

Abstract
Toll–IL-1–resistance (TIR) domain–containing adaptor-inducing IFN-β (TRIF)–related adaptor molecule (TRAM) is the fourth TIR domain–containing adaptor protein to be described that participates in Toll receptor signaling. Like TRIF, TRAM activates interferon regulatory factor (IRF)-3, IRF-7, and NF-κB-dependent signaling pathways. Toll-like receptor (TLR)3 and 4 activate these pathways to induce IFN-α/β, regulated on activation, normal T cell expressed and secreted (RANTES), and γ interferon–inducible protein 10 (IP-10) expression independently of the adaptor protein myeloid differentiation factor 88 (MyD88). Dominant negative and siRNA studies performed here demonstrate that TRIF functions downstream of both the TLR3 (dsRNA) and TLR4 (LPS) signaling pathways, whereas the function of TRAM is restricted to the TLR4 pathway. TRAM interacts with TRIF, MyD88 adaptor–like protein (Mal)/TIRAP, and TLR4 but not with TLR3. These studies suggest that TRIF and TRAM both function in LPS-TLR4 signaling to regulate the MyD88-independent pathway during the innate immune response to LPS.
Keywords: innate immunity, endotoxin, interferon, signal transduction, host defense
Introduction
The Toll-like receptor (TLR) family is the essential recognition and signaling component of mammalian host defense (1–3). At least 10 TLRs have been cloned in mammals, which recognize molecular products derived from all the major classes of pathogens. (1–3). Toll signaling to NF-κB originates from the conserved Toll–IL-1–resistance (TIR) domain, which mediates recruitment of the TIR domain–containing adaptor molecule, myeloid differentiation factor 88 (MyD88 [4]), a critical adaptor molecule used by all TLRs (5). The recruitment of MyD88 to proximal TIR domains of activated TLRs allows for the interaction and activation of the IL-1R–associated kinase (IRAK) family members (6, 7) and the subsequent activation of TNF receptor–associated factor (TRAF)-6 (8). These events, at a minimum, result in NF-κB activation via the IκB kinase (IKK)α−β−γ complex (9).
Whereas most of the TLRs seem to be absolutely dependent on the expression of MyD88 for all of their functions, TLR3 and TLR4 are unique in their ability to activate both MyD88-dependent and MyD88-independent responses (10–13). A feature of MyD88-independent signaling is the induction of a DC maturation pathway and the induction of the type 1 interferon (IFN-β) (12–16). The transcription enhancer of the IFN-β promoter binds NF-κB, interferon regulatory factor (IRF)-3, and ATF-2–c-Jun. Whereas all TLRs activate NF-κB and ATF2–c-Jun, not all TLRs induce IFN-β because not all TLRs induce IRF-3 activation. Thus, TLR3 and TLR4 appear to have evolutionarily diverged from other TLRs to activate gene expression programs and trigger antiviral responses by a mechanism involving the coordinate activation of NF-κB and IRF-3 (17).
MyD88 adaptor–like protein (Mal) (18), also called TIRAP (19), is a related MyD88-like protein, which was discovered based on its ability to mediate TLR4 signaling. Mal/TIRAP has been implicated in LPS-induced IFN-β induction in vitro (12, 20). However, studies with Mal/TIRAP gene–targeted mice show that Mal/TIRAP functions in the MyD88-dependent NF-κB activation pathway after LPS stimulation, and engagement of TLR2 by its ligands (21, 22). A third adaptor protein, TIR domain–containing adaptor–inducing IFN-β (TRIF) (16), also known as TIR-containing adaptor molecule (TICAM)-1 (13), interacts with TLR3 and mediates the TLR3-dependent induction of IFN-β via NF-κB and IRF-3 activation.
Constitutively expressed IRF-3 has been implicated in the induction of IFN-β (23–25), RANTES (26, 27), and ISG-54/56 expression (28). IRF-3 is activated after phosphorylation on a cluster of specific COOH-terminal serine residues (23, 29, 30), facilitating its dimerization and interaction with the coactivators CBP and p300 (31–34). The activated IRF-3 complex then translocates to the nucleus where it regulates the transcription of target genes (27, 31). IRF-7 is a related transcriptional regulator that is expressed mostly in lymphoid cells and is essential for IFN-α gene expression (35, 36). The transcription of IRF-7 is induced by IFN and posttranslationally activated by phosphorylation on its COOH-terminal serine residues, some of which are conserved with IRF-3 (35, 37). IKKɛ (38, 39) and TANK-binding kinase (TBK)1 (40–42) are key regulators of the IRF-3 and IRF-7 activation pathways in cells that have been exposed to some viruses and/or activated by dsRNA via TLR3 (43, 44). IKKɛ and TBK1 are also required components of the TRIF signaling pathway, resulting in IRF-3 activation (43).
Studies with IRF3-deficient mice have established an essential role for IRF-3 in LPS-induced IFN-β gene expression and endotoxin shock (45). However, the molecular mechanisms regulating the MyD88-independent LPS–TLR4 pathway to IRF-3 and NF-κB activation are unknown. Here, we have identified a fourth TIR domain–containing adaptor molecule, which we have named TRIF-related adaptor molecule (TRAM). TRAM, like all of the TIR domain–containing adaptor molecules, activates NF-κB. In addition, TRAM, like TRIF, activates IRF-3 and IRF-7. Unlike any of the other known TIR domain–containing adapters, TRAM appears to be restricted to the LPS activation (TLR4) pathway, whereas TRIF plays a role in both TLR3 and TLR4 pathways leading to IRF target gene expression. Our findings suggest that TRAM may have evolved to mediate TLR4-specific signals, resulting in a gene expression profile that is not shared by TLR3.
Materials and Methods
Reagents.
The IRF-3–ΔN, Gal4–IRF-3, and Gal4–luciferase reporter gene were from T. Fujita (The Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). IKKɛ–k38a and TBK1–k38a were as described (38, 43). IRF-7, IRF-7–ΔN, and Gal4–IRF-7 were from P. Pitha (Johns Hopkins University, Baltimore, MD). The RANTES reporter construct was as described (26). The γ interferon–inducible protein 10 (IP-10) reporter construct was from A. Luster (Massachusetts General Hospital, Boston, MA). The NF-κB–luciferase construct (18), pEF-Bos-Flag Mal, and Flag-TRIF were as described (43). The plasmids pEF-Bos-Flag-TRAM, TRAM-CFP, TRIF-CFP, and Mal-CFP were generated by PCR cloning from a human PBMC cDNA library. pEF-Bos-TRAM-TIR (aa 63–235), pEF-Bos-TRAM-C113H, TRAM-P112H, and TRIF-P434H were generated using the Quick Change site-directed mutagenesis kit (Stratagene). Polyclonal antibodies to IRF-3 were from Zymed Laboratories, and CBP were from Santa Cruz Biotechnology, Inc. pCMV-TRIFΔNΔC and MyD88-deficient mice were gifts from S. Akira (Osaka University, Osaka, Japan); the MyD88 knockout mice used were backbred onto a C57BL6 background for five generations. LPS-derived from Escherichia coli strain 011:B4 was purchased from Sigma-Aldrich, dissolved in deoxycholate, and reextracted by phenolchloroform as described (46). Poly IC was from Amersham Biosciences.
Stable Cell Lines.
We engineered clonal stable cell lines by transfecting HEK293 cells with chimeric fluorescent protein TLR constructs as described (47). A HEK293 cell line stably expressing both TLR4 and MD-2 was generated by retroviral transduction of HEK-TLR4 cells with a retrovirus encoding human MD2 (48). HEK-TLR3, HEK–IRF-3–GFP (43), and U373–CD14 cells (49) were as described.
Electrophoretic Mobility Shift Assays.
BM-derived macrophages were cultured from C57Bl6 mice or age- and sex-matched MyD88−/− mice for 8 d in M-CSF (10 ng/ml). Nuclear extracts from 5 × 105 cells were purified after LPS (10 ng/ml), Malp-2 (1 nM), or Poly I:C (50 μg/ml) stimulation for the times indicated. The extracts were incubated with a specific probe for the interferon-stimulated response element (ISRE) consensus sequence (Promega), electrophoresed, and visualized by autoradiography (50). Supershift analysis was performed with antibodies to mouse IRF-3, p65, or IgG control.
ELISA.
Macrophages (5 × 104 cells per well) were seeded into 96-well plates for 24 h before stimulation with LPS, poly IC, or medium for 12 h. Cell culture supernatants were removed and analyzed for the presence of RANTES, IP-10, or TNFα by ELISA (R&D Systems).
Transfection Assays.
Cells were seeded into 96-well plates at a density of 1.5 × 104 cells per well and transfected 24 h later with 40 ng of the indicated luciferase reporter genes using Genejuice (Novagen). The thymidine kinase Renilla-luciferase reporter gene (Promega) (40 ng/well) was cotransfected in order that the data could be normalized for transfection efficiency. Cell lysates were prepared, and reporter gene activity was measured using the Dual Luciferase Assay System (Promega). Data are expressed as the mean relative stimulation ± SD. All of the experiments described were performed a minimum of three occasions and gave similar results.
Immunofluorescense and Confocal Microscopy.
A HEK293–IRF–GFP stable cell line was transiently transfected with Flag-tagged constructs as indicated. After allowing 2 d for protein expression to occur, the transfected cells were fixed, permeabilized, and stained with Cy3-conjugated anti-Flag antibody (clone M2; Sigma-Aldrich). DRAQ5 was added to counter stain nuclei. Cells were imaged by confocal microscopy using a Leica TCS SP2 AOBS microscope.
RNA Interference.
siRNA duplexes targeting the coding region of TRAM and lamin A/C were from Dharmacon: TRAM-siRNA sequences, GGAAGAAAGTCGTGGATT (product no D-004334–01™) and lamin A/C, CTGGACTTCCAGAAGAACA. siRNA duplexes targeting the 3′ UTR of TRIF were as described (13). To determine the efficiency of gene silencing, 293T cells (24 well plates, 4 × 104 cells/well) were transfected with 0.5 μg of plasmids encoding TRAM–CFP, TRIF–CFP, or Mal–CFP expression vectors. These cells were cotransfected with TRAM or lamin A/C siRNA duplexes (50 nM) using Mirus TransIT® TKO and TransIT-LT1® transfection reagents in a combination protocol exactly according to the manufacturers recommendations (Mirus). CFP fluorescence was quantified by flow cytometry (LSR; Becton Dickinson) 24 h later. For reporter assays, U373–CD14 cells or TLR3–expressing HEK293 cells (4 × 104 cells/well) were transfected with 0.5 μg of the RANTES reporter gene and 0.25 μg of a thymidine kinase–renilla reporter gene and cotransfected with 50 nM of siRNA targeting vectors as described above in 24-well tissue culture dishes. 36 h after transfection, cells were stimulated with LPS or dsRNA for ∼8 h before luciferase activity was measured.
Coimmunoprecipitation.
293T cells or TLR-expressing cells (10 cm plates) were transfected using GeneJuice (Novagen) with 4 μg of the indicated plasmids. Cells were lysed in 1 ml of lysis buffer (20 mM Tris-HCl, 2 mM EDTA, 137 mM NaCl, 0.5% Triton X-100, glycerol 10%, with protease inhibitors) 1-2 d after transfection. Polyclonal anti-GFP (Molecular Probes), anti–IRF-3, or anti-CBP antibodies were incubated with the cell lysates in protein A sepharose overnight. The immune complexes were precipitated and subjected to 4–15% SDS-PAGE and immunoblotted for Flag- or CFP/YFP-tagged adapters using the anti-Flag mAb M2 (Sigma-Aldrich) or anti-GFP mAb (CLONTECH Laboratories), which also recognizes the spectral variants of GFP.
Results
LPS and dsRNA Activate IRF-3 and IRF-7.
The promoters of RANTES and IP-10, like that of IFN-β, contains transcription factor binding elements for NF-κB and IRF-3 (26, 27). The expression of RANTES and IP-10 represent downstream targets of Toll receptors that are entirely independent of MyD88 expression after stimulation by LPS or dsRNA. Stimulation of mouse BM-derived macrophages with LPS (TLR4) or dsRNA (TLR3) induced RANTES secretion, an effect that was observed equally in BM macrophages deficient in MyD88 (Fig. 1 a). This was also true for IP-10 levels as measured by ELISA (unpublished data). In contrast, TLR2 signaling via lipopeptides absolutely requires MyD88 and does not lead to RANTES expression. TLR2-mediated production of TNFα was entirely absent in MyD88-deficient macrophages (unpublished data), in agreement with published studies (11, 51, 52).
Figure 1.

LPS and dsRNA activate IRF-3 and IRF-7. (a) BM-derived macrophages from WT and MyD88-deficient mice were stimulated with LPS (0.1–100 ng/ml), Malp-2 (5 nM), and dsRNA (1–100 μg/ml) for 12 h. The concentration of RANTES was measured by ELISA. (b) Nuclear extracts were isolated from WT and MyD88-deficient macrophages stimulated with LPS (10 ng/ml), Malp-2 (5 nM), and dsRNA (50 μg/ml) for 1 h and subjected to EMSA using a 32P-labeled ISRE consensus sequence (ISG-15) as a probe. Activated complexes were visualized by autoradiography. Activated ISRE DNA-binding complexes were preincubated with polyclonal antibody to IRF-3 or two control antibodies before incubation with the ISRE probe (right). (c) TLR3 and TLR4/MD2-expressing HEK293 cell lines were transfected with a luciferase reporter gene containing the Gal4 upstream activation sequence and with Gal4-DBD, Gal4–IRF-3, or Gal4–IRF-7 (40 ng). After 24 h, cells were stimulated with LPS (10 ng/ml), dsRNA (50 μg poly IC/ml), IL-1β (10 ng/ml), or left untreated for ∼8 h, and luciferase reporter gene activity was measured.
We next examined the effect of LPS and dsRNA on IRF-3 DNA binding activity. IRF-3 DNA binding activity was induced in both WT and MyD88-deficient macrophages after LPS and dsRNA stimulation (Fig. 1 b). The presence of IRF-3 in this ISRE DNA binding complex was confirmed by depletion (“gel shift”) analysis using antibodies to IRF-3 (Fig. 1 b, bottom). Stimulation of cells with the TLR2 ligand, Malp-2, did not result in IRF-3 activation. NF-κB was activated in WT cells by all stimuli and in MyD88-deficient macrophages after LPS or dsRNA stimulation (unpublished data).
We next addressed the question of whether the related transcriptional regulator IRF-7 was a target of TLR signaling. We employed an in vivo assay for IRF-7 activation, which utilizes a hybrid protein consisting of the yeast Gal4 DNA-binding domain (DBD) fused to IRF-7 lacking its own DBD (53). Reporter gene expression from the Gal4 upstream activation sequence in this assay requires IRF-7 activation (31). IRF-3 activation was also measured in this assay using a Gal4–IRF-3 fusion protein. Stimulation of TLR3 or TLR4/MD2-expressing HEK293 cells with dsRNA or LPS but not IL1β activated both IRF-3 and IRF-7 (Fig. 1 c). IRF7 plays a critical role in regulating IFN-α1 expression. Exogenously expressed IRF7 increased the activation of an IFN-α1 reporter construct when TLR4/MD2- or TLR3-expressing HEK293 cells were stimulated with LPS or dsRNA, whereas a dominant negative IRF7 mutant inhibited the effect (unpublished data). These observations are strong evidence that TLR3 and TLR4 activate IRF-3 and IRF-7 and, as a result, induce IRF target genes such as RANTES and IFNα/β.
Discovery of a Fourth TIR Domain–containing Adaptor Molecule, TRAM.
A search of the human genome for additional TIR domain–containing adaptor molecules resulted in the identification of a small protein fragment that shares sequence similarity with other TIR domain–containing proteins, most notably with TRIF/TICAM-1. A set of overlapping EST sequences were subsequently identified and used to clone the full-length cDNA of human and mouse TRAM, which share 75% sequence identity (sequence data available from GenBank/EMBL/DDBJ under accession nos. AY232653 and AY268050, respectively). The TRAM gene is located on human chromosome 5 (ENSEMBL ID: ENSG00000164226). TRAM is a 235 aa protein with a COOH-terminal TIR domain. Fig. 2 a shows a multiple sequence alignment of human and mouse TRAM with other human adapters and TLRs. The crystal structure of the TIR domain of TLR2 has been resolved. The TIR domain “BB loop” is an essential part of its structure, and this portion of the molecule appears to engage downstream elements such as adaptor molecules or other TLRs (3, 54). Most TIR domain BB loop sequences contain a conserved proline residue in the BB loop. When this residue is mutated to histidine, the mutant protein is typically unable to signal and may even function as a dominant suppressing mutation (18, 19, 55). Unlike the other known adaptor proteins, both human and mouse TRAM contain a cysteine residue at this position (Fig. 2 a, #). A proline residue resides directly adjacent to this residue in TRAM, at position 112.
Figure 2.
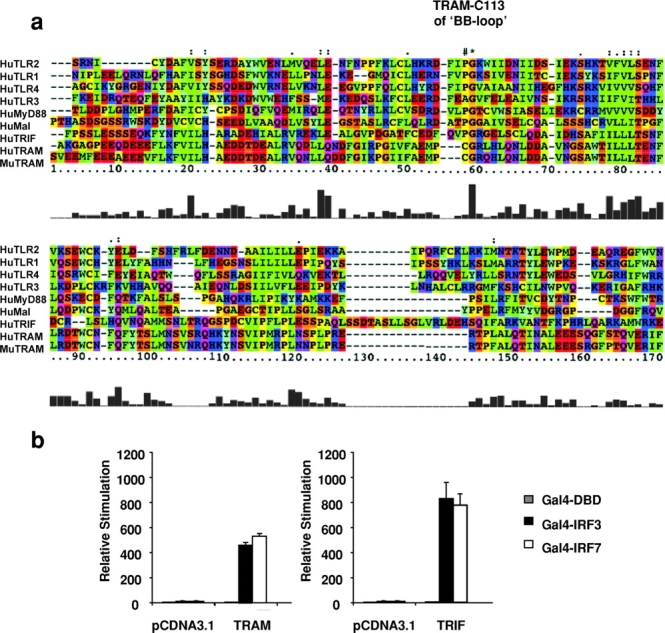
TRAM activates IRF-3 and IRF-7. (a) Alignment of TIR domains of TRAM, TRIF, MyD88, and Mal with TLR1, TLR2, TLR3, and TLR4. The amino acid colors are based on their physico-chemical properties where yellow = small, green = hydrophobic, turquoise = aromatic, blue = positively charged, and red = negatively charged. (b) HEK293 cells were transfected as in Fig. 1 c and cotransfected with 40 ng of TRAM or TRIF. After 24 h, luciferase reporter gene activity was measured.
Because of the similarity in the sequence of the TIR domain of TRAM and TRIF, we first compared the effect of TRIF and TRAM on IRF-3 and IRF-7 activation. Overexpression of TRAM activated the IRF-3 and IRF-7 response (Fig. 2 b). TRIF also activated both transcription factors (Fig. 2 b). As a consequence, TRAM and TRIF also induced the IFN-β, RANTES, IP-10, and IFN-α1/α2 promoters, all of which contain ISRE elements (unpublished data). These data imply that TRAM and TRIF also activate NF-κB, as some of these promoters (IFN-β, RANTES, and IP-10) also require NF-κB for full activity (see TRAM Also Activates NF-kB).
As a further test of TRAM- and TRIF-dependent IRF-3 activation, we examined their effects on the nuclear translocation of IRF-3. Overexpression of TRAM and TRIF in a stable cell line expressing a GFP chimera of IRF-3 resulted in the nuclear translocation of this IRF-3–GFP fusion protein (Fig. 3 a). TRIF has been shown recently to coimmunoprecipitate with IRF-3 (16). We were interested in determining if TRAM might also associate with IRF-3. When HEK293 cells were transfected with Flag-tagged TRAM and immunoprecipitated with antibody to endogenous IRF-3, Flag-tagged TRAM could be detected in the immunoprecipitated complex (Fig. 3 b, top). Immunoprecipitation with an anti-Flag antibody confirmed this interaction; endogenous and cotransfected IRF-3 could be detected in the immunoprecipitated complexes (Fig. 3 b, second panel). TRIF also interacted with endogenous and transfected IRF-3 in agreement with published studies (unpublished data). There were no nonspecific associations detected in cells lacking the transfected adaptor constructs. Although not shown, we also performed similar experiments with IRF-7. IRF-7 also interacted with TRAM and TRIF and vice versa.
Figure 3.
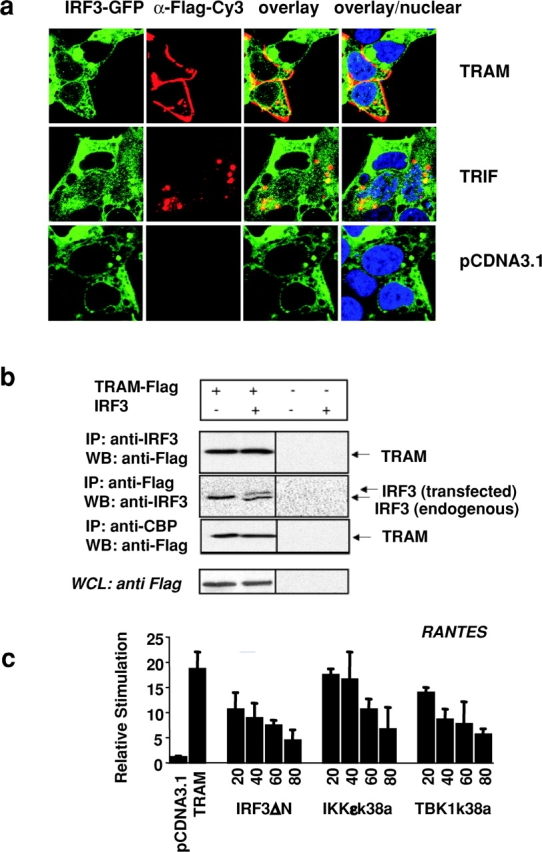
TRAM interacts with IRF-3 and CBP and signals via IKKɛ and TBK1. (a) IRF-3–GFP–expressing HEK293 cells were plated on 35-mm glass-bottom sterile tissue culture dishes and transiently transfected with 1 μg of Flag-tagged TRAM, TRIF, or pCDNA3.1 and visualized 24 h later by confocal microscopy. (b) 293T cells were transfected with 4 μg of Flag-TRAM with or without a plasmid encoding IRF-3 (untagged) as indicated. 24 h later, whole cell lysates were immunoprecipitated with anti–IRF-3, anti-Flag, or anti-CBP, and the immunoprecipitated complexes were immunoblotted for Flag-tagged TRAM and IRF-3. Whole cell lysates (WCL) were also analyzed for Flag-tagged proteins. (c) HEK293 cells were transfected with the RANTES luciferase reporter gene and TRAM (20 ng) and cotransfected with increasing concentrations of IKKɛ-k38a, TBK1-k38a, or IRF3-ΔN from 10, 20, 30, 40, 60, and 80 ng. Luciferase reporter gene activity was measured 24 h after transfection.
Activated IRF-3 must associate with the coactivators CBP and p300 in order to enhance target gene expression (31–34). When endogenous CBP was immunoprecipitated from cell lysates expressing transfected Flag-tagged TRAM, TRAM could be detected in these immunoprecipitated complexes (Fig. 3 b, third panel). This was also true for transfected TRIF (unpublished data).
The noncanonical IκB kinases, IKKɛ (38, 39) and TBK1 (40–42), are key regulators of the IRF3 activation pathway resulting from viral exposure and activation of TLR3 or TRIF-signaling cascades (43, 44). IKKɛ has also been implicated in LPS signaling (56). We next examined the effect of dominant negative mutants of IKKɛ and TBK1 on TRAM signaling. We used the RANTES reporter gene construct to address this issue. Cells were cotransfected with TRAM, which activates downstream molecules as a result of overexpression, and the kinase inactive mutants of IKKɛ (IKKɛ-k38a) or TBK1 (TBK1-k38a). Both mutants inhibited TRAM-induced RANTES reporter activation in a dose-dependent manner, suggesting that these two kinases may also function downstream of TRAM. Together these observations provide strong evidence that TRAM and TRIF are important components of the IRF3 signaling pathway and suggest that these adaptor proteins form a multiprotein complex with IRF-3/7, CBP, and the IRF-3/7 kinases (IKKɛ and TBK1) during signal transduction.
TRAM Activates the IRF Pathway in the TLR4 but Not the TLR3 Signaling Pathway.
We next generated a series of TRAM mutants and examined their ability to activate the RANTES reporter gene. Transfection of HEK293 cells with a plasmid encoding the TIR domain alone of TRAM (TRAM-TIR) induced the RANTES reporter, although this response was considerably less than that observed with the full-length TRAM cDNA (Fig. 4 a). TRAM contains a cysteine residue (C113) in the BB loop with an adjacent proline residue (P112). Mutation of the proline residue to histidine (TRAM-P112H) significantly impaired the RANTES-inducing activity of TRAM, whereas mutation of the cysteine residue at position 113 (TRAM-C113H) completely abrogated all activity (Fig. 4 a). This suggested that either TRAM-C113H or TRAM-P112H might function as a dominant interfering mutant of TRAM activity. The effect of these TRAM constructs was similar when an IP-10 promoter–based reporter construct was assessed (unpublished data).
Figure 4.

TRAM mediates the TLR4 pathway to IRF-3 and IRF-7. (a) HEK293 cells were transfected with 40 ng of a RANTES reporter construct and cotransfected with TRAM, TRAM-TIR, TRAM-C113H, or TRAM-P112H. (b) TLR4/MD2- and TLR3-expressing HEK293 cell lines were transfected with a luciferase reporter gene containing the Gal4 upstream activation sequence and cotransfected with Gal4–DBD, Gal4–IRF-3, or Gal4–IRF-7 or the RANTES luciferase reporter gene as well as TRAM-C113H or TRIF-ΔNΔC. The next day, cells were stimulated with LPS (10 ng/ml) or dsRNA (50 μg/ml poly IC) or left untreated for ∼8 h, and luciferase reporter gene activity was measured.
We subsequently examined the effect of these TRAM mutants on TLR-mediated signaling that culminates in RANTES promoter activation or the activation of the transcription factors IRF-3 and IRF-7. We focused on TLR3 and TLR4 because of their unique abilities to activate both NF-κB and IRF-3. Neither the TRAM-TIR domain nor the TRAM-P112H mutants had any dominant negative inhibitory activity on either TLR-dependent IRF-3 pathway tested (unpublished data). Transfection of HEK-TLR3 cells with TRAM-C113H had no inhibitory effect on dsRNA-induced RANTES response (Fig. 4 b). In contrast, LPS-induced activation of the RANTES reporter via TLR4 was impaired by the TRAM-C113H mutant (Fig. 4 b, left). The LPS-dependent induction of the RANTES reporter gene was considerably less potent than that observed after TLR3 stimulation. The TRAM-C113H mutant also inhibited the TLR4- but not the TLR3-dependent activation of IRF-3 and IRF-7 (Fig. 4 c). The TRAM-C113H mutant also inhibited the TLR4- but not the TLR3-dependent activation of an IP-10 reporter construct (unpublished data). We also examined the role of TRIF in the TLR3- and TLR4-dependent pathways in parallel by expressing a dominant negative mutant of TRIF lacking both the NH2-terminal and COOH-terminal regions surrounding the TIR domain (TRIFΔNΔC) [16]). As expected, this mutant completely suppressed the TLR3-dependent response (Fig. 4 b). The TRIFΔNΔC mutant also inhibited the TLR4 response, although the effect was less potent than that observed in the TLR3 pathway under identical experimental conditions (Fig. 4 d, right). Together, these observations suggest that TRIF regulates the TLR3 and TLR4 pathways to IRF target genes, whereas TRAM appears to be TLR4 specific.
TRAM Also Activates NF-κB.
We subsequently addressed the role of TRAM in the NF-κB activation pathway. Transfection of HEK293 cells with TRAM resulted in a potent NF-κB activation response (Fig. 5 a). The isolated TIR domain of TRAM also induced a robust NF-κB response, though this was considerably less than that observed with the full-length gene (Fig. 5 a). Neither the TRAM-P112H nor the TRAM-C113H mutants induced NF-κB activation. Thus, like all of the other known adapters, TRAM is also an NF-κB activator.
Figure 5.

TRAM activates NF-κB and is specific to the TLR4 pathway. (a) HEK293 cells were transfected with 40 ng of an NF-κB reporter construct and cotransfected with TRAM, TRAM-TIR, TRAM-C113H, and TRAM-P112H. (b) TLR-expressing HEK293 stable cell lines were transfected with 40 ng of an NF-κB reporter gene and cotransfected with increasing concentrations of TRAM-C113H. 1 d after transfection, TLR-expressing cells were stimulated with Malp-2 (2 nM), dsRNA (100 μg/ml poly IC), LPS (10 ng/ml), R-848 (10 μM), IL1β (10 ng/ml) or TNFα (10 ng/ml) or left untreated or 8 h, and luciferase reporter gene activity was measured.
The TRAM-C113H–negative interfering mutant was next tested for its ability to inhibit TLR-dependent signaling to NF-κB. TLR2-, TLR3-, TLR4/MD2-, TLR7-, and TLR8-expressing HEK293 cells were transfected individually with an NF-κB reporter gene and cotransfected with increasing concentrations of TRAM-C113H. After stimulation with their cognate TLR agonists, luciferase reporter gene activity was measured. NF-κB activation induced by the TLR2 agonist Malp-2, the TLR3 agonist dsRNA, the TLR7 and TLR8 agonists, R-848, IL-1β, or TNFα were all unaffected when cells were cotransfected with the suppressing TRAM-C113H mutant (Fig. 5 b). In striking contrast to these negative results, the TRAM-C113H mutant inhibited LPS-induced NF-κB activity in HEK–TLR4–MD2 cells. The TRAM-P112H had no inhibitory activity on any TLR pathway to NF-κB, including the TLR4 pathway, confirming the importance of the C113 residue for this response. These observations suggest that TRAM regulates NF-κB and IRF-3/7 in the LPS/TLR4 signaling pathway.
TRIF and TRAM Cooperate in the IRF-3 Activation Pathway.
We examined the effect of the TRIFΔNΔC mutant on TRAM-induced RANTES promoter activation in order to define the relationship between TRIF, TRAM, and the TLR4 pathway. The TRIFΔNΔC construct inhibited the TRIF-induced RANTES reporter gene response (Fig. 6 a, hatched bars). The TRIFΔNΔC mutant completely abrogated the TRAM-induced RANTES reporter gene response (Fig. 6 a). The TRAM-C113H mutant also abrogated the induction of the RANTES reporter gene in response to TRAM overexpression (Fig. 6 a, far right) but had no effect on the response to TRIF overexpression (Fig. 6 a, hatched bars). The observation that a TRIF dominant negative construct blocked TRAM activity but not vice versa suggests that TRAM signaling requires TRIF.
Figure 6.
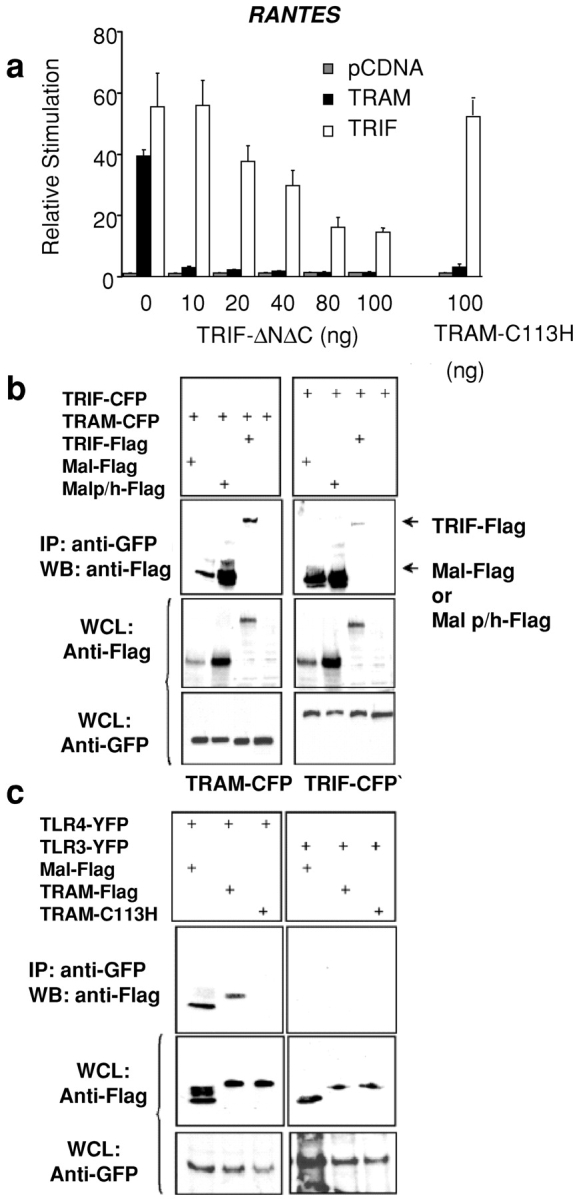
TRAM signaling requires the expression of TRIF. (a) HEK293 cells were transfected with the RANTES luciferase reporter gene, TRAM or TRIF (40 ng), and cotransfected with TRIF-ΔNΔC or TRAM-C113H. Luciferase reporter gene activity was measured 24 h later. (b) 293T cells were transfected with 4 μg of TRAM-CFP or TRIF-CFP and cotransfected with Flag-Mal, Flag-Mal-P125H, or Flag-TRIF. Whole cell lysates were harvested 48 h later and immunoprecipitated with anti-GFP antibody (which also immunoprecipitates cyan fluorescent protein [CFP] or yellow fluorescent protein [YFP]). Immunoprecipitated complexes were resolved by SDS-PAGE and immunoblotted for Flag-tagged adapters. Whole cell lysates (WCL) were also analyzed for CFP- and Flag-tagged proteins by immunoblotting. (c) Stable TLR4YFP- or TLR3YFP-expressing HELA cells were transfected with 4 μg of plasmid encoding Flag-Mal, Flag-TRAM, or Flag TRAM-C113H. 48 h later, whole cell lysates were immunoprecipitated with anti-GFP antibody, and immunoprecipitated complexes were immunoblotted for Flag-tagged adapters. Western blotting of lysates demonstrates expression of stable TLRs and transfected adaptor proteins.
Subsequently, we performed coimmunoprecipitation experiments on cells that heterologously expressed both of these gene products and the related adaptor molecule Mal/TIRAP. These immunoprecipitation studies demonstrated that TRAM interacted with both TRIF and Mal/TIRAP (Fig. 6 b, left). TRIF also interacted with Mal (Fig. 6 b, right). Finally, both TRIF and TRAM interacted with the Mal-P125H (dominant negative) mutant. The stronger interaction of TRIF and TRAM observed with the Mal-P125H mutant does not reflect a higher avidity for this interaction but rather was a consequence of the higher expression level of the MAL-P125H mutant in whole cell lysates, compared with the expression level of Mal or TRIF (Fig. 6 b, middle). These data may explain a previously unexplained finding, i.e., that the Mal/TIRAP dominant negative mutant powerfully inhibited LPS-induced signaling to NF-κB (18, 19) and IFN-β expression (12, 20), whereas the Mal/TIRAP knockout mouse both retained the ability to induce NF-κB (21, 22) and IFN-β expression (22). It is likely that the more profound effect of the dominant negative construct is due to its ability to limit the function of other adaptor molecules involved in LPS signaling such as TRAM and TRIF. Furthermore, these data suggest that TRIF and TRAM interact with Mal at a site distinct from the TLR4 interaction site of Mal (19).
Coimmunoprecipitation studies were next performed to determine if TRAM interacts with TLR4. Stable TLR3- or TLR4-expressing cell lines were transiently transfected with Flag-tagged expression vectors for TRAM, TRAM-C113H, and Mal, and coimmunoprecipitation experiments were performed. These experiments indicated that TRAM interacts with TLR4 but not with TLR3 (Fig. 6 c), one more indication of the specificity of TRAM for the TLR4 pathway. The dominant negative mutant TRAM-C113H failed to immunoprecipitate with TLR4, suggesting that the C113 residue is critical for this interaction. Mal also interacted with TLR4 and not TLR3, providing additional evidence that Mal has a role in the TLR4 but not the TLR3 signaling pathway.
TRIF and TRAM Are Essential for TLR4 Signaling.
The data obtained by testing dominant negative constructs and assessing protein–protein interactions suggest that TRIF and TRAM both function in the TLR4 signal transduction pathway. Dominant negative constructs, when highly expressed, have the potential to bind (as seen in Fig. 6 b) and interfere with proteins that might otherwise not be related to a specific signal transduction pathway. Therefore, we performed siRNA-silencing experiments as an additional methodology to delineate the relationship between TRIF and TRAM in the TLR4 and TLR3 signaling pathways.
To assess the gene-silencing activity of siRNA duplexes we selected, cells were transiently transfected with a fluorescent chimeric construct of TRAM (TRAM–CFP) and cotransfected with siRNA duplexes targeting the coding region of TRAM or a control siRNA, lamin A/C. These siRNA duplexes can therefore be used to assess the silencing effect of a fluorescent chimeric construct of TRAM. This methodology has been used extensively to assess siRNA efficiency and provides a quantitative assessment of silencing efficiency (57). We found that siRNA duplexes targeting the TRAM coding region completely ablated the expression of the TRAM–CFP chimeric fusion protein, whereas lamin A/C siRNA duplexes were without effect (Fig. 6 a). We also examined the effect of these TRAM siRNA duplexes on TRIF and Mal expression in order to ensure the specificity of the TRAM siRNA duplexes. This is particularly important as TRIF and TRAM are most closely related in sequence. TRAM siRNA duplexes had no effect on chimeric constructs of TRIF or Mal expressed as CFP fusion proteins (Fig. 7 a).
Figure 7.

TRAM and TRIF are required for RANTES activation by LPS. (a) 293T cells plated in 24-well plates were transfected with 1 μg of plasmids encoding TRAM-CFP, TRIF-CFP, or Mal-CFP and cotransfected with 50 nM siRNA-TRAM or lamin A/C as indicated. 24 h later, CFP fluorescence was measured by flow cytometry using a 405 nm laser for excitation of CFP. The siRNA-TRAM nearly completely abolished expression of the subpopulation of cells that express CFP. (b) U373–CD14 or TLR3-expressing HEK293 cells were transfected with a RANTES reporter gene and cotransfected with siRNA duplexes as indicated for 36 h. Cells were then stimulated for 8 h with LPS or dsRNA, and luciferase reporter gene activity was measured.
Having determined that the siRNA duplexes chosen for TRAM effectively and specifically suppressed TRAM expression, we examined the effect of these siRNA duplexes on the LPS and dsRNA signaling pathways. Native macrophages and macrophage cells lines are extraordinarily difficult to transfect with siRNA. In contrast, U373–CD14 cells resemble CNS macrophages, are easily transfectable, and are highly inducible by treatment with LPS. Thus, we tested the effect of TRAM siRNA duplexes on the LPS response in U373–CD14 cells. In comparison, we used HEK-TLR3 cells to test the effects of TRAM and TRIF in pIC-stimulated RANTES expression. The response of each of these cell lines to these TLR ligands is comparable.
TRAM siRNA duplexes inhibited the LPS-dependent induction of the RANTES reporter gene in U373–CD14 cells, whereas siRNA targeting of lamin A/C had no effect (Fig. 7 b, top). We also examined the effect of published TRIF siRNA duplexes, which target the 3′ untranslated region of TRIF on this response. These TRIF siRNA duplexes have been shown to completely silence endogenous TRIF mRNA expression (13). TRIF siRNA duplexes also inhibited the LPS response to RANTES induction (Fig. 7 b, top). In striking contrast to LPS, when the TLR3-mediated response to dsRNA was analyzed, the TRAM siRNA duplexes had no inhibitory effect on the dsRNA response, whereas TRIF siRNA duplexes inhibited dsRNA-dependent RANTES induction, in agreement with published reports (13). As with RANTES, siRNA silencing of TRAM prevented LPS but not poly IC induction of the IP-10 promoter (unpublished data). These observations confirm the studies with TRIF and TRAM dominant negative mutants and demonstrate that both adaptor molecules are required for full LPS-TLR4 signaling to IRF target genes.
Discussion
The ability of individual TLRs to discriminate between invading pathogens is an important determinant of the unique gene expression profiles activated by different microorganisms. Whereas the specificity of microbe detection begins with the ligand recognition features of one or more TLR, the discovery of a family of TIR domain–containing adaptor molecules, including MyD88 (4), Mal (18, 19), TRIF (13, 16), and TRAM, suggest that the outcome of induced pathogen recognition also depends on the TLR-restricted utilization of these molecules, alone and in combination, to drive a stimulus-specific response. The TLR3- and TLR4-restricted utilization of IRF-inducing adaptor molecules such as TRAM and TRIF induces not only the cytokines, costimulatory molecules, and antimicrobial peptides that are induced by all TLRs but also anti–viral type I interferons and specific chemokines including IP-10 and RANTES.
The dominant negative, siRNA, and protein–protein interaction data presented here demonstrate that TRAM is specifically required for LPS signaling, whereas TRIF is required for signaling by both TLR3 and TLR4. Both MyD88 and Mal/TIRAP also have a role in more than one TLR. Thus, the activity of TRAM, unlike any of the other known adaptor molecules, may be restricted to a single TLR.
Defining the constituents of the TLR4 pathway activated by LPS has proven to be a complicated process. First, it was believed that MyD88 was the only adaptor molecule needed for the full extent of the LPS response (11). However, data rapidly emerged that showed that at least part of the LPS response also involves Mal/TIRAP (21, 22). Thus, both MyD88 and Mal/TIRAP are involved in LPS signal transduction but cannot account for all, or even most, of the observed signaling traffic. The existence of TRAM and the potential cooperativity of TRAM and TRIF in the TLR4 pathway may explain how double MyD88–Mal-null cells are still capable of responding to endotoxin. It is intriguing that the TLR4 pathway requires TRIF, TRAM, MyD88, and Mal/TIRAP, whereas TLR3 signaling appears to require only TRIF and MyD88. The gene expression profiles activated after dsRNA and LPS stimulation of cells, though similar, are not identical (17, 58). The utilization of TRAM by TLR4 and not TLR3 may allow LPS-TLR4 to induce signaling pathways and gene expression programs not possible by TLR3–TRIF-mediated signaling. Thus, the combinatorial utilization of TRIF and TRAM by TLR4 may allow a specific tailoring of the immune response to the pathogens that activate TLR4.
Both functional and direct biochemical studies indicate that the adaptor molecules used for the LPS response are interacting with one another and with TLR4. One obvious question that needs to be addressed is how these types of observations, which were made in a few types of transfected cell lines and primary BM-derived macrophages, apply to the myriad of cell types that respond to LPS. The response to LPS is not uniform, an observation that is generally attributed to the cell surface density of the LPS receptor components. However, simple receptor density clearly does not always explain these differences and many different mechanisms are undoubtedly at work. Based on scanning electron micrographs of LPS-exposed cells, it is likely that activated TLR4 forms a large “signalosome” involving multiple molecules of TLR4 (unpublished data). The cytoplasmic face of the activated LPS receptor is likely to have a large surface upon which these adaptor molecules may sit. We propose that cell-specific differences in the response to LPS may also involve differences in the number and the composition of adaptor molecules that assemble on TLR4. Cocrystalization of the cytoplasmic face of TLRs in complexes with the various adaptor molecules or combinations of adaptor molecules is going to be necessary to fully understand the physical basis of how an activated Toll receptor actually collaborates with these molecules to propagate an intracellular signal. We predict that TRAM will be a portion of a large platform of adaptor molecules bound to TLR4 upon which a variety of kinases and other molecules can effectively function to initiate LPS responses.
While this manuscript was in preparation, Shu and colleagues reported on the identification of TRAM, to which they gave the name TIRP, as an adaptor molecule that interacts with the IL-1R, Mal, IRAK, and TRAF-6 (59). These authors reported that TRAM/TIRP functions exclusively in IL-1 signaling (59). In agreement with Shu and colleagues, we have also detected TRAM in association with Mal (Fig. 4 b), TRAF-6 and the IL1RAcP (unpublished data). Furthermore, we detected no inhibition of the IL1-induced NFκB response when cells were transfected with the TRAM–C113H mutant (Fig. 6). Since submission of this paper, two articles have reported on the role of TRIF in LPS signaling (60, 61). One of these articles (61), postulates that there may be another TLR4 adaptor molecule, which is designated “X.” TRAM may represent a potential candidate for such a molecule, although the precise mechanism whereby the four known LPS adapters interact and contribute to MyD88-dependent and MyD88-independent signaling remains to be determined.
Acknowledgments
This work was supported by a grant from the Wellcome Trust (to K.A. Fitzgerald) and National Institutes of Health grants DK50305, GM54060, and GM63244 (to D.T. Golenbock) and IR21 A1055453701 (to P.M. Pitha). D.C. Rowe is supported by National Institutes of Health training grant T32AIO7349-14.
K.A. Fitzgerald and D.C. Rowe contributed equally to this work.
Abbreviations used in this paper: DBD, DNA-binding domain; IKK, IκB kinase; IP-10, γ interferon–inducible protein 10; IRAK, IL-1R–associated kinase; IRF, interferon regulatory factor; ISRE, interferon-stimulated response element; Mal, MyD88 adaptor–like protein; MyD88, myeloid differentiation factor 88; RANTES, regulated on activation, normal T cell expressed and secreted; TBK, TANK-binding kinase; TICAM, TIR-containing adaptor molecule; TIR, Toll–IL-1 resistance; TLR, Toll-like receptor; TRAM, TRIF-related adaptor molecule; TRAF, TNF receptor–associated factor; TRIF, TIR domain–containing adaptor-inducing IFN-β.
References
- 1.Medzhitov, R., P. Preston-Hurlburt, and C.A. Janeway, Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 388:394–397. [DOI] [PubMed] [Google Scholar]
- 2.Akira, S. 2001. Toll-like receptors and innate immunity. Adv. Immunol. 78:1–56. [DOI] [PubMed] [Google Scholar]
- 3.Dunne, A., and L.A. O'Neill. 2003. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE. 2003:re3. [DOI] [PubMed]
- 4.Muzio, M., J. Ni, P. Feng, and V.M. Dixit. 1997. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL- 1 signaling. Science. 278:1612–1615. [DOI] [PubMed] [Google Scholar]
- 5.Janssens, S., and R. Beyaert. 2002. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem. Sci. 27:474–482. [DOI] [PubMed] [Google Scholar]
- 6.Cao, Z., W.J. Henzel, and X. Gao. 1996. IRAK: a kinase associated with the interleukin-1 receptor. Science. 271:1128–1131. [DOI] [PubMed] [Google Scholar]
- 7.Li, S., A. Strelow, E.J. Fontana, and H. Wesche. 2002. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. USA. 99:5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao, Z., J. Xiong, M. Takeuchi, T. Kurama, and D.V. Goeddel. 1996. TRAF6 is a signal transducer for interleukin-1. Nature. 383:443–446. [DOI] [PubMed] [Google Scholar]
- 9.Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 18:621–663. [DOI] [PubMed] [Google Scholar]
- 10.Kaisho, T., O. Takeuchi, T. Kawai, K. Hoshino, and S. Akira. 2001. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J. Immunol. 166:5688–5694. [DOI] [PubMed] [Google Scholar]
- 11.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 11:115–122. [DOI] [PubMed] [Google Scholar]
- 12.Toshchakov, V., B.W. Jones, P.Y. Perera, K. Thomas, M.J. Cody, S. Zhang, B.R. Williams, J. Major, T.A. Hamilton, M.J. Fenton, and S.N. Vogel. 2002. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 3:392–398. [DOI] [PubMed] [Google Scholar]
- 13.Oshiumi, H., M. Matsumoto, K. Funami, T. Akazawa, and T. Seya. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol.4:161–167. [DOI] [PubMed] [Google Scholar]
- 14.Kaisho, T., and S. Akira. 2001. Dendritic-cell function in Toll-like receptor- and MyD88-knockout mice. Trends Immunol. 22:78–83. [DOI] [PubMed] [Google Scholar]
- 15.Kawai, T., O. Takeuchi, T. Fujita, J. Inoue, P.F. Muhlradt, S. Sato, K. Hoshino, and S. Akira. 2001. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167:5887–5894. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto, M., S. Sato, K. Mori, K. Hoshino, O. Takeuchi, K. Takeda, and S. Akira. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the toll-like receptor signaling. J. Immunol. 169:6668–6672. [DOI] [PubMed] [Google Scholar]
- 17.Doyle, S., S. Vaidya, R. O'Connell, H. Dadgostar, P. Dempsey, T. Wu, G. Rao, R. Sun, M. Haberland, R. Modlin, and G. Cheng. 2002. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 17:251–263. [DOI] [PubMed] [Google Scholar]
- 18.Fitzgerald, K.A., E.M. Palsson-McDermott, A.G. Bowie, C.A. Jefferies, A.S. Mansell, G. Brady, E. Brint, A. Dunne, P. Gray, M.T. Harte, et al. 2001. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 413:78–83. [DOI] [PubMed] [Google Scholar]
- 19.Horng, T., G.M. Barton, and R. Medzhitov. 2001. TIRAP: an adapter molecule in the Toll signaling pathway. Nat. Immunol. 2:835–841. [DOI] [PubMed] [Google Scholar]
- 20.Shinobu, N., T. Iwamura, M. Yoneyama, K. Yamaguchi, W. Suhara, Y. Fukuhara, F. Amano, and T. Fujita. 2002. Involvement of TIRAP/MAL in signaling for the activation of interferon regulatory factor 3 by lipopolysaccharide. FEBS Lett. 517:251–256. [DOI] [PubMed] [Google Scholar]
- 21.Horng, T., G.M. Barton, R.A. Flavell, and R. Medzhitov. 2002. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 420:329–333. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto, M., S. Sato, H. Hemmi, H. Sanjo, S. Uematsu, T. Kaisho, K. Hoshino, O. Takeuchi, M. Kobayashi, T. Fujita, et al. 2002. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 420:324–329. [DOI] [PubMed] [Google Scholar]
- 23.Hiscott, J., P. Pitha, P. Genin, H. Nguyen, C. Heylbroeck, Y. Mamane, M. Algarte, and R. Lin. 1999. Triggering the interferon response: the role of IRF-3 transcription factor. J. Interferon Cytokine Res. 19:1–13. [DOI] [PubMed] [Google Scholar]
- 24.Juang, Y.T., W. Lowther, M. Kellum, W.C. Au, R. Lin, J. Hiscott, and P.M. Pitha. 1998. Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc. Natl. Acad. Sci. USA. 95:9837–9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juang, Y.T., W.C. Au, W. Lowther, J. Hiscott, and P.M. Pitha. 1999. Lipopolysaccharide inhibits virus-mediated induction of interferon genes by disruption of nuclear transport of interferon regulatory factors 3 and 7. J. Biol. Chem. 274:18060–18066. [DOI] [PubMed] [Google Scholar]
- 26.Lin, R., C. Heylbroeck, P. Genin, P.M. Pitha, and J. Hiscott. 1999. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol. Cell. Biol. 19:959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Genin, P., M. Algarte, P. Roof, R. Lin, and J. Hiscott. 2000. Regulation of RANTES chemokine gene expression requires cooperativity between NF-kappa B and IFN-regulatory factor transcription factors. J. Immunol. 164:5352–5361. [DOI] [PubMed] [Google Scholar]
- 28.Guo, J., K.L. Peters, and G.C. Sen. 2000. Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology. 267:209–219. [DOI] [PubMed] [Google Scholar]
- 29.Yoneyama, M., W. Suhara, and T. Fujita. 2002. Control of IRF-3 activation by phosphorylation. J. Interferon Cytokine Res. 22:73–76. [DOI] [PubMed] [Google Scholar]
- 30.Servant, M.J., B. ten Oever, C. LePage, L. Conti, S. Gessani, I. Julkunen, R. Lin, and J. Hiscott. 2001. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J. Biol. Chem. 276:355–363. [DOI] [PubMed] [Google Scholar]
- 31.Wathelet, M.G., C.H. Lin, B.S. Parekh, L.V. Ronco, P.M. Howley, and T. Maniatis. 1998. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell. 1:507–518. [DOI] [PubMed] [Google Scholar]
- 32.Lin, R., C. Heylbroeck, P.M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoneyama, M., W. Suhara, Y. Fukuhara, M. Fukuda, E. Nishida, and T. Fujita. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17:1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weaver, B.K., K.P. Kumar, and N.C. Reich. 1998. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol. Cell. Biol. 18:1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Au, W.C., P.A. Moore, D.W. LaFleur, B. Tombal, and P.M. Pitha. 1998. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J. Biol. Chem. 273:29210–29217. [DOI] [PubMed] [Google Scholar]
- 36.Yeow, W.S., W.C. Au, Y.T. Juang, C.D. Fields, C.L. Dent, D.R. Gewert, and P.M. Pitha. 2000. Reconstitution of virus-mediated expression of interferon alpha genes in human fibroblast cells by ectopic interferon regulatory factor-7. J. Biol. Chem. 275:6313–6320. [DOI] [PubMed] [Google Scholar]
- 37.Marie, I., J.E. Durbin, and D.E. Levy. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters, R.T., S.M. Liao, and T. Maniatis. 2000. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell. 5:513–522. [DOI] [PubMed] [Google Scholar]
- 39.Shimada, T., T. Kawai, K. Takeda, M. Matsumoto, J. Inoue, Y. Tatsumi, A. Kanamaru, and S. Akira. 1999. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int. Immunol. 11:1357–1362. [DOI] [PubMed] [Google Scholar]
- 40.Pomerantz, J.L., and D. Baltimore. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18:6694–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonnard, M., C. Mirtsos, S. Suzuki, K. Graham, J. Huang, M. Ng, A. Itie, A. Wakeham, A. Shahinian, W.J. Henzel, et al. 2000. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 19:4976–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tojima, Y., A. Fujimoto, M. Delhase, Y. Chen, S. Hatakeyama, K. Nakayama, Y. Kaneko, Y. Nimura, N. Motoyama, K. Ikeda, et al. 2000. NAK is an IkappaB kinase-activating kinase. Nature. 404:778–782. [DOI] [PubMed] [Google Scholar]
- 43.Fitzgerald, K.A., S.M. McWhirter, K.L. Faia, D.C. Rowe, E. Latz, D.T. Golenbock, A.J. Coyle, S.M. Liao, and T. Maniatis. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. [DOI] [PubMed] [Google Scholar]
- 44.Sharma, S., B.R. tenOever, N. Grandvaux, G.P. Zhou, R. Lin, and J. Hiscott. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science. 300:1148–1151. [DOI] [PubMed] [Google Scholar]
- 45.Sakaguchi, S., H. Negishi, M. Asagiri, C. Nakajima, T. Mizutani, A. Takaoka, K. Honda, and T. Taniguchi. 2003. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem. Biophys. Res. Commun. 306:860–866. [DOI] [PubMed] [Google Scholar]
- 46.Hirschfeld, M., Y. Ma, J.H. Weis, S.N. Vogel, and J.J. Weis. 2000. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J. Immunol. 165:618–622. [DOI] [PubMed] [Google Scholar]
- 47.Latz, E., A. Visintin, E. Lien, K.A. Fitzgerald, B.G. Monks, E.A. Kurt-Jones, D.T. Golenbock, and T. Espevik. 2002. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J. Biol. Chem. 277:47834–47843. [DOI] [PubMed] [Google Scholar]
- 48.Visintin, A., A. Mazzoni, J.A. Spitzer, and D.M. Segal. 2001. Secreted MD-2 is a large polymeric protein that efficiently confers lipopolysaccharide sensitivity to Toll-like receptor 4. Proc. Natl. Acad. Sci. USA. 98:12156–12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lien, E., J.C. Chow, L.D. Hawkins, P.D. McGuinness, K. Miyake, T. Espevik, F. Gusovsky, and D.T. Golenbock. 2001. A novel synthetic acyclic lipid A-like agonist activates cells via the lipopolysaccharide/toll-like receptor 4 signaling pathway. J. Biol. Chem. 276:1873–1880. [DOI] [PubMed] [Google Scholar]
- 50.Fitzgerald, K.A., A.G. Bowie, B.S. Skeffington, and L.A. O'Neill. 2000. Ras, protein kinase C zeta, and I kappa B kinases 1 and 2 are downstream effectors of CD44 during the activation of NF-kappa B by hyaluronic acid fragments in T-24 carcinoma cells. J. Immunol. 164:2053–2063. [DOI] [PubMed] [Google Scholar]
- 51.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 52.Takeuchi, O., K. Takeda, K. Hoshino, O. Adachi, T. Ogawa, and S. Akira. 2000. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int. Immunol. 12:113–117. [DOI] [PubMed] [Google Scholar]
- 53.Maniatis, T., J.V. Falvo, T.H. Kim, T.K. Kim, C.H. Lin, B.S. Parekh, and M.G. Wathelet. 1998. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 63:609–620. [DOI] [PubMed] [Google Scholar]
- 54.Xu, Y., X. Tao, B. Shen, T. Horng, R. Medzhitov, J.L. Manley, and L. Tong. 2000. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 408:111–115. [DOI] [PubMed] [Google Scholar]
- 55.Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C.V. Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 56.Kravchenko, V.V., J.C. Mathison, K. Schwamborn, F. Mercurio, and R.J. Ulevitch. 2003. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J. Biol. Chem. 278:26612–26619. [DOI] [PubMed] [Google Scholar]
- 57.Brummelkamp, T.R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296:550–553. [DOI] [PubMed] [Google Scholar]
- 58.Doyle, S.E., R. O'Connell, S.A. Vaidya, E.K. Chow, K. Yee, and G. Cheng. 2003. Toll-like receptor 3 mediates a more potent antiviral response than Toll-like receptor 4. J. Immunol. 170:3565–3571. [DOI] [PubMed] [Google Scholar]
- 59.Bin, L.H., L.G. Xu, and H.B. Shu. 2003. TIRP, a novel Toll/interleukin-1 receptor (TIR) domain-containing adapter protein involved in TIR signaling. J. Biol. Chem. 278:24526–24532. [DOI] [PubMed] [Google Scholar]
- 60.Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo, O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, and S. Akira. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 301:640–643. [DOI] [PubMed] [Google Scholar]
- 61.Hoebe, K., X. Du, P. Georgel, E. Janssen, K. Tabeta, S.O. Kim, J. Goode, P. Lin, N. Mann, S. Mudd, et al. 2003. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 424:743–748. [DOI] [PubMed] [Google Scholar]