

Abstract
During development, the stochastic process assembling the genes encoding antigen receptors invariably generates B and T lymphocytes that can recognize self-antigens. Several mechanisms have evolved to prevent the activation of these cells and the concomitant development of autoimmune disease. One such mechanism is the induction of apoptosis in developing or mature B cells by engagement of the B cell antigen receptor (BCR) in the absence of T cell help. Here we report that B lymphocytes lacking the pro-apoptotic Bcl-2 family member Bim are refractory to apoptosis induced by BCR ligation in vitro. The loss of Bim also inhibited deletion of autoreactive B cells in vivo in two transgenic systems of B cell tolerance. Bim loss prevented deletion of autoreactive B cells induced by soluble self-antigen and promoted accumulation of self-reactive B cells developing in the presence of membrane-bound self-antigen, although their numbers were considerably lower compared with antigen-free mice. Mechanistically, we determined that BCR ligation promoted interaction of Bim with Bcl-2, inhibiting its survival function. These findings demonstrate that Bim is a critical player in BCR-mediated apoptosis and in B lymphocyte deletion.
Keywords: apoptosis, transgenic mice, tolerance, gene knockout, autoimmunity
Introduction
The combinatorial nature of the genetic rearrangements that mediate the formation of the Ig genes ensures diversity in the B lymphocyte repertoire and the ability to recognize numerous foreign antigens. However, It also entails the development of B lymphocytes expressing self-reactive Ig molecules. Whereas a degree of self-reactivity may be essential for the development and survival of B lymphocytes (1), interactions of too high an affinity may be deleterious to the well being of the organism. Thus, several mechanisms have evolved to ensure that B cells bearing potentially harmful antiself specificities are removed from the system (2). These mechanisms, so far defined on the basis of experimental observation, include the functional silencing of the lymphocytes in a process termed anergy (3, 4), the reinduction of gene rearrangement at the Ig heavy and light chain loci to generate edited receptors with nonself reactivity (5, 6), and finally the deletion of autoreactive B cells at specific points during development (7–9). Although the precise mechanisms ensuring these various forms of negative selection remain undefined, it is clear that the induction of apoptosis is critical (10, 11).
Two distinct but convergent apoptosis signaling pathways activate the aspartate-specific cysteine proteases (caspases) that mediate cell death (12, 13). Ligation of certain receptors belonging to the TNF receptor (TNF-R) family, such as CD95 (Fas/APO-1) or TNF-R1, activates caspase-8 via the adaptor protein FADD/MORT1. The other pathway is regulated by the interplay of pro- and anti-apoptotic members of the Bcl-2 family and is amplified by, but not fully dependent on, the action of the cytochrome c/Apaf-1–caspase-9 apoptosome(12–14).
Deletion of activated B cells can be mediated through interaction with FasL on CD4+ T lymphocytes (15, 16), which explains why not only T cells but also B cells contribute to lymphadenopathy in mutant Fas- (lpr) and FasL-deficient (gld) mice (17). The death receptor pathway is, however, dispensable for BCR ligation–induced deletion of immature and mature resting B lymphocytes in vivo (15, 18). Moreover, BCR ligation–induced apoptosis of B lymphoma–derived cell lines and primary B cells is independent of Fas (19) and not inhibited by blocking the function of FADD or caspase-8 (19, 20). In contrast, this death can be prevented by overexpression of Bcl-2 or Bcl-xL (19), indicating that pro-apoptotic Bcl-2 family members might be the critical initiators of apoptosis in B lymphocyte–negative selection. Recent genetic and biochemical studies have shown that apoptosis is initiated by BH3-only Bcl-2 family members, which share with their relatives only the short BH3 interaction domain (21) and that pro-apoptotic Bax and Bak, which are structurally more akin to the anti-apoptotic Bcl-2 family members, act downstream in the cell death program (21). We were prompted to investigate the role of the BH3-only protein Bim (22) in B lymphocyte–negative selection when disruption of the bim gene showed it to be a critical regulator of lymphocyte homeostasis, a barrier against autoimmune disease (23), and necessary for killing of autoreactive thymocytes (24) and mature, antigen-activated T cells (25, 26).
Materials and Methods
Mice.
The generation and genotyping of bim −/− mice (23), MD4 transgenic mice expressing anti-HEL Ig (specific for hen egg lysozyme) in their B cells (3), soluble HEL (sHEL) (3) or membrane-bound HEL (mHEL) (8) in a wide range of cell types, and vav–bcl-2 transgenic mice (27), which express human Bcl-2 under the control of the vav promoter region in all hemopoietic cells, have been described. All animals were on a C57BL/6 (N > 10) background and were analyzed at 5–10 wk of age, except some vav–bcl-2 mice that were analyzed at 6 mo. Some experiments used C57BL/6 mice congenic for Ly5.1, with or without the MD4 Ig transgene. These latter mice were bred and maintained at the Walter and Eliza Hall Institute. BM reconstitutions were as described (28), and recipients were analyzed at least 8 wk post reconstitution.
Anti-IgM Antibody Treatment.
Immature and mature B cells from control (WT), bim −/−, and vav–bcl-2 transgenic mice were purified by positive or negative sorting from spleen and LNs, respectively (see Immunofluorescence Staining, Flow Cytometric Analysis, and Cell Isolation) and then cultured in the presence of graded concentrations of F(ab′)2 goat anti–mouse IgM antibodies (Jackson ImmunoResearch Laboratories), either soluble or plate bound via biotin-avidin. B cells were harvested 15 h later, and the proportion of live and apoptotic cells was determined by staining with FITC-coupled annexin V (PharMingen) and propidium iodide (PI) followed by analysis in a FACScan® (Becton Dickinson). Specific cell viability after culture with anti-IgM antibodies was calculated by dividing the cell survival in the stimulated culture with that from the unstimulated culture and expressing the result as a percentage. Thus, the unstimulated culture is always 100% survival and the stimulated culture some fraction of this.
Immunofluorescence Staining, Flow Cytometric Analysis, and Cell Isolation.
B lymphocytes were stained in 10% 2.4G2 (anti–mouse FcγRII) hybridoma supernatant plus 1% normal rat serum with mAbs RA3–6B2 (anti–CD45-R/B220), 1D3 (anti-CD19), 331.12 (anti-IgM), 11.2C (anti-IgD), 187.1 (anti-Igκ light chain), LS132 (anti-Igλ light chain), 7G6 (anti-CD21), and AMS9.1 (anti-IgDa) conjugated with Cy5, FITC, R-PE, or biotin. Antibodies were produced and conjugated to fluorochromes in our laboratory. Immature B cells were collected as the B220losIgM+ fraction from among BM cells stained with 331.12 (anti-IgM) and RA3–6B2 (anti-B220). For sorting mature B cells, LN cells were stained with 30H12 (anti-Thy1), M1/70 (anti-Mac1), RB6–8C5 (anti-Gr1), and Ter119 (anti-erythroid cell marker), all labeled with FITC, and B cells were collected as the FITC-negative fraction using a high speed cell sorter. Efficiency of the sort was determined by staining an aliquot of the sorted cells with anti-B220 mAb and was found on analysis to always exceed 95%. Biotinylated antibodies and biotinylated HEL were detected with R-PE- or TRI-COLOR streptavidin (Caltag). Flow cytometric analysis of viable cells (those excluding PI) used a FACScan® or LSR (Becton Dickinson), whereas cell sorting used either a MoFlo (Cytomation) or FACS® Diva (Becton Dickinson) high speed cell sorter.
Mature B Cell Transfer.
Mature B cells were prepared from pooled spleens and LNs of two Ly5.2 bim −/− and four Ly5.1 bim +/+ Ig transgenic mice. After red cell lysis, T cells were depleted using complement mediated killing of cells labeled with antibodies specific for Thy1, CD4, and CD8 following established protocols. The resulting enriched B cell suspensions (70–80% B220+ HEL binding) were mixed in a ratio of 2 bim −/− to 1 bim +/+ transgenic B cells and injected i.v. into C57BL/6 recipients that were either soluble HEL (sHEL)+ or sHEL− (control) recipients. At 2 and 5 d post transfer, recipients were killed, had their spleens removed, and lymphocytes were prepared and analyzed by flow cytometry for the presence of HEL-binding IgDa+ B cells that were either Ly5.1+ or Ly5.2+. Staining with fluorescent antibodies and subsequent analysis was as described above. Uninjected C57BL/6 mice were used to define the level of detection of such B cells.
Cell Stimulation, Lysis, Immunoprecipitation, and Western Blotting.
B cells purified from spleen using anti-B220 MACS beads (Miltenyi Biotechnology GmbH) were cultured overnight at a concentration of 107/mL with or without 10 μg/mL F(ab′)2 goat anti–mouse IgM antibodies. Subsequent lysis was performed as described (29) in a final volume of 200 μL which was then divided equally in two. Bcl-2 was immunoprecipitated from each 100 μL of extract with a hamster anti–mouse Bcl-2 mAb (clone 3F11) conjugated and cross-linked to protein G sepharose. Bound proteins were eluted with 200 μL of 0.1 M glycine-HCl, pH 2.7, then precipitated with 3 vol of chilled acetone and washed with 70% ethanol. The precipitates were each dissolved in 20 μL SDS sample buffer and size fractionated by SDS-PAGE. Duplicate Western blots were probed with rabbit anti-Bim (Stressgen) or hamster anti–mouse Bcl-2 (clone 3F11) antibodies. Bound antibodies were revealed with HRP-conjugated goat anti–rabbit Ig (Silenus) or goat anti–hamster Ig (Southern Biotechnology Associates, Inc.) antibodies, respectively, followed by enhanced chemiluminescence (Amersham Biosciences).
Results and Discussion
One widely used model of B lymphocyte negative selection is stimulation in vitro with anti-IgM antibody F(ab′)2 fragments (30). As previously shown (30), BCR ligation caused apoptosis of many immature and mature B cells from control mice within 16 h. Specific survival (calculated by dividing the percentage of viable cells from treated cultures by the percentage of viable cells from control cultures; see Materials and Methods) ranged from 10 to 50% (Fig. 1, A–C) , depending on the concentration of the anti-IgM antibodies. Anti-IgM antibodies were more potent inducers of apoptosis when applied in plate-bound form (Fig. 1 C), presumably because of enhanced BCR cross-linking. B cell death induced by BCR ligation was completely prevented by expression of a bcl-2 transgene and was almost abrogated by loss of Bim (Fig. 1, A–C). The inhibition of BCR-induced cell death in bim −/− B cells was found to be cell intrinsic when apoptosis of bim −/− and bim +/+ B cells in a mixed culture was compared with that in individual cultures. A Ly5 allotypic difference was used to distinguish bim +/+ and bim −/− mature B cells, negatively sorted from LNs, and cocultured for 15 h in the presence or absence of 5.0 μg/mL F(ab′)2 anti-IgM. This revealed 72.4 ± 4.7% specific survival of control B cells compared with 88.6 ± 2.0% in Bim-deficient B cells. In parallel single cultures, B cell survival after BCR ligation was 72.4 ± 2.1% and 89.0 ± 0.7% for bim +/+ and bim −/− cells, respectively. These results demonstrate that Bim is required in immature and mature B cells for the apoptosis induced by BCR ligation in a cell intrinsic manner.
Figure 1.
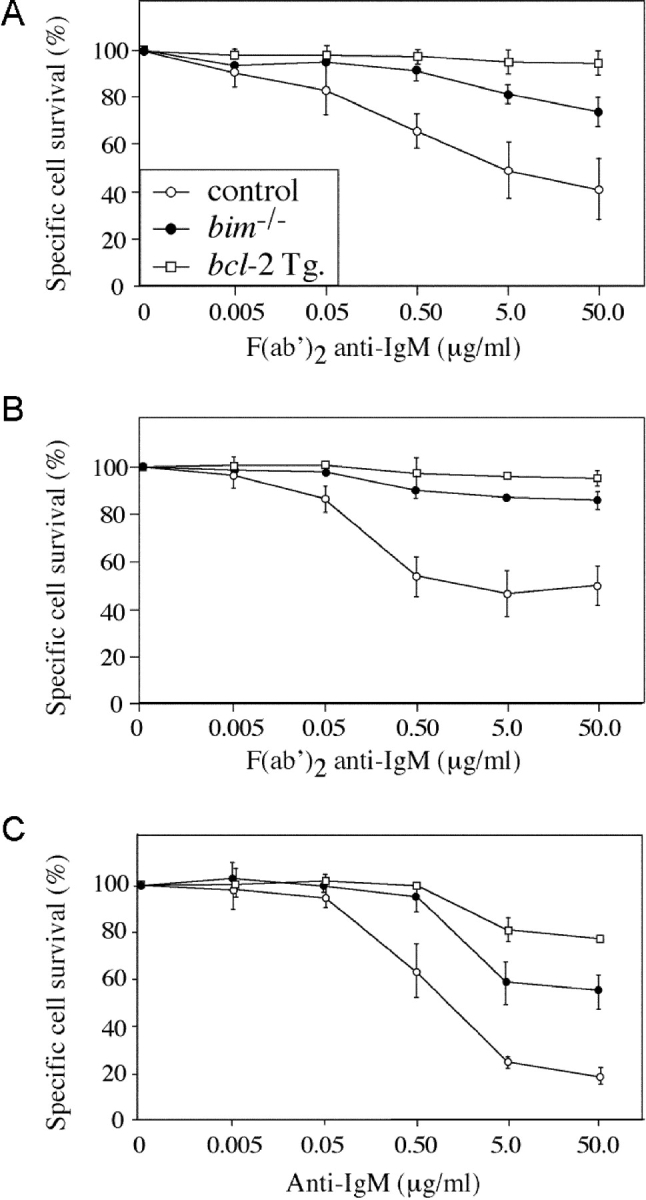
Bim is required for BCR ligation–induced killing of immature and mature B lymphocytes. (A) Immature B cells sorted from spleen as B220+IgM+IgD− were cultured overnight in the presence of F(ab′)2 anti-IgM antibodies and then assessed for viability. Cell survival after 15 h BCR ligation was calculated as a percentage of B cell survival in unstimulated cultures. B cells show a dose-dependent killing reaching 60% at 50 μg/mL for controls, 30% for bim −/−, and <10% for bcl-2 transgene-expressing cells. (B) Same as for A, except mature B cells were negatively sorted from LNs as Thy1−Mac-1−Gr-1−Ter119− cells. Control B cells show 50% death at 50 μg/mL F(ab′)2 anti-IgM antibodies, whereas bim −/− and bcl-2 transgenic B cells show 15 and 5%, respectively. (C) Same as for B, except B cells were stimulated with plate-bound F(ab′)2 anti-IgM antibodies, inducing more severe cell death as evidenced by the 80% cell death of control B cells, 40% death of bim −/− B cells, and 20% death of bcl-2 transgene-expressing B cells at 50 μg/mL anti-IgM antibody concentration.
Negative selection of B cells in vivo can be studied using Igha allotype transgenic mice producing B cells expressing IgM and IgD BCR specific for HEL on an Ighb allotype background. In doubly transgenic animals expressing both anti-HEL Ig and HEL as either a membrane-bound (mHEL) or a soluble (sHEL) neo-self antigen, developing B cells are, respectively, either deleted in the BM with few mature B cells appearing in peripheral lymphoid organs (8), or persist in the periphery in a functionally inactivated state (3). Similar B lymphocyte tolerance is seen when HEL transgenic mice are lethally irradiated and reconstituted with hemopoietic stem cells from BM or fetal liver of anti-HEL Ig transgenic mice (28). Indeed, we found on average 0.5 × 106 transgenic IgH chain–expressing B cells per femur and 0.3 × 106 per spleen of mHEL transgenic animals reconstituted with control (bim +/+anti-HEL Ig) stem cells (Table I). In contrast, when irradiated mHEL transgenic mice were reconstituted with Bim-deficient anti-HEL Ig transgenic stem cells, twofold more IgDa+-expressing B cells were recovered from BM and sixfold more from spleen (Table I and Fig. 2) . The down-regulation of IgM on the persisting IgDa+ B cells is consistent with exposure to antigen (3). None of the IgDa B cells from either BM or spleen bound HEL immediately ex vivo (Fig. 2), indicating either receptor editing (5) or receptor blockade by endogenous HEL. These possibilities were resolved by sorting IgDa+ B cells from the BM and spleen of the reconstituted mHEL mice, culturing them overnight, and then staining to test for a restoration of HEL binding. Sorted IgDa+ B cells from BM of both genotypes (bim +/+ or bim −/−) recovered >85% HEL binding after culture (Fig. 2 C). Culturing IgDa+ B cells purified from the spleens of bim +/+ recipients, however, restored HEL binding in only 15%, suggesting a permanent alteration to the BCR specificity of the remainder (Fig. 2 C). In contrast, ∼60% of IgDa+ B cells from bim −/− spleen recovered HEL binding. Collectively, these results show that despite a high affinity interaction, self-reactive bim −/− B cells are produced and exist in the periphery at ∼25 times the number of bim +/+ controls (an average of 1.0 × 106/spleen compared with 0.04 × 106 in controls; Table II). This is a substantial enhancement of survival of autoreactive B cells, but the numbers of B cells are still considerably less than those produced in mice that do not express the self-antigen HEL. This may indicate that B cell production and survival is also controlled by Bim-independent mechanisms. These might include activation of other BH3-only proteins, additional cell death mechanisms, development arrest, anergy, or inhibition of cell proliferation.
Table I.
Composition of Bone Marrow and Spleen in Control and mHEL+ Reconstituted Mice
| Bone marrow
|
Spleen
|
||||||
|---|---|---|---|---|---|---|---|
| Donor
|
Recipienta
|
Cellularityb
|
IgDa+
c
|
HEL bindingc
|
Cellularityb
|
IgDa+
c
|
HEL bindingc
|
| (n) | 106 | 106 | 106 | 106 | 106 | 106 | |
| Tg+ bim+/+ | Control (4) | 14.9 ± 2.5 | 1.2 ± 0.3 | 1.1 ± 0.4 | 17.8 ± 5.6 | 10.8 ± 3.5 | 10.4 ± 3.5 |
| mHEL+ (8) | 10.3 ± 2.3 | 0.5 ± 0.2d | 0.4 ± 0.1e | 8.1 ± 6.1 | 0.3 ± 0.2f | 0.01 ± 0.03 | |
| Tg+ bim−/− | Control (4) | 15.5 ± 3.0 | 4.0 ± 0.6 | 3.6 ± 0.5 | 83.5 ± 68.3 | 66.1 ± 58.7 | 63.6 ± 57.3 |
| mHEL+ (8) | 11.4 ± 3.2 | 1.1 ± 0.5d | 0.9 ± 0.3e | 20.6 ± 11.9 | 1.7 ± 0.9f | 0.07 ± 0.1 | |
Recipient mice were C57BL/6 (control) or C57BL/6 mHEL transgene positive.
Cellularity was determined by viable counts after red cell removal and are given per femur and per spleen.
IgDa and HEL-binding cell counts are based on the proportion of cells staining positive by flow cytometry immediately ex vivo and whole organ cell counts.
P = 0.01.
P < 0.001.
P = 0.001.
Figure 2.
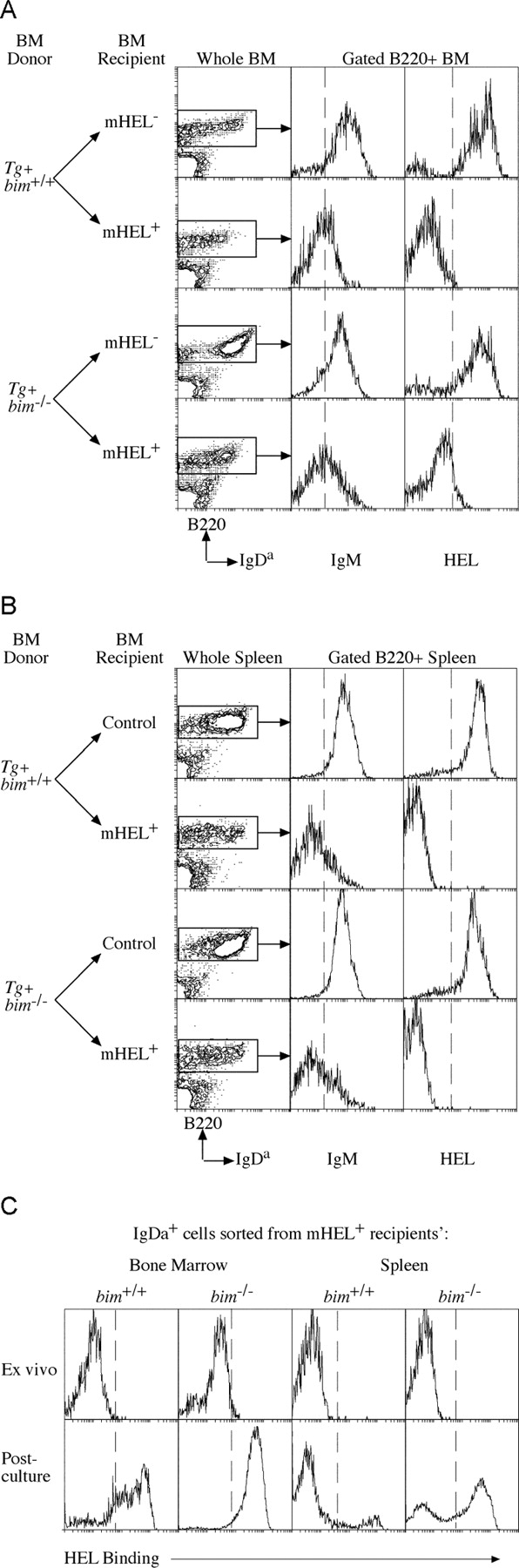
Bim is required for BCR stimulation–induced deletion of self-reactive B cells in the BM. Lethally irradiated control and mHEL transgenic mice, reconstituted with either bim −/− or bim +/+ Ig transgenic BM at least 8 wk previously, were analyzed for the presence of IgH transgene+ B cells. Representative FACS® profiles of BM (A) and spleen (B) from the indicated reconstituted mice, stained with antibodies specific for CD45R(B220) and IgDa, show the enhanced persistence of bim −/− IgDa+ B cells relative to controls (see Table I for quantification and statistics). Histograms show the level of IgM expression and HEL binding on the B220+ cells from the corresponding tissue. (C) Transgenic IgDa+ cells purified from the BM and spleens of mHEL transgenic mice reconstituted with bim −/−anti-HEL Ig transgenic stem cell recover HEL binding after overnight culture, whereas those from the spleens of bim +/+ anti-HEL Ig transgenic stem cell reconstituted mice do not. IgDa+ cells from BM of both types of donor stem cells recover HEL binding (see Table II for quantification and statistics).
Table II.
Restoration of HEL Binding after Culture of Bim-deficient Splenic B Cells Sorted from mHEL + Mice
| Tissue
|
mHEL+ recipient reconstituted with: |
IgDa+ cells/organ
|
Restored HEL binding after culturea |
HEL reactive cellsb
|
|---|---|---|---|---|
| 106 | % | 106 | ||
| Bone marrow | Tg+ bim+/+ | 0.5 ± 0.2 | 86.5 ± 4.9 | 0.43 |
| Tg+ bim−/− | 1.1 ± 0.5 | 95.3 ± 3.1 | 1.05 | |
| Spleen | Tg+ bim+/+ | 0.3 ± 0.2 | 14.0 ± 2.8 | 0.04 |
| Tg+ bim−/− | 1.7 ± 0.9 | 58.7 ± 6.0 | 1.00 |
HEL binding is measured by flow cytometric staining after overnight culture, shown in Fig. 2 C.
Calculates number of cells retaining HEL specificity as revealed after cell culture.
The role of Bim in negative selection of mature, splenic B cells was examined by reconstituting lethally irradiated sHEL mice with BM from anti-HEL Ig transgenic bim +/+ and bim −/− mice (Fig. 3) . This revealed 17-fold more self-reactive B cells in the spleens of bim −/− recipients compared with bim +/+ controls (Table III). Remarkably, an equal number of IgH transgenic Bim-deficient B cells was found in the spleens of self-antigen positive and negative mice, whereas control bim +/+ B cells showed a 10-fold reduction in absolute number or a threefold reduction in proportion in the presence of self-antigen (Table III); both changes were within the range reported for this model of B cell tolerance (31). The larger reduction in B cell number compared with proportion reflects the reduced cellularity of the spleens of sHEL+ mice reconstituted with bim +/+ BM rather than the loss of only B lymphocytes (Table III), again a previously reported property of this system (31). Irrespective of their bim genotype, the persistent IgDa+ B cells in the sHEL+ recipients retained the ability to bind HEL and expressed unaltered levels of IgDa, showed down-regulation of IgM, and did not acquire high levels of CD21 (Fig. 3), all indicative of chronic exposure to antigen (3, 32). In addition, the titers of anti-HEL antibody in sHEL+ recipients of both genotypes were the same (unpublished data). Thus, the persistence of these IgDa+ bim −/− B cells was not due to either enhanced receptor editing or masking of the self epitope by excessive secretion of specific antibody. Furthermore, these results indicated that although autoreactive Bim-deficient B cells were refractory to apoptosis, systemic tolerance was maintained. This presumably reflects in part the maintenance of T cell tolerance to HEL in the bim −/− mice, a result consistent with our previous studies showing peripheral T cell tolerance to be normal despite significant abnormalities in the thymus of bim −/− mice (24).
Figure 3.
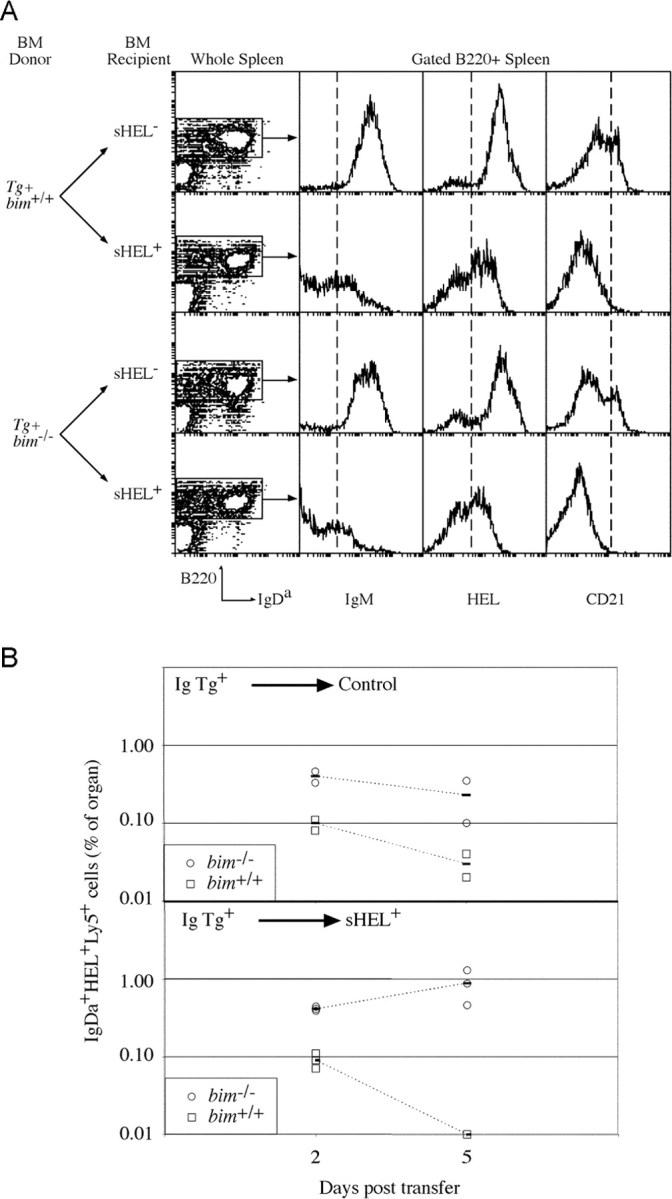
Bim is required for BCR stimulation–induced deletion of mature B cells in the absence of T cell help. (A) Lethally irradiated control and sHEL-expressing mice, reconstituted with either bim −/− or bim +/+ anti-HEL Ig transgenic BM previously at least 8 wk, were analyzed for the persistence of transgenic IgH B cells in the spleen. Shown are contour plots of the expression of B220 and IgDa and histograms depicting expression of IgM, HEL binding, and CD21 on those cells within the gated regions defining B220+ cells. Quantification of the B cell populations in the spleens of these mice is given in Table III. (B) Mature bim +/+ (□) and bim −/− (○) B cells were transferred into sHEL− (top) and sHEL+ (bottom) recipients, and their representations were determined 2 and 5 d later. The proportion of each B cell type in recipient's spleens, as assessed by flow cytometry following staining for IgDa, Ly5.2, and HEL binding, is plotted. At each time point, two sHEL− and three sHEL+ recipients were examined, and the solid bar is the average of the mice of that genotype.
Table III.
Persistence of Bim-deficient HEL-binding B Cells in the Presence of Soluble Self-antigen
| Donora
|
Recipientb
|
Cellularityc
|
IgDa+
|
IgDa+
d
|
HEL bindingd
|
|---|---|---|---|---|---|
| (n) | 106 | % of organ | 106 | 106 | |
| Tg+ bim+/+ | Control (3) | 29.3 ± 9.5 | 55.9 ± 2.3 | 16.4 ± 5.6 | 15.0 ± 4.7 |
| sHEL+ (3) | 8.1 ± 3.1 | 17.3 ± 7.5 | 1.4 ± 0.9e | 1.2 ± 0.9f | |
| Tg+ bim−/− | Control (3) | 51.5 ± 17.3 | 51.8 ± 12.3 | 26.7 ± 14.6 | 23.3 ± 14.7 |
| sHEL+ (3) | 86.7 ± 54.3 | 29.3 ± 11.4 | 25.4 ± 13.8e | 20.2 ± 13.9f |
The failure to delete Bim-deficient B cells in the presence of their cognate self-antigen could result from changes to any of several mechanisms associated with the imposition and maintenance of B cell tolerance (33). To investigate further the basis of the bim −/− B cell persistence, a mixture of mature, peripheral B cells from bim −/− and bim +/+ Ig transgenic mice marked by a Ly5 allotypic difference was transferred into recipients that either expressed sHEL or did not. 2 and 5 d later, groups of such recipients were killed, and the representation of Ig transgenic B cells remaining was determined. The bim +/+ and bim −/− B cells were resolved by their Ly5 allotypes in the recipients. In control recipients, both types of B cells remained detectable during the time course of the assay despite diminishing in representation (Fig. 3). The rate of loss of bim −/− B cells appeared to be less than that of bim +/+ B cells—not an unexpected result given the enhanced survival of bim −/− B cells under conditions of limiting availability of growth factors (23). In sHEL+ recipients, control Ig transgenic B cells were detectable at day 2 posttransfer but not by day 5 (Fig. 3 B), indicating an accelerated turnover (death) in the presence of their cognate antigen, a result consistent with previous reports (34, 35). Bim-deficient B cells, on the other hand, remained detectable during this period and, if anything, appeared to increase in representation (Fig. 3 B). Thus, mature bim −/− B cells persist in the periphery in the presence of antigen due to extended survival.
In view of the critical role of Bim in triggering deletion of autoreactive B lymphocytes, we investigated how BCR ligation affected its association with other components of the cell death pathway. In healthy cells, most BimL and BimEL molecules are sequestered to the microtubular dynein motor complex by binding to the dynein light chain DLC1/LC8 (29). Certain apoptotic stimuli release Bim from this complex, allowing it to translocate and bind to Bcl-2 or its homologues, thereby blocking their survival function (29). Therefore, we investigated whether BCR ligation triggered translocation of Bim to Bcl-2, the dominant prosurvival Bcl-2 family member expressed in B lymphocytes. Lysates were prepared from B cells that had been stimulated for 16 h with anti-IgM antibodies or left untreated, and Bcl-2 immunoprecipitates subjected to Western blot analysis with anti-Bim antibodies. The levels of BimL and BimEL complexed with Bcl-2 increased substantially upon BCR stimulation, whereas the total amount of Bim increased (approximately twofold) and Bcl-2 levels dropped (approximately twofold) (Fig. 4, A and B) . Thus, increased association of Bim with Bcl-2 in activated B cells supports its role in BCR-mediated apoptosis.
Figure 4.
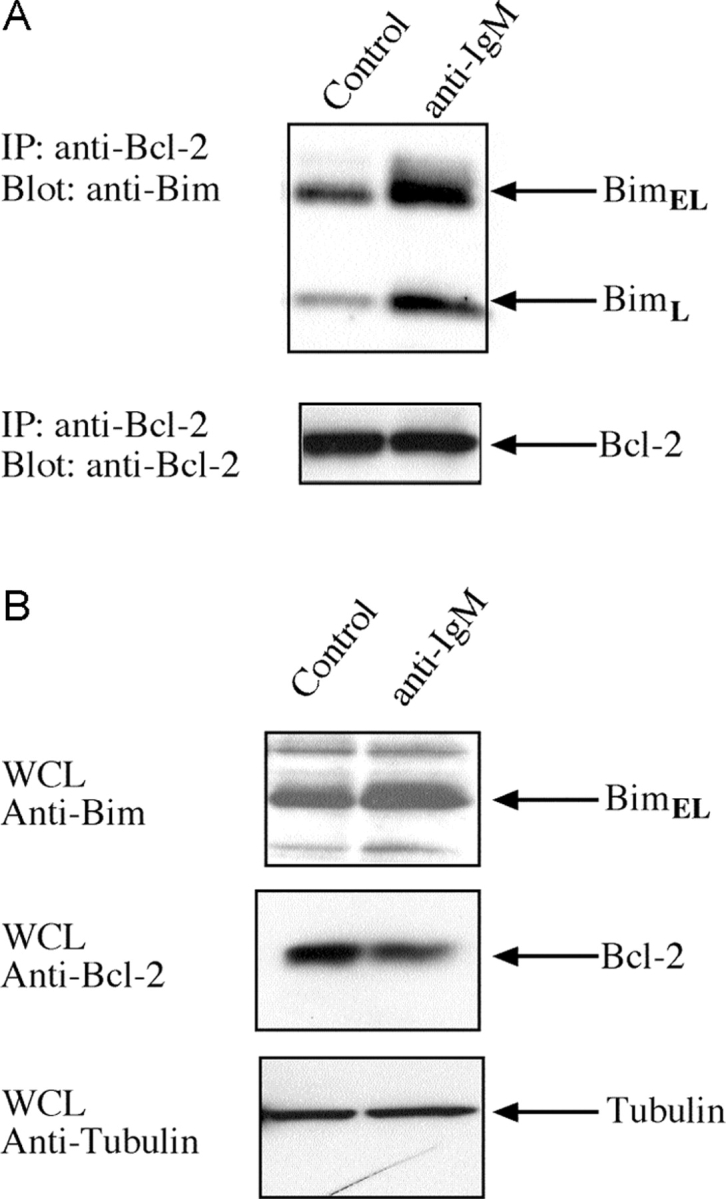
BCR ligation promotes binding of Bim to Bcl-2. (A) Coimmunoprecipitation of Bim with Bcl-2. After lysis proteins were immunoprecipitated with Bcl-2 antibody, the immunoprecipitated proteins were fractionated by SDS-PAGE, blotted onto membranes, and probed with antibodies to BimL and BimEL (top) or Bcl-2 (bottom). (B) Western blot analysis of extracts from B cells that had been stimulated in culture with cross-linking F(ab′)2 anti-IgM antibodies or left untreated, using antibodies to Bim and Bcl-2.
The biochemical data presented in this report showing that Bim mediates BCR-induced apoptosis by sequestering Bcl-2 suggests an important role for Bcl-2 itself in B cell tolerance. Perhaps not surprisingly this was found to be the case in several diverse situations. First, constitutive, B cell–restricted expression of a human bcl-2 transgene abrogated the normal mechanisms of B cell tolerance in that these mice developed an eventually lethal antibody-mediated autoimmune condition reminiscent of human system lupus erythematosus (33). Second, crossing these bcl-2 transgene onto the HEL/anti-HEL Ig self-antigen/antiself antibody system used in this study produced a phenotype similar to that reported here for the Bim-deficient mice. Hartley and colleagues found that Bcl-2 overexpression increased the number of Ig transgenic B cells in the periphery of mHEL+ mice by approximately fivefold (10), just as we find a fivefold increase in IgDa transgenic bim −/− B cells in the spleens of mHEL+ recipients (Table I). This indicates that the majority of the effects seen in the bcl-2 transgenic mice are directly due to its inhibition of Bim's pro-apoptotic activity and that consequently Bim is a major mediator of tolerance induction in B cells.
Collectively, these studies from four models of negative selection indicate that activation of Bim is a primary trigger for killing autoreactive B cells during their development in the BM and at the mature stage in the spleen. It is noteworthy that Bim deficiency inhibits BCR ligation–induced apoptosis of cultured B cells less potently than overexpression of Bcl-2 (Fig. 1), indicating that BH3-only proteins in addition to Bim may also be involved in this death pathway. Blk is a possible candidate because its expression was found to increase upon BCR ligation in a human B lymphoma–derived cell line (34). Although the detailed mechanisms of Bim activation remain unclear, our results identify Bim as an essential initiator of apoptosis in autoreactive B cells. Bim is also critical for negative selection of autoreactive thymocytes (24) and for deletion of antigen-activated mature T cells (25, 26). Thus, Bim appears essential for apoptosis induction of B and T lymphocytes in both central and peripheral tolerance. The retention of autoreactive lymphocytes in Bim-deficient animals is presumably the basis of the SLE-like autoimmune disease they develop (23) and suggests that diminished Bim activity may underlie some forms of human autoimmune disease.
Acknowledgments
We are grateful to Profs. C. Goodnow (John Curtis School of Medical Research, Canberra, Australia) and A. Basten (Centenary Institute, Sydney, Australia) for anti-HEL Ig and sHEL and mHEL transgenic mice. We thank M. James for animal care, Drs. A. Harris, J. Adams, S. Cory, J. Miller, D. Huang, D. Vaux and L. O'Reilly for insightful discussions and critical comments on the manuscript, and A. Light for expert technical assistance.
This work was supported by fellowships and grants from the National Health and Medical Research Council (Canberra, Australia), the Dr. Josef Steiner Cancer Research Foundation (Bern, Switzerland), the Boehringer Ingelheim Stiftung, the Leukemia and Lymphoma Society of America, the Juvenile Diabetes Foundation, and the National Institutes of Health.
A. Strasser and D.M. Tarlinton are senior authors on this paper.
Abbreviations used in this paper: BCR, B cell antigen receptor; HEL, hen egg lysozyme; mHEL, membrane-bound HEL; sHEL, soluble HEL; TNF-R, TNF receptor.
References
- 1.Lam, K.P., R. Kuhn, and K. Rajewsky. 1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 90:1073–1083. [DOI] [PubMed] [Google Scholar]
- 2.Nossal, G.J.V. 1994. Negative selection of lymphocytes. Cell. 76:229–239. [DOI] [PubMed] [Google Scholar]
- 3.Goodnow, C.C., J. Crosbie, H. Jorgensen, R.A. Brink, and A. Basten. 1989. Induction of self-tolerance in mature peripheral B lymphocytes. Nature. 342. [DOI] [PubMed] [Google Scholar]
- 4.Erikson, J., M.Z. Radic, S.A. Camper, R.R. Hardy, C. Carmack, and M. Weigert. 1991. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 349:331–334. [DOI] [PubMed] [Google Scholar]
- 5.Tiegs, S.L., D.M. Russell, and D. Nemazee. 1993. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 177:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gay, D., T. Saunders, S. Camper, and M. Weigert. 1993. Receptor editing: an approach by autoreactive B cells to escape tolerance. J. Exp. Med. 177:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nemazee, D.A., and K. Burki. 1989. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody gene. Nature. 337:562–566. [DOI] [PubMed] [Google Scholar]
- 8.Hartley, S.B., J. Crosbie, R. Brink, A.B. Kantor, A. Basten, and C.C. Goodnow. 1991. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 353:765–769. [DOI] [PubMed] [Google Scholar]
- 9.Russell, D.M., Z. Dembic, G. Morahan, J.F. Miller, K. Burki, and D. Nemazee. 1991. Peripheral deletion of self-reactive B cells. Nature. 354:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartley, S.B., M.P. Cooke, D.A. Fulcher, A.W. Harris, S. Cory, A. Basten, and C.C. Goodnow. 1993. Elimination of self-reactive B lymphocytes proceeds in two stages: arrested development and cell death. Cell. 72:325–335. [DOI] [PubMed] [Google Scholar]
- 11.Lang, J., M. Jackson, L. Teyton, A. Brunmark, K. Kane, and D. Nemazee. 1996. B cells are exquisitely sensitive to central tolerance and receptor editing induced by ultralow affinity, membrane-bound antigen. J. Exp. Med. 184:1685–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gross, A., J.M. McDonnell, and S.J. Korsmeyer. 1999. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 13:1899–1911. [DOI] [PubMed] [Google Scholar]
- 13.Strasser, A., L. O'Connor, and V.M. Dixit. 2000. Apoptosis signaling. Annu. Rev. Biochem. 69:217–245. [DOI] [PubMed] [Google Scholar]
- 14.Marsden, V., L. O'Connor, L.A. O'Reilly, J. Silke, D. Metcalf, P. Ekert, D.C.S. Huang, F. Cecconi, K. Kuida, K.J. Tomaselli, et al. 2002. Apoptosis initiated by Bcl–2-regulated caspase activation independently of the cytochrome c/Apaf–1/caspase–9 apoptosome. Nature. 419:634–637. [DOI] [PubMed] [Google Scholar]
- 15.Rathmell, J.C., and C.C. Goodnow. 1994. Effects of the lpr mutation on elimination and inactivation of self-reactive B cells. J. Immunol. 153:2831–2842. [PubMed] [Google Scholar]
- 16.Rathmell, J.C., M.P. Cooke, W.Y. Ho, J. Grein, S.E. Townsend, M.M. Davis, and C.C. Goodnow. 1995. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 376:181–184. [DOI] [PubMed] [Google Scholar]
- 17.Nagata, S. 1999. Fas ligand-induced apoptosis. Annu. Rev. Genet. 33:29–55. [DOI] [PubMed] [Google Scholar]
- 18.Rubio, C.F., J. Kench, D.M. Russell, R. Yawger, and D. Nemazee. 1996. Analysis of central B cell tolerance in autoimmune-prone MRL/lpr mice bearing autoantibody transgenes. J. Immunol. 157:65–71. [PubMed] [Google Scholar]
- 19.Yoshida, T., T. Higuchi, H. Hagiyama, A. Strasser, K. Nishioka, and T. Tsubata. 2000. Rapid B cell apoptosis induced by antigen receptor ligation does not require fas (CD95/APO-1), the adaptor protein FADD/MORT1 or CrmA- sensitive caspases but is defective in both MRL-+/+ and MRL-lpr/lpr mice. Int. Immunol. 12:517–526. [DOI] [PubMed] [Google Scholar]
- 20.Lens, S.M., B.F. den Drijver, A.J. Pötgens, K. Tesselaar, M.H. van Oers, and R.A. van Lier. 1998. Dissection of pathways leading to antigen receptor-induced and Fas/CD95-induced apoptosis in human B cells. J. Immunol. 160:6083–6092. [PubMed] [Google Scholar]
- 21.Huang, D.C.S., and A. Strasser. 2000. BH3-only proteins—essential initiators of apoptotic cell death. Cell. 103:839–842. [DOI] [PubMed] [Google Scholar]
- 22.O'Connor, L., A. Strasser, L.A. O'Reilly, G. Hausmann, J.M. Adams, S. Cory, and D.C.S. Huang. 1998. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouillet, P., D. Metcalf, D.C.S. Huang, D.M. Tarlinton, T.W.H. Kay, F. Köntgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- 24.Bouillet, P., J.F. Purton, D.I. Godfrey, L.-C. Zhang, L. Coultas, H. Puthalakath, M. Pellegrini, S. Cory, J.M. Adams, and A. Strasser. 2002. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 415:922–926. [DOI] [PubMed] [Google Scholar]
- 25.Hildeman, D.A., Y. Zhu, T.C. Mitchell, P. Bouillet, A. Strasser, J. Kappler, and P. Marrack. 2002. Activated T cell death in vivo mediated by pro-apoptotic Bcl-2 family member, Bim. Immunity. 16:759–767. [DOI] [PubMed] [Google Scholar]
- 26.Davey, G.M., C. Kurts, J.F. Miller, P. Bouillet, A. Strasser, A.G. Brooks, F.R. Carbone, and W.R. Heath. 2002. Peripheral deletion of autoreactive CD8 T cells by cross presentation of self-antigen occurs by a Bcl-2–inhibitable pathway mediated by Bim. J. Exp. Med. 196:947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogilvy, S., D. Metcalf, C.G. Print, M.L. Bath, A.W. Harris, and J.M. Adams. 1999. Constitutive bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc. Natl. Acad. Sci. USA. 96:14943–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cyster, J.G., and C.C. Goodnow. 1995. Protein tyrosine phosphatase 1C negatively regulates antigen receptor signaling in B lymphocytes and determines thresholds for negative selection. Immunity. 2:13–24. [DOI] [PubMed] [Google Scholar]
- 29.Puthalakath, H., D.C.S. Huang, L.A. O'Reilly, S.M. King, and A. Strasser. 1999. The pro-apoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell. 3:287–296. [DOI] [PubMed] [Google Scholar]
- 30.Tsubata, T., J. Wu, and T. Honjo. 1993. B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature. 364:645–648. [DOI] [PubMed] [Google Scholar]
- 31.Fulcher, D.A., and A. Basten. 1994. Reduced life span of anergic self-reactive B cells in a double-transgenic model. J. Exp. Med. 179:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cyster, J.G., S.B. Hartley, and C.C. Goodnow. 1994. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. 371:389–395. [DOI] [PubMed] [Google Scholar]
- 33.Strasser, A., S. Whittingham, D.L. Vaux, M.L. Bath, J.M. Adams, S. Cory, and A.W. Harris. 1991. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA. 88:8661–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang, A., and E.A. Clark. 2001. Involvement of Bik, a proapoptotic member of the Bcl-2 family, in surface IgM-mediated B cell apoptosis. J. Immunol. 166:6025–6033. [DOI] [PubMed] [Google Scholar]