA. 2–3-d later, mice were killed, and organs were collected after perfusion of lung with ∼5 ml PBS. Isolation of small intestinal intraepithelial lymphocytes (IELs) and lymphocytes from LNs, spleen and lung was performed as previously described (7).
Results
Dendritic Cells from MLN, but Not from Spleen, Are Necessary and Sufficient for Antigen-dependent Generation of α4β7 +, CCR9+, and CD62Llow CD8+ T Cells In Vitro.
The selective generation of CCR9+ (7) and α4β7 + (8) T cells in the MLN during in vivo primary immune responses suggests that APCs residing in the intestinal tissues are functionally distinct from APCs present in nonintestinal sites. To address this point, we stimulated OVA-specific TCR transgenic CD8+ T (OT-1) cells in vitro with OVA peptide-loaded DCs from MLN and spleen, respectively. Since naive murine CD8+ T cells express CCR9 (7, 11), we labeled the purified OT-1 cells with CFSE before culture to enable analysis of responding T cells only. When cultured with MLN DCs for 4 d, 39 ± 3% (mean value ± SD, n = 6) of responding OT-1 cells expressed CCR9 (Fig. 1, A and B) . Parallel cultures with spleen DCs gave rise to only 2.9 ± 0.9% (n = 6) CCR9+ cells among responding T cells. In agreement with a recent report, expression of α4β7 was also selectively induced by MLN DCs (Fig. 1 B) (12). OT-1 cells stimulated with anti-CD3 plus anti-CD28 mAbs were CCR9−α4β7 −. Similarly, T cells responding to peptide-loaded, DC-depleted APCs from MLN were CCR9−, and compared with MLN DCs these DC-depleted APCs were also poor in supporting T cell expression of α4β7 (Fig. 1, B and C). In contrast, DC and DC-depleted APCs from both MLN and spleen were potent inducers of CXCR3 on OT-1 cells (Fig. 1, B and C). Finally, OT-1 cells activated by MLN DCs, but not by spleen DCs, migrated to CCL25, whereas both populations migrated to the CXCR3 ligand CXCL10 (Fig. 1 D). Together these results demonstrate an explicit requirement for gut-associated lymphoid tissue (GALT) DCs in the generation of CCR9+α4β7 +CD8+ effector T cells.
Figure 1.

DCs from MLN, but not from spleen, are necessary and sufficient to induce a CCR9+α4β7 +CD62Llow phenotype on Ag-specific CD8+ T cells. CFSE-labeled OT-1 cells were stimulated with SIINFEKL peptide-pulsed CD11c+ DCs and CD11c− non-DCs or anti-CD3/CD28 mAbs. After 4 d of culture, the fraction of responding OT-1 cells expressing CCR9, CD62L, α4β7, and CXCR3 was determined by flow cytometry. (A) Representative results obtained for CCR9 and CD62L. (B) Percentages ± SD of responding OT-1 cells expressing indicated phenotype after stimulation with DCs (DC, n = 6), DC-depleted APCs (−DC, n = 3), or anti-CD3/CD28 mAbs (n = 3–5). Gray and white bars represent APCs from MLN and spleen, respectively. (C) Percentages ± SD of OT-1 cells expressing indicated phenotype as a function of cell division (n = 3). (D) Responsiveness of MLN DC (gray bars) and spleen DC (white bars) stimulated OT-1 cells to CCL25 and CXCL10. One representative experiment is shown.
We also investigated the role of APC in the down-regulation of CD62L on OT-1 cells. Both MLN and spleen DC populations efficiently induced a CD62Llow phenotype on activated OT-1 cells, a regulatory property not shared by DC-depleted APCs from MLN or spleen (Fig. 1, A and B). This was not due to differences in cell cycle progression, since OT-1 cells that had undergone a large number of divisions in the absence of DCs maintained high levels of CD62L expression (Fig. 1 C). Thus, although DCs are also required to support an efficient loss of CD62L on OT-1 cells, this capacity is not restricted to GALT DCs.
Both CD8α+ and CD8α− MLN DC Support the Antigen-dependent Generation of CCR9+α4β7 +CD62L− T Cells.
DCs can be divided into discrete subsets based on phenotypic and functional differences. Since the relative number of these DC subsets differs in various lymphoid organs, the selective capacity of MLN DCs to induce CCR9 and α4β7 could potentially reflect differences in DC subset composition in MLN and spleen, respectively. Therefore, we activated CFSE-labeled OT-1 cells with peptide-loaded CD8α+ and CD8α− MLN DCs, respectively. OT-1 cells stimulated by both DC subsets expressed CCR9 and α4β7 and down-regulated CD62L (Fig. 2) . Thus, the selective capacity of DCs in the MLN to instruct T cells to express CCR9 and α4β7 is not related to a prevalence of a particular DC subset in this lymphoid organ.
Figure 2.
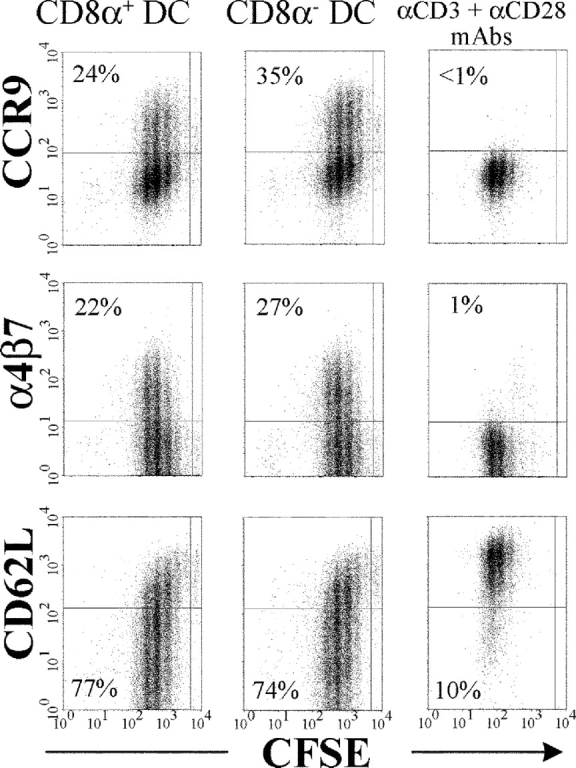
MLN CD8α1 and CD8α− DC subsets regulate OT-1 cell expression of CCR9, α4β7, and CD62L in a similar manner. CFSE-labeled OT-1 cells (105) were stimulated for 4 d with Ag-pulsed CD8α+ and CD8α− DC subsets from MLN (5 × 104 of each) or by anti-CD3/CD28 mAbs, and CCR9, α4β7, and CD62L were analyzed by flow cytometry. The percentage of responding OT-1 cells expressing a CCR9+, α4β7 +, or a CD62Llow phenotype is indicated for each analysis.
In Vivo Gut-homing CD8+ T Lymphocytes Selectively Arise in the GALT and Are Efficiently Generated Only in the Presence of Adjuvant.
Next, we performed OT-1 cell adoptive transfer experiments to investigate the nature of, and requirements for, the in vivo differentiation of gut-homing CD8+ T cells. In this transfer model, OT-1 cells start to proliferate ∼24 h after immunization, are retained within the lymphoid organ for up to ∼48 h, and start to appear in the peripheral tissues 3-d postimmunization (not depicted and reference 7). To enable identification of the donor cells, Ly5.2+ OT-1 cells were adoptively transferred to C57BL/6J-Ly5.1 recipient mice, and the phenotype of Ly5.2+ donor cells in the PLN, MLN, spleen, lung, and the IEL compartment was analyzed by flow cytometry 3 d after i.p. immunization with OVA plus pI:C. The majority of OT-1 cells in MLN, but not PLN or spleen, expressed CCR9 and α4β7 (Fig. 3 A). In all of these lymphoid organs, OT-1 cells had acquired expression of CXCR3 (not depicted) and down-regulated CD62L, indicating a similar degree of activation in these tissues. OT-1 cells that had migrated to the IEL compartment displayed a highly homogenous phenotype, expressing α4β7 and CCR9 but not CD62L. In contrast, OT-1 cells accumulating in the lung were a more heterogeneous population that appeared to phenotypically reflect OT-1 cells generated in both MLN, PLN, and spleen (Fig. 3 A). Thus, during an ongoing systemic immune response, MLN, but not PLN or spleen, support the differentiation of Ag-specific CD8+ T cells into activated cells, displaying the phenotypic signature of CD8+ T cells migrating into the small intestinal epithelium.
Figure 3.
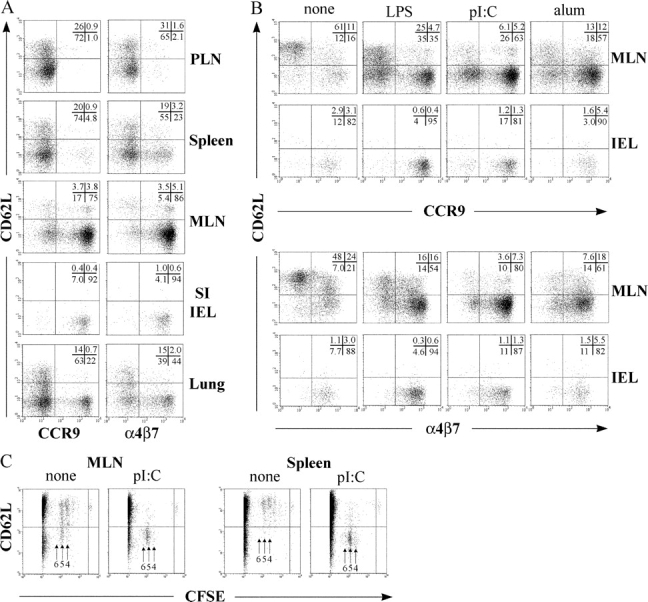
In vivo gut-homing OT-1 cells express a CCR9+α4β7 +CD62Llow phenotype which is induced efficiently only in GALT and after adjuvant-triggered inflammation. OT-1 cells were injected i.v. into C57BL/6J-Ly5.1 mice, and 2 d later mice received OVA i.p., either alone or in combination with adjuvant. (A) Flow cytometry analysis of OT-1 cells in indicated organs 3 d after immunization with OVA and pI:C. (B) Phenotype of OT-1 cells in the MLN and IEL compartment 3 d after immunization with OVA alone or OVA with adjuvant. (C) CFSE-labeled OT-1 cells were transferred into C57BL/6J-Ly5.1 mice, and donor cells in MLN and spleen were analyzed by flow cytometry 2 d after immunization with OVA or OVA plus pI:C. Results are representative of two to three performed experiments.
Since DCs tend to undergo maturation during isolation procedures, we investigated the in vivo requirements for adjuvant in the regulation of these homing receptors. OT-1 recipient mice were immunized i.p. with either OVA alone or in combination with LPS, pI:C, or alum. Whereas LPS and pI:C represent toll-like receptor (TLR)–dependent adjuvants (TLR4 and TLR3, respectively), the inflammatory response elicited by alum is TLR independent (13). Irrespective of the adjuvant used, a large number OT-1 cells in MLN 3 d after immunization were CCR9+α4β7 +CD62L− (Fig. 3 B). In mice receiving OVA alone, the majority of OT-1 cells in MLN maintained expression of CD62L and were CCR9−. The fraction of α4β7 + OT-1 cells was also relatively low compared with mice receiving OVA in the presence of adjuvant. In contrast, adjuvant was not required for induction of CXCR3 (not depicted). To rule out the possibility that the adjuvant-dependent regulation of CCR9, α4β7, and CD62L reflected increased cell division of OT-1 cells in the presence of adjuvant, CFSE-labeled OT-1 cells were transferred into recipient mice, and their expression of CD62L and intensity of CFSE fluorescence were determined 2 d after immunization. Both in the presence of pI:C and in the absence of adjuvant, the majority of OT-1 cells had undergone four to five cell divisions; however, only immunization with adjuvant supported efficient cell cycle–dependent down-regulation of CD62L (Fig. 3 C). Finally, irrespective of immunization strategy being employed, OT-1 cells migrating to the small intestinal epithelium expressed CCR9 and α4β7 and had down-regulated CD62L (Fig. 3 B). These results demonstrate that the in vivo acquisition of a gut-homing phenotype on Ag-activated CD8+ T cells is restricted to the GALT and is highly dependent on the presence of adjuvants.
OT-1 Lymphocyte Localization to the Small Intestinal Epithelium After Immunization with OVA and Adjuvant Is CCR9 Dependent.
To determine the importance of CCR9 in the localization of recently activated CD8+ T cells to the small intestinal epithelium during an Ag plus adjuvant-driven immune response, CCR9−/−OT-1 (Ly5.2+) cells (see Materials and Methods) were coinjected with WT OT-1 (Ly5.1+Ly5.2+) cells into C57BL/6J-Ly5.1 recipient mice, and the percentage of each population in the MLN, lung, and small intestine was determined 3 d after immunization with OVA and LPS. As shown in Fig. 4 , the CCR9−/− to WT OT-1 cell ratio in the MLN and lung remained similar to the input ratio. In marked contrast, the CCR9−/− to WT OT-1 cell ratio in the small intestinal epithelium was reduced ∼10-fold compared with the input ratio (Fig. 4, B and C). Thus, CCR9 plays a critical and selective role in CD8+ T cell localization to the small intestinal epithelium. The role of CCR9 was far greater than that implied from previous studies using neutralizing anti-CCL25 antibody (7), potentially due to an inability of the antibody to fully neutralize epithelial-derived CCL25 in vivo.
Figure 4.

OT-1 lymphocyte localization to the small intestinal epithelium after immunization with OVA and adjuvant is CCR9 dependent. CCR9−/− (Ly5.1−Ly5.2+) and WT (Ly5.1+Ly5.2+) OT-1 cells were coinjected (3–5 × 106 total cells) i.v. into C57BL/6J-Ly5.1 (Ly5.1+Ly5.2−) mice. Mice received OVA and LPS i.p. 2 d after cell transfer, and the percentage of CCR9−/− and WT OT-1 cells among CD8β1T cells was determined in each organ by flow cytometry analysis 3 d later. (A and B) Representative flow cytometry analysis from one experiment of three performed. (C). The CCR9−/− to WT OT-1 cell ratio in the MLN and IEL. Organ ratio was determined by dividing the percentage of CCR9−/− (Ly5.2+) OT-1 cells with the percentage of WT (Ly5.1+Ly5.2+) OT-1 cells in each organ. Results are mean (SEM) of four mice in each group and from one representative experiment of three performed. *P = 0.0286. Dotted line represents the CCR9−/− to WT OT-1 cell input ratio.
Discussion
In the course of DC–T cell interactions, DCs have proven instrumental in shaping the magnitude and nature of the induced adaptive response. For example, DCs are involved in the regulation of immunogenic versus tolerogenic responses and in the development of CD4+ Th1 versus Th2-associated immunity (14). In the present study, we demonstrate an additional and central role for DCs in the generation of tissue tropic effector T lymphocyte subsets. Our results reveal that DCs (but not other APCs) residing in the MLN, but not in spleen, directly support the generation of Ag-responding T cells expressing a CCR9+ α4β7 + phenotype, identical to that of T cells entering into the small intestinal epithelium. Since DCs in PP were also recently shown to imprint expression of α4β7 and responsiveness to CCL25 among responding T cells (9), it appears that GALT DCs share a capacity to induce a gut-homing phenotype on effector T cell populations. Differences in DC subset composition in GALT, compared with other secondary lymphoid organs, are unlikely to account for this capacity, since both CD8α+ and CD8α− DCs in MLN were able to support T cell expression of CCR9 and α4β7. Rather, since immature PP DCs are in close proximity to the intestinal surface (15), and MLN DCs are thought to derive from the intestinal mucosa, it seems likely that the intestinal mucosal microenvironment is conferring on intestinal DCs the ability to generate gut-homing T cell populations. Our results also demonstrate a critical role for DCs in down-regulating CD62L on responding T cells. Thus, DCs appear to play a dual role in the generation of gut-homing T cell populations, first in providing GALT-specific signals, leading to the generation of CCR9+α4β7 + T cells, and second in a nontissue selective manner, by down-regulating CD62L and thus preventing T cell reentry into secondary lymphoid organs (16).
We also demonstrated that an efficient generation of gut tropic T cells in vivo requires adjuvant. Adjuvants are potent in inducing DC maturation, and although DCs can support initial T cell activation after immunization with protein Ags only, such a response is driven by immature DCs, as evidenced by a tolerogenic rather than an immunogenic outcome (17). Hence, our results indicate that only mature DCs can support the generation of gut-homing T cells in vivo and are in agreement with previous reports demonstrating an important role for CD40 (18) and CD80/CD86 (19) for CD8+ T cells localization to the intestinal epithelium, as these costimulatory molecules are expressed at high levels only by mature DCs (14). However, since engagement of CD40L by CD40 and CD28 by CD80/CD86 is not confined to the GALT, other molecular interactions must operate in parallel or in a sequential manner in order to drive the GALT-selective generation of gut tropic T cells. Such sequential cross-regulation by DCs and T cells operates, for example, during the adjuvant-dependent differentiation of naive CD4+ T cells into CXCR5+ follicular helper T cells (20). A few OT-1 cells expressing a gut tropic phenotype were also generated after immunization with OVA alone, and cells localizing to the small intestinal epithelium under these conditions were invariably CCR9+α4β7 +CD62L−, suggesting that there may be low grade homeostatic inflammation in GALT, potentially driven by adjuvants provided by the enteric microflora. However, the efficient generation of gut tropic T cells in the presence of adjuvant indicates that the majority of T cells within the normal intestinal mucosa are specific to Ag that has been presented in the context of an inflammatory response.
In conclusion, the current study provides several important insights into the generation of the intestinal T cell compartment and intestinal acquired immune responses. First, we provide compelling evidence for the importance of GALT DCs in the generation of gut tropic T cells; second, we demonstrate a requirement for adjuvant for the efficient DC-dependent generation of gut tropic T cells and finally we show, to our knowledge, the first direct evidence for a role of CCR9 in T cell localization to the small intestinal epithelium. Together, these results suggest that targeting GALT DCs and CCR9 will provide a mechanism for modulating the generation of gut tropic T cells and T cell entry to the intestinal mucosa, respectively—physiological processes highly relevant to the development of mucosal vaccines and treatment of inflammatory bowel disease.
Acknowledgments
We would like to thank Dominic Picarella and Dulce Soler (Millenium Pharmaceuticals, Inc., Cambridge, MA) for kindly providing the anti-CXCR3 antibody.
This work was supported by grants to W. Agace from the Swedish Medical Research Council (MFR 3131), the Crafoordska, Österlund, Åke Wiberg, Richard and Ruth Julins, Nanna Svartz and Kocks Foundations, the Swedish Foundation for Strategic Research “Microbes and Man” research program, the Lund Family American Cancer Society, and the Royal Physiographic Society. Construction of the CCR9-deficient mice was supported by institutional grants from Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, and a specific grant from the European Commission (project QLG1-CT1999-00202) to B. Malissen. W. Agace is an Assistant Professor with the Swedish Medical Research Council.
B. Johannsson-Lindbom and M. Svensson contributed equally to this work.
References
- 1.Kunkel, E.J., and E.C. Butcher. 2002. Chemokines and the tissue-specific migration of lymphocytes. Immunity. 16:1–4. [DOI] [PubMed] [Google Scholar]
- 2.Berlin, C., R.F. Bargatze, J.J. Campbell, U.H. von Andrian, M.C. Szabo, S.R. Hasslen, R.D. Nelson, E.L. Berg, S.L. Erlandsen, and E.C. Butcher. 1995. alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 80:413–422. [DOI] [PubMed] [Google Scholar]
- 3.Zabel, B.A., W.W. Agace, J.J. Campbell, H.M. Heath, D. Parent, A.I. Roberts, E.C. Ebert, N. Kassam, S. Qin, M. Zovko, et al. 1999. Human G protein–coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J. Exp. Med. 190:1241–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kunkel, E.J., J.J. Campbell, G. Haraldsen, J. Pan, J. Boisvert, A.I. Roberts, E.C. Ebert, M.A. Vierra, S.B. Goodman, M.C. Genovese, et al. 2000. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J. Exp. Med. 192:761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wurbel, M.A., J.M. Philippe, C. Nguyen, G. Victorero, T. Freeman, P. Wooding, A. Miazek, M.G. Mattei, M. Malissen, B.R. Jordan, et al. 2000. The chemokine TECK is expressed by thymic and intestinal epithelial cells and attracts double- and single-positive thymocytes expressing the TECK receptor CCR9. Eur. J. Immunol. 30:262–271. [DOI] [PubMed] [Google Scholar]
- 6.Lefrancois, L., C.M. Parker, S. Olson, W. Muller, N. Wagner, M.P. Schon, and L. Puddington. 1999. The role of beta7 integrins in CD8 T cell trafficking during an antiviral immune response. J. Exp. Med. 189:1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Svensson, M., J. Marsal, A. Ericsson, L. Carramolino, T. Broden, G. Marquez, and W.W. Agace. 2002. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J. Clin. Invest. 110:1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell, D.J., and E.C. Butcher. 2002. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J. Exp. Med. 195:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mora, J.R., M.R. Bono, N. Manjunath, W. Weninger, L.L. Cavanagh, M. Rosemblatt, and U.H. Von Andrian. 2003. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 424:88–93. [DOI] [PubMed] [Google Scholar]
- 10.Wurbel, M.A., M. Malissen, D. Guy-Grand, E. Meffre, M.C. Nussenzweig, M. Richelme, A. Carrier, and B. Malissen. 2001. Mice lacking the CCR9 CC-chemokine receptor show a mild impairment of early T- and B-cell development and a reduction in T-cell receptor gammadelta(+) gut intraepithelial lymphocytes. Blood. 98:2626–2632. [DOI] [PubMed] [Google Scholar]
- 11.Carramolino, L., A. Zaballos, L. Kremer, R. Villares, P. Martin, C. Ardavin, A.C. Martinez, and G. Marquez. 2001. Expression of CCR9 beta-chemokine receptor is modulated in thymocyte differentiation and is selectively maintained in CD8(+) T cells from secondary lymphoid organs. Blood. 97:850–857. [DOI] [PubMed] [Google Scholar]
- 12.Stagg, A.J., M.A. Kamm, and S.C. Knight. 2002. Intestinal dendritic cells increase T cell expression of alpha4beta7 integrin. Eur. J. Immunol. 32:1445–1454. [DOI] [PubMed] [Google Scholar]
- 13.Barton, G.M., and R. Medzhitov. 2002. Toll-like receptors and their ligands. Curr. Top. Microbiol. Immunol. 270:81–92. [DOI] [PubMed] [Google Scholar]
- 14.Guermonprez, P., J. Valladeau, L. Zitvogel, C. Thery, and S. Amigorena. 2002. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 20:621–667. [DOI] [PubMed] [Google Scholar]
- 15.Iwasaki, A., and B.L. Kelsall. 2000. Localization of distinct Peyer's patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3alpha, MIP-3beta, and secondary lymphoid organ chemokine. J. Exp. Med. 191:1381–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steeber, D.A., N.E. Green, S. Sato, and T.F. Tedder. 1996. Lymphocyte migration in L-selectin-deficient mice. Altered subset migration and aging of the immune system. J. Immunol. 157:1096–1106. [PubMed] [Google Scholar]
- 17.Hawiger, D., K. Inaba, Y. Dorsett, M. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lefrancois, L., S. Olson, and D. Masopust. 1999. A critical role for CD40–CD40 ligand interactions in amplification of the mucosal CD8 T cell response. J. Exp. Med. 190:1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim, S.K., D.S. Reed, S. Olson, M.J. Schnell, J.K. Rose, P.A. Morton, and L. Lefrancois. 1998. Generation of mucosal cytotoxic T cells against soluble protein by tissue-specific environmental and costimulatory signals. Proc. Natl. Acad. Sci. USA. 95:10814–10819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker, L.S., A. Gulbranson-Judge, S. Flynn, T. Brocker, C. Raykundalia, M. Goodall, R. Forster, M. Lipp, and P. Lane. 1999. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5–positive CD4 cells and germinal centers. J. Exp. Med. 190:1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]