

Abstract
Macrophages are activated from a resting state by a combination of cytokines and microbial products. Microbes are often sensed through Toll-like receptors signaling through MyD88. We used large-scale microarrays in multiple replicate experiments followed by stringent statistical analysis to compare gene expression in wild-type (WT) and MyD88−/− macrophages. We confirmed key results by quantitative reverse transcription polymerase chain reaction, Western blot, and enzyme-linked immunosorbent assay. Surprisingly, many genes, such as inducible nitric oxide synthase, IRG-1, IP-10, MIG, RANTES, and interleukin 6 were induced by interferon (IFN)-γ from 5- to 100-fold less extensively in MyD88−/− macrophages than in WT macrophages. Thus, widespread, full-scale activation of macrophages by IFN-γ requires MyD88. Analysis of the mechanism revealed that MyD88 mediates a process of self-priming by which resting macrophages produce a low level of tumor necrosis factor. This and other factors lead to basal activation of nuclear factor κB, which synergizes with IFN-γ for gene induction. In contrast, infection by live, virulent Mycobacterium tuberculosis (Mtb) activated macrophages largely through MyD88-independent pathways, and macrophages did not need MyD88 to kill Mtb in vitro. Thus, MyD88 plays a dynamic role in resting macrophages that supports IFN-γ–dependent activation, whereas macrophages can respond to a complex microbial stimulus, the tubercle bacillus, chiefly by other routes.
Keywords: macrophage activation, Toll-like receptors, innate immunity, NF-κB, microarray gene expression analysis
Introduction
In a standard view of macrophage activation, macrophages start out in a resting state and are driven synergistically by host- and pathogen-derived signals to a state of enhanced antimicrobial activity against facultative or obligate intracellular pathogens (1–5). IFN-γ is the principal but not the sole host-derived signal for macrophage activation (6). Mycobacterium tuberculosis (Mtb) is a prime example of a pathogen whose control by the host is a hallmark of macrophage activation, and the protective role of IFN-γ in experimental tuberculosis is well established (7). Host products that can synergize with IFN-γ to activate macrophages include IFN-α, IFN-β (8, 9), membrane-associated or soluble TNF (9–11), and CD40 ligand (12). Pathogen-derived macrophage-activating stimuli are diverse and signal via numerous receptors including Toll-like receptors (TLRs) to activate transcription of genes that regulate innate and adaptive immune responses. Such genes encode cytokines, chemokines, costimulatory molecules, and enzymes like inducible nitric oxide synthase (iNOS; 13).
The intracellular adaptor molecules MyD88, TIRAP/MAL, and TICAM-1 transduce signals from TLRs by linking TLRs to IL-1R–associated protein kinase. IL-1R–associated protein kinase initiates a signal cascade culminating in activation of MAP kinases, PI3 kinase, and nuclear factor (NF)-κB (14–18). MyD88 integrates signals from multiple TLRs. MyD88 deficiency impairs the macrophage response to several bacterial products (19) and MyD88-deficient mice were highly susceptible to infection with Listeria monocytogenes, Staphylococcus aureus, and Toxoplasma gondii (20–22). However, killing of L. monocytogenes by activated macrophages occurred by MyD88-independent mechanisms (23). Moreover, MyD88 deficiency improved resistance against polymicrobial sepsis, indicating both that MyD88-dependent responses can be deleterious and that MyD88-independent antibacterial mechanisms exist (24). Mycobacterial cell wall glycolipid lipoarabinomannan (LAM), mannosylated phosphatidylinositol, and a 19-kD Mtb lipoprotein were characterized as TLR2 agonists, whereas an undefined cell-associated and heat-labile moiety of live Mtb acted via TLR4 (25, 26).
There have been many studies of the regulation of individual genes in macrophages by subcellular microbial products via the TLR/MyD88 signal transduction pathway. However, there have apparently been no studies of the role of MyD88 in macrophage activation defined in terms of antimicrobial activity, induced by intact, virulent bacteria, and monitored by analysis of overall gene regulation. Here we compared the expression of ∼11,000 genes by primary bone marrow–derived macrophages (BMMφ) from both WT and MyD88-deficient (MyD88−/−) mice in response to IFN-γ alone and in response to infection with live, virulent Mtb in the presence or absence of IFN-γ. Three surprising findings emerged that suggest the need to revise the current understanding of macrophage activation. First, macrophages operationally considered to be “resting” in culture undergo active self-priming for activation, and this process is dependent on MyD88. Second, although the canonical IFN-γ signaling pathway does not involve MyD88, the expression of many genes in macrophages in response to IFN-γ is extensively dependent on MyD88, at least in part due to the impact of self-priming. Third, the majority of transcriptional responses of macrophages to live, virulent Mtb do not require MyD88. This implies either that TLRs are not the major receptors for recognizing intact, live Mtb, or that TLR-dependent responses to Mtb involve mostly MyD88-independent signaling pathways.
Materials and Methods
Activation and Infection of Macrophages.
Macrophages were collected in six independent experiments from 8–10-wk-old WT C57BL/6 × 129/SvJ mice and from MyD88−/− mice on the same background (six mice per experiment). The mice have been backcrossed six times to the C57BL/6 background. Bone marrow cells were differentiated into macrophages, treated with IFN-γ or not, and infected with Mtb from early log phase cultures of a low passage clinical isolate (strain 1254; American Type Culture Collection 51910) as previously described (27). Intracellular survival of Mtb and measurement of nitrite in the conditioned media were as reported (27). Bone marrow cells were seeded in 175 cm2 tissue culture flasks (Nunclon; Nalgene), 25 cm2 tissue culture flasks (Corning), or 24-well tissue culture plates (Corning) at 105 cells/cm2 and 0.25 ml medium/cm2, unless stated otherwise. DMEM was purchased from GIBCO BRL and FBS was purchased from Hyclone. All media, FBS, and buffers were tested for LPS contamination using a Limulus amebocyte lysate test kit (Bio Whittaker Inc.) and the LPS content was ≤20 pg/ml.
Array Hybridization.
24 h after infection, monolayers were lysed with Trizol (GIBCO BRL) and total RNA was isolated. After treatment with DNase I (Ambion) and purification (RNAeasy; QIAGEN), 2–3 μg RNA was reverse transcribed (Superscript II; GIBCO BRL) with a T7-polyT primer and cDNA was transcribed in the presence of biotinylated UTP and CTP (Enzo). Hybridization to GeneChip oligonucleotide arrays (Mu11KsubA, B) and scanning (Gene-Array Scanner) followed Affymetrix, Inc. protocols.
Data Processing.
Primary image analysis of the arrays was performed using GeneChip Microarray Analysis Suite version 5.0 (Affymetrix, Inc.) and images were scaled to an average hybridization intensity (average difference) of 250. Data analysis was performed using GeneSpring 4.1 software (Silicon Genetics). All measurements on each chip were divided by the 50th percentile value of that chip. Each gene was compared with its control by dividing its intensity by the average intensity of that gene in the six control samples (untreated WT macrophages). Data from six independent replicates were used to perform a Wilcoxon two-sample rank test for each gene. Only genes with an Affymetrix, Inc. “present call” in at least one of the two conditions compared were included in the analysis. A gene was considered “regulated” compared with control if its expression changed across the six experiments with P ≤ 0.05. To identify MyD88-dependent and MyD88-independent genes, expression levels (absolute signal intensities of normalized samples) and regulation factors (absolute signal intensities in response to a stimulus divided by signal intensities of untreated samples) were tested with a Wilcoxon two-sample rank test for each gene in WT versus MyD88−/− macrophages.
Immunocytology.
2 × 104/ml macrophages in 1 ml were cultured on 13-mm diameter glass coverslips in 24-well plates containing complete medium. 0.6 μg/ml anti–mouse TNF antibody (R&D Systems) was added to WT cells, 50 pg/ml mouse TNF (R&D Systems) was added to MyD88−/− cells, or cells were left untreated. 24 h after incubation at 37°C, medium was removed and cells were washed with PBS and fixed with 1% paraformaldehyde in cacodylate buffer, pH 7.4 (75 mM sodium cacodylate, 0.72% sucrose), for 10 min at room temperature. Cells were then washed with PBS and permeabilized with 0.05% Triton X-100 in PBS for 4 min at room temperature. Coverslips were washed twice in PBS and once in distilled water, inverted, and mounted in 90% glycerol in water on a glass slide.
Western Blot.
Cells cultured and stimulated with 100 U/ml IFN-γ for the indicated time were lysed with buffer containing 25 mM TrisCl, 1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 100 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM PMSF, and 5 μg/ml each of leupeptin, aprotinin, and pepstatin. Cell lysates were boiled for 5 min in reducing SDS-PAGE sample buffer, subjected to 7.5% SDS-PAGE, and transferred to a 0.2-μm pore nitrocellulose membrane (Schleicher & Schuell) in 20% methanol, 25 mM Tris, and 192 mM glycine, pH 8.3. The membrane was blocked with 5% milk, blotted with anti–mouse iNOS (28), or anti–mouse pY-STAT1 and anti–mouse STAT1 antibody (Cell Signaling Technology), followed by secondary antibody coupled to horseradish peroxidase (1:10,000; Amersham Biosciences). Bound antibody was detected by enhanced chemiluminescence (NEN Life Science Products).
Real-Time Quantitative RT-PCR (qRT-PCR).
The 5′ 3′ nuclease activity of Taq polymerase was applied for the detection of PCR-amplified nucleic acids. Standard curves were generated for each gene product using plasmid dilution series containing the target sequences. Probes were synthesized by Biosearch Technologies and labeled with the reporter dye FAM at the 5′ end and the quencher Black Hole Quencher at the 3′ end. Primer and probe sequences are listed in Table S3, which is available at http://www.jem.org/cgi/content/full/jem.20030603/DC1. 100 ng RNA was transcribed into cDNA with gene-specific primers in 20 μl using 50 U MuLV reverse transcriptase (PerkinElmer). cDNA was diluted to 1,000 μl. PCR was performed in a volume of 15 μl on the ABI PRISM 7900HT sequence detection system (PerkinElmer).
ELISA.
Multiplex cytokine detection for IL-10, MIP1α, MCP-5, and RANTES was performed by Pierce Chemical Co. with 24-h culture supernatants. The same supernatants were used for mouse TNF ELISA (Duoset; R&D Systems).
Electrophoretic Mobility Shift Assays (EMSAs) and Supershift Analyses.
EMSA was performed as previously described (29). For supershift experiments, nuclear proteins were incubated with polyclonal antibodies against mouse protein p50 and p65 (Santa Cruz Biotechnology, Inc.) for 30 min on ice before the addition of radiolabeled oligonucleotide and gel electrophoresis.
Online Supplemental Material.
Table I shows MyD88-dependent gene regulation in response to IFN-γ, Table S2 shows gene regulation in response to Mtb, and Table S3 shows sequences of primers and probes for qRT-PCR. Fig. S1 shows gene regulation in WT and MyD88−/− macrophages in response to activation with IFN-γ, infection with Mtb, or both. Tables S1–S3 and Fig. S1 are available at http://www.jem.org/cgi/content/full/jem.20030603/DC1.
Results
Functional Activation of MyD88−/− Macrophages.
We began by testing MyD88's contribution to macrophage activation at the functional level. Mouse macrophages activated with IFN-γ can kill Mtb through a nitric oxide (NO)-dependent mechanism. In contrast, nonactivated macrophages produce little NO and exert a bacteriostatic effect (7, 27). Accordingly, we examined nitrite accumulation and bacterial survival in both resting and IFN-γ–activated BMMφ after infection with a disease-causing clinical isolate of Mtb. In response to IFN-γ and Mtb, WT macrophages produced similar amounts of NO per cell when cells were plated at two different cell densities (≤105 cells/cm2 and 1.5–2 × 105 cells/cm2; Fig. 1, A and B) . As expected, activated WT macrophages killed intracellular Mtb (Fig. 1 C). In contrast, production of NO by IFN-γ–treated, Mtb-infected MyD88−/− cells was dependent on cell density (Fig. 1, A and B). At the higher cell density, MyD88−/− cells produced nearly normal amounts of NO and killed Mtb. Thus, macrophages can be functionally activated by the combination of IFN-γ and Mtb in the absence of MyD88 (Fig. 1 C). However, the effect of cell density suggested that the response to exogenous activating signals may depend on an autocrine factor whose amount becomes limiting in the absence of MyD88.
Figure 1.

Activation of WT and MyD88−/− BMMφ in response to IFN-γ and Mtb. BMMφ were seeded at a cell density of ≤105/cm2 (A) or 1.5–2 × 105/cm2 (B), activated with 100 U/ml IFN-γ for 24 h, and infected with Mtb at a multiplicity of infection (MOI) of 5. Production of nitrite, an oxidation product of NO, is shown from WT macrophages (open bars) and MyD88−/− macrophages (solid bars; means ± SE of seven to eight independent experiments). (C) WT and MyD88−/− macrophages at a cell density of 2 × 105/cm2 were infected with Mtb at an MOI of 5. ▪, CFUs from nonactivated (resting) macrophages; •, CFUs from macrophages activated for 48 h with 100 U/ml IFN-γ (means ± SD of triplicates in one experiment representative of three).
Abnormal Phenotype of MyD88−/− Macrophages at Rest.
In the foregoing experiments, strikingly different morphologies of WT versus MyD88−/− macrophages were evident under resting conditions, i.e., before the addition of IFN-γ or Mtb. WT macrophages after 6 d in culture were well spread with the irregular, crescentic, or polygonal outline typical of BMMφ. In contrast, MyD88−/− macrophages cultured at lower density (∼104 cells/cm2) spread less well and tended to adopt a spindle shape (to be illustrated subsequently). Flow cytometric analysis revealed that both WT and MyD88−/− macrophages were homogeneous cell populations with respect to expression of the following surface markers, and moreover, the markers were expressed at similar levels: CD14, Mac-1, CD18, CD16/32, and F4/80 (unpublished data). Thus, the different phenotypes of WT and MyD88−/− macrophages could not be attributed to a failure of MyD88−/− bone marrow cells to differentiate into macrophages.
To develop a hypothesis for the mechanistic basis for the phenotypic difference between WT and MyD88−/− macrophages, we turned to global gene expression analysis, comparing resting macrophages of both genotypes. All statements regarding differential gene expression in this paper refer to statistically significant differences in expression levels for a given gene in two different macrophage populations or for two different treatments of the same macrophage population, based on uncensored data from six independent experiments. Statistically significant differences in the ratios of expression for a given gene in treated versus untreated samples (fold change) were used as an additional criterion where so stated.
578 genes were differentially expressed between WT and MyD88−/− macrophages at rest. Of these, 88 genes were at least twofold more strongly expressed in resting WT than resting MyD88−/− macrophages, and 31 genes were expressed at least twofold less. The impact of MyD88 on expression of 12 annotated genes that were among those with the biggest differences in expression between resting WT and resting MyD88−/− macrophages was confirmed by independent methods (Fig. 2) . By real-time qRT-PCR, immune responsive gene 1 (IRG1), serum amyloid A (SAA3), IL-1β, IL-6, IL-10, TNF, IP10, RANTES, MIG, formyl peptide receptor (FPR), and macrophage receptor with collagenous domain (MARCO) were subnormally expressed in resting MyD88−/− macrophages, whereas the chemokine receptor CXCR4 was more highly expressed in MyD88−/− macrophages than in the WT cells (Fig. 2 A). ELISA confirmed that less IL-10, TNF, and MIP1α were released by resting MyD88−/− macrophages than by WT cells (Fig. 2 B). To exclude the possibility that the observed differences between WT and MyD88−/− macrophages were due to MyD88-independent genetic differences in the strain background (C57BL/6 × 129/SvJ, backcrossed six times to C57BL/6), we analyzed gene expression of resting macrophages and IFN-γ–activated macrophages from C57BL/6 and 129Sv mice by qRT-PCR. The mRNA levels of IRG1, FPR, MARCO, SAA3, IP10, MIG, and TNF were similar in resting C57BL/6 and 129/SvJ macrophages and treatment with IFN-γ resulted in a similar induction of iNOS, MIG, IP10, and IRG1 in WT macrophages of both strains (unpublished data). In addition, WT macrophages of both strains resembled WT C57BL/6 × 129/SvJ macrophages morphologically, showing none of the phenotypic features of MyD88−/− macrophages. We conclude that the observed differences between WT and MyD88−/− macrophages were MyD88 dependent.
Figure 2.
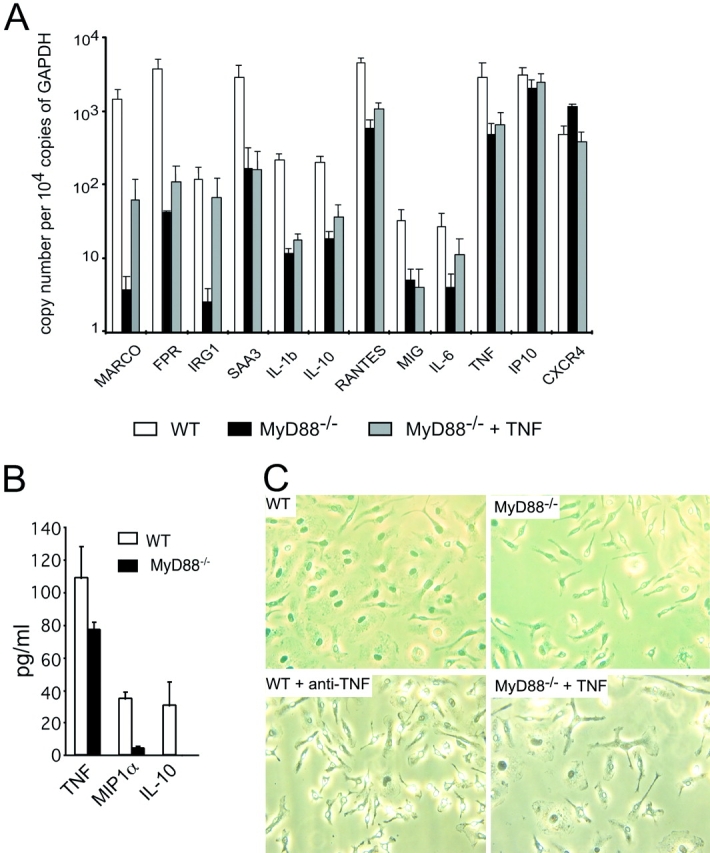
Comparison of resting WT and MyD88−/− BMMφ. (A) Differential gene expression between resting WT and MyD88−/− macrophages (qRT-PCR). RNA harvested from resting WT (open bars) and MyD88−/− (solid bars) BMMφ and MyD88−/− BMMφ treated with 50 pg/ml TNF for 1 wk (gray bars) was tested for gene expression by qRT-PCR (means ± SE of three independent experiments). (B) Differential cytokine/chemokine production between resting WT and MyD88−/− macrophages. Cell culture supernatant was collected from resting WT (open bars) and MyD88−/− (solid bars) BMMφ. Production of TNF, MIP1α, and IL-10 was tested by ELISA (means ± SE of three independent experiments; P < 0.05 for each cytokine/chemokine). (C) Morphology of WT and MyD88−/− BMMφ. Bone marrow cells were isolated from WT and MyD88−/− mice and cultured in the presence of 20% L929 cell–conditioned medium for 7 d. Cells were either left untreated (WT and MyD88−/−) or treated with 50 pg/ml TNF (MyD88−/−) or 0.6 μg/ml anti-TNF (WT) for 48 h.
Because expression of IL-1β, IL-10, and TNF was lower in resting MyD88−/− cells, we tested whether the poor spreading of MyD88−/− macrophages might be a consequence of insufficient basal production of one of these cytokines. We tested this by adding to MyD88−/− macrophage cultures the amounts of IL-10 or TNF that were lacking in comparison to WT cultures, and by adding neutralizing anti–IL-1α, anti–IL-1β, or anti-TNF antibodies to WT cultures. Because MyD88−/− cells do not respond to IL-1 (30), we did not add IL-1 to MyD88−/− cells. Addition of IL-10 had no effect on the morphology of MyD88−/− cells, nor did neutralizing anti–IL-1 antibodies affect the morphology of WT cells (unpublished data). In contrast, anti-TNF antibody caused WT cells to spread less and adopt the shape of untreated MyD88−/− cells. Conversely, 50 pg/ml TNF improved the spreading of MyD88−/− macrophages (Fig. 2 C). The ability of TNF to enhance spreading of WT macrophages has been described (31). Culturing the cells at a higher density (105/cm2 instead of 104/cm2) also restored normal morphology (unpublished data). Moreover, the addition of low dose TNF enhanced the expression of genes in otherwise untreated MyD88−/− macrophages (Fig. 2 A). Thus, WT macrophages released low but functionally important amounts of TNF under resting conditions. MyD88−/− cells were impaired in release of this cytokine.
Gene Induction and Suppression by IFN-γ and/or Mtb.
Even though MyD88-deficient macrophages started out differently than WT macrophages, if cultured at high density and exposed to IFN-γ and Mtb they came to resemble WT cells functionally with respect to anti-mycobacterial activity and NO secretion (Fig. 1). However, the functional assays measured only a few of the many changes that macrophages undergo during activation. To monitor activation more broadly, we compared expression of ∼11,000 genes (about one third of the mouse genome) between WT and MyD88−/− macrophages in response to IFN-γ, Mtb, and both stimuli together.
Microarray analysis of WT macrophages indicated that >800 genes were regulated in response to IFN-γ and >600 genes were regulated in response to Mtb infection (see Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20030603/DC1). A similar number of genes were regulated in MyD88−/− macrophages, and the fold change of regulation was similar in both genotypes (see Fig. S1 B, available at http://www.jem.org/cgi/content/full/jem.20030603/DC1). In macrophages of both genotypes, the largest number of genes was regulated in response to the combination of IFN-γ and Mtb to an extent that indicated synergy between the cytokine and the bacteria. For WT macrophages, the number of genes up-regulated and down-regulated by each stimulus or their combination was almost equal. The same was true for MyD88−/− cells. These findings extend a report with macrophages of other genetic backgrounds (27), showing that in addition to their well-known ability to activate gene expression, IFN-γ and Mtb are comparably active at suppressing gene expression.
We then compared the regulation of individual genes by IFN-γ or Mtb in macrophages of the two genotypes. As shown above, many genes were expressed differently in resting WT and MyD88−/− macrophages. This resulted in different expression levels in response to stimulation when the fold change of regulation was similar in both macrophage genotypes. We therefore used two approaches to identify genes that do or do not depend in part on MyD88 for their response to IFN-γ and Mtb. One approach was based on the comparison of gene expression levels in WT and MyD88−/− macrophages after treatment with IFN-γ and Mtb (Fig. 3 A). Genes whose expression levels were less than twofold different between WT and MyD88−/− macrophages upon stimulation were classified as MyD88 independent. In contrast, genes whose expression levels upon treatment were statistically significantly different and more than twofold different in WT compared with MyD88−/− macrophages, regardless of the fold regulation within one genotype, were classified as MyD88 dependent. By applying these criteria nearly half of the ∼800 genes that were statistically significantly regulated in response to IFN-γ were categorized as MyD88 dependent or MyD88 independent. The remaining ∼400 genes were statistically indeterminate with respect to their (in)dependence on MyD88. “MyD88 dependent” in this context means that the stimulus-induced change in expression was significantly limited in the absence of MyD88, without implying an absolute requirement for MyD88. In contrast, our second approach directly compared the fold change of expression of significantly regulated genes in macrophages of the two genotypes without regard to the expression levels attained (Fig. 3 B). To our surprise, both of these approaches identified many genes that were MyD88 dependent in their response to IFN-γ. This group of genes was larger when defined by the impact of MyD88 on gene expression levels (105 genes; Fig. 3 A and see Table S1, available at http://www.jem.org/cgi/content/full/jem.20030603/DC1) than when defined by the impact of MyD88 on fold regulation (50 genes; Fig. 3 B). Next, we turned to independent experimental approaches to confirm these observations and unveil mechanisms underlying the impact of MyD88 on gene regulation in response to IFN-γ.
Figure 3.

Specificity of gene regulation in WT and MyD88−/− macrophages. (A) Specificity based on expression level: Numbers within the common sectors (red) of each Venn diagram represent the count of individual genes whose expression level upon treatment was not statistically significantly different between WT and MyD88−/− macrophages and was less than twofold different between WT and MyD88−/− macrophages. Numbers in the blue and green sectors represent genes whose expression level upon treatment was statistically significantly different (P ≤ 0.05) and greater than twofold different between WT and MyD88−/− macrophages. Genes represented by blue sectors were regulated in WT macrophages but not or significantly less regulated in MyD88−/− macrophages. Genes represented in green sectors were regulated in MyD88−/− macrophages but not or significantly less regulated in WT macrophages. (B) Specificity based on fold regulation: Numbers within the common sectors (red) of each Venn diagram represent genes whose fold change in expression upon treatment was not statistically significantly different between WT and MyD88−/− macrophages and whose fold change in expression in response to treatment was less than twofold different between WT and MyD88−/− macrophages. Numbers in the blue and green sectors represent genes whose fold change in expression upon treatment was statistically significantly different (P ≤ 0.05) and greater than twofold different between WT and MyD88−/− macrophages upon treatment. Genes represented by blue sectors were regulated in WT macrophages but not or significantly less regulated in MyD88−/− macrophages. Genes represented in green sectors were regulated in MyD88−/− macrophages but not or significantly less regulated in WT macrophages.
MyD88-dependent Gene Regulation by IFN-γ: Role of TNF.
Fig. 4 shows the extent of regulation of genes whose responses to IFN-γ illustrate partial dependence on, or independence of, MyD88. Many of these genes were induced by IFN-γ in MyD88−/− macrophages with a fold change comparable to that of WT cells (Fig. 4 A). Nonetheless, their expression in response to IFN-γ remained significantly and substantially subnormal in MyD88−/− macrophages (Fig. 4 B) as confirmed by qRT-PCR, Western blot, and ELISA (Fig. 5) . Thus, qRT-PCR confirmed induction of iNOS, IRG1, IP10, MIG, RANTES, and IL-6 by IFN-γ in WT macrophages. Induction of these genes was severely reduced in MyD88−/− macrophages (Fig. 5 A). Western blot showed iNOS induction in WT but not MyD88−/− macrophages after 72 h of incubation with IFN-γ alone (Fig. 5 B), as opposed to the result when cells were treated with IFN-γ in combination with Mtb (Fig. 1). ELISA demonstrated MCP-5 induction by IFN-γ in WT macrophages but not in MyD88−/− macrophages (Fig. 5 C).
Figure 4.

Quantitative regulation of individual genes in WT and MyD88−/− macrophages in response to IFN-γ. Data are means from six microarray experiments. RNA was isolated from resting macrophages and from macrophages treated for 48 h with 100 U/ml IFN-γ. Red squares indicate genes regulated in both WT and MyD88−/− macrophages. Blue squares indicate genes whose regulation was dependent on MyD88. MyD88-dependent genes were defined as genes whose expression level in response to IFN-γ in MyD88−/− macrophages was statistically different (P ≤ 0.05) from that in WT macrophages and at least twofold less than in WT macrophages. Genes regulated with high fold changes and genes of special interest are named. In A, the fold regulation in response to IFN-γ in WT macrophages is compared with fold regulation in response to IFN-γ in MyD88−/− macrophages. Fold regulation is calculated as ratio of mean signal intensity of expression of the gene in question in IFN-γ–treated WT or MyD88−/− macrophages over its mean signal intensity in untreated macrophages of the same genotype. In B, gene expression levels in IFN-γ–treated WT and IFN-γ–treated MyD88−/− macrophages are compared. Gene expression levels are normalized by taking the mean signal intensity of expression of the gene in question in IFN-γ–treated WT or MyD88−/− macrophages and dividing that value by the mean signal intensity in untreated WT macrophages. GBP, guanylate nucleotide binding protein; H1, MHC class I Qa-TIa; OSF2, osteoblast-specific factor 2; SPI2, serine protease inhibitor 2; Upase, uridine phosphorylase. See text for other abbreviations.
Figure 5.

Confirmation of MyD88-dependent gene regulation in response to IFN-γ. (A) Cells were left untreated (WT and MyD88−/−) or treated with 50 pg/ml TNF (MyD88−/−) or 0.6 μg/ml anti-TNF (aTNF; WT) for 1 wk followed by IFN-γ treatment (100 U/ml) for 4 h. RNA was assayed by qRT-PCR. Gene expression is reported as copy number per 10,000 copies of GAPDH on a log10 scale (means of three independent experiments ± SE). Numbers atop each bar indicate the fold change of gene expression after treatment compared with expression of the same gene in untreated WT cells. If expression was not detectable (for example, iNOS in untreated WT cells), the value of 1 copy per 10,000 GAPDH was assigned and fold change was conservatively estimated based on this value. All qRT-PCR data in this paper are presented in the same way. Open bars depict gene expression in WT cells and solid bars depict gene expression in MyD88−/− cells. (B) Cell lysates were collected over a 72-h time course of IFN-γ treatment. Western blot was performed with anti-iNOS antibody. The stripped membrane was blotted with anti-HSP90 antibody as a loading control. Data are representative of three experiments. (C) MCP-5 production was measured by ELISA in cell culture supernatant 24 h after treatment with 100 U/ml IFN-γ (means of three experiments ± SE; P < 0.05). Open bars depict gene expression in WT cells and solid bars depict gene expression in MyD88−/− cells.
Next, we tested whether basal TNF deficiency contributed to the hyporesponsiveness of MyD88−/− macrophages to IFN-γ. We added 50 pg/ml TNF to MyD88−/− macrophages and 0.6 μg/ml anti-TNF to WT macrophages before IFN-γ treatment, and then tested gene expression by qRT-PCR. For most of the genes tested, the addition of low dose TNF potentiated their expression in MyD88−/− macrophages after IFN-γ treatment (Fig. 5 A). Conversely, WT cells pretreated with TNF neutralizing antibody responded to IFN-γ with reduced gene induction (Fig. 5 A). Thus, basal expression of TNF markedly affected macrophage responses to IFN-γ.
Hyporesponsiveness of MyD88−/− Macrophages to IFN-γ Correlates with Subnormal Basal NF-κB Activity.
To address the mechanism by which MyD88 enhances macrophage responses to IFN-γ, we tested the impact of MyD88 on STAT1 activation after IFN-γ treatment. Tyr-701 phosphorylation of STAT1 in response to IFN-γ was normal in MyD88−/− macrophages (Fig. 6 A), demonstrating the integrity of early IFN-γ signaling events in these cells. This suggests that although STAT1 activation is necessary, it is not sufficient for full-scale gene regulation by IFN-γ. Transcription factors other than STAT1 also contribute, and a relative lack of such factors might account for the hyporesponsiveness of MyD88−/− macrophages to IFN-γ. With this in mind, we inspected the promoter regions of 16 annotated genes that were among the most strongly regulated by IFN-γ in WT macrophages and whose response to IFN-γ was subnormal in MyD88−/− cells. All of these promoters contained binding sites for NF-κB as well as for IFN-γ–recruited or –induced transcription factors such as STAT1, IFN regulatory factor 1, or IFN regulatory factor 3. We hypothesized that basal NF-κB activity in resting WT macrophages could synergize with IFN-γ for maximal gene expression. In contrast, MyD88−/− macrophages might be deficient in this basal NF-κB activity (Fig. 6 B). This would contribute to subnormal gene expression in resting MyD88−/− macrophages and impaired gene induction by IFN-γ.
Figure 6.

One potential mechanism of hyporesponsiveness of MyD88−/− BMMφ to IFN-γ. (A) Western blot to detect phosphorylated STAT1 (pY-STAT1). WT and MyD88−/− BMMφ were untreated or treated with 100 U/ml IFN-γ. Cell lysates were harvested 15 min and 2 h after IFN-γ treatment. Western blot was performed with anti-pTyr701 STAT1. (B) Hypothetical model for hyporesponsiveness of MyD88−/− BMMφ to IFN-γ. The horizontal line represents the plasma membrane. Endogenous ligands that activate the MyD88 pathway might be produced by the macrophage (e.g., IL-1, IL-18, heat shock proteins) or might be produced by the action of macrophages on the extracellular matrix (e.g., fibronectin and fibronectin fragments). MyD88-dependent signaling in response to these ligands may drive the NF-κB–dependent expression of additional stimuli (e.g., TNF, SAA3), which can also help sustain NF-κB activation. The resulting activation of NF-κB in ostensibly resting macrophages can synergize with IFN-γ–activated signals (e.g., STAT1) in driving IFN-γ–dependent gene expression. (C) Detection of NF-κB by EMSA. Nuclear extracts were harvested from resting WT and MyD88−/− macrophages and equal amounts of protein were subjected to EMSA. Data are representative of three experiments.
To test this hypothesis, EMSAs were performed on nuclear extracts from resting WT and MyD88−/− macrophages. WT cells indeed displayed a higher basal activity of NF-κB than MyD88−/− macrophages (Fig. 6 C). Supershift analyses with monospecific antibodies identified p65/p50 heterodimers and p50 homodimers.
Mtb Regulates Macrophage Gene Expression by Both MyD88-dependent and MyD88-independent Pathways.
Despite their defect in IFN-γ–induced gene expression, MyD88−/− macrophages responded to Mtb far better than anticipated. As shown in Fig. 7 and Table S2, available at http://www.jem.org/cgi/content/full/jem.20030603/DC1, the vast majority of genes showing substantial induction by Mtb in WT macrophages were similarly induced in macrophages lacking MyD88. These observations were confirmed by qRT-PCR and ELISA. Thus, iNOS, COX-2, IP10, MIG, RANTES, IRG-1, argininosuccinate synthetase 1, and chemokines JE and KC were up-regulated by Mtb in both WT and MyD88−/− macrophages to a similar expression level (Fig. 8 A), as confirmed for RANTES by ELISA (Fig. 8 C). In contrast, MyD88 was required for Mtb to induce IL-1β and IL-6 (Figs. 7 and 8 B), cytokines that play a critical role in the immune response to and control of Mtb (32–35). Similarly, cell surface receptors FPR and MARCO and acute phase reactant SAA3 were up-regulated by Mtb in WT macrophages, but their expression in MyD88−/− macrophages in response to Mtb was minimal (Fig. 8 B). The situation was similar for MCP-5, as confirmed by ELISA (Fig. 8 C).
Figure 7.

Comparison of gene regulation in WT and MyD88−/− macrophages in response to Mtb. Data are averages of six microarray experiments. RNA was isolated from resting macrophages and from macrophages 24 h after infection with Mtb. Red squares indicate genes regulated in WT and MyD88−/− macrophages. Blue squares indicate genes whose regulation was dependent on MyD88. MyD88-dependent genes were defined as genes whose expression level in response to Mtb in MyD88−/− macrophages was statistically different (P ≤ 0.05) from that in WT macrophages and at least twofold less than in WT macrophages. Genes regulated with high fold changes and genes of special interest are named. In A, the fold regulation in response to Mtb in WT macrophages is compared with fold regulation in response to Mtb in MyD88−/− macrophages. Fold regulation is calculated as ratio of mean signal intensity of expression of the gene in question in Mtb-treated WT and MyD88-deficient macrophages over its mean signal intensity in uninfected macrophages of the same genotype. In B, gene expression levels are normalized for Mtb-infected macrophages by taking the mean signal intensity of expression of the gene in question in infected WT or MyD88−/− macrophages and dividing that value by the mean signal intensity in uninfected WT macrophages. DAF, decay-accelerating factor; IGFBP, insulin-like growth factor binding protein; GARG-16, glucocorticoid-attenuated response gene 16; LIX, LPS-induced C-X-C chemokine. See text for other abbreviations.
Figure 8.

MyD88-independent and MyD88-dependent gene regulation in response to Mtb. (A and B) Gene regulation measured by qRT-PCR. RNA was harvested from WT and MyD88−/− BMMφ 4 h after Mtb infection and assayed for gene expression by qRT-PCR. Data are means of three independent experiments ± SE. Open bars depict gene expression in WT cells and solid bars depict gene expression in MyD88−/− cells. MyD88-independent (A) and MyD88-dependent (B) gene regulation in response to Mtb. (C) MyD88-independent production of RANTES and MyD88-dependent production of MCP-5 in infected macrophages. Cells were infected with Mtb for 24 h. Cell culture supernatants were assayed for RANTES and MCP-5 by ELISA (means of three experiments ± SE; P < 0.05). Open bars depict gene expression in WT cells and solid bars depict gene expression in MyD88−/− cells.
Discussion
These results reveal an MyD88-dependent process of self-priming in resting macrophages, the importance of MyD88-dependent autocrine signals in macrophage activation by IFN-γ, and the ability of macrophages to sense the presence of viable Mtb with little help from MyD88. Taken together, these three observations suggest that MyD88 plays a different role in macrophage activation than previously understood. The essentiality of MyD88 for full-scale activation by IFN-γ and the relative dispensability of MyD88 for activation by Mtb were equally unexpected.
Compared with resting WT macrophages, nonactivated MyD88-deficient macrophages appeared morphologically quiescent and displayed subnormal expression of many genes in the face of reduced basal NF-κB activity. NF-κB activity has been observed previously in unstimulated macrophages (36). Our study suggests that impaired NF-κB activity in resting MyD88−/− macrophages might be one explanation for the functional differences between nonactivated WT and MyD88−/− cells.
Lower basal NF-κB activity in MyD88−/− macrophages also appeared to contribute to the impaired transcriptional response to IFN-γ in MyD88−/− macrophages. MyD88−/− cells had no defect in early steps in IFN-γ signaling. However, maximal activation of IFN-γ–induced genes often results from synergy between NF-κB–mediated and IFN-γ–dependent signals. Preincubation with TNF, an NF-κB activating stimulus, potentiated gene expression in MyD88−/− macrophages in response to IFN-γ. A contribution of spontaneously produced, autocrine-acting cytokines to macrophage activation has been noted before (37), but a central role for MyD88 in this process was not recognized.
Two interrelated mechanisms may account for MyD88-dependent NF-κB activity in resting WT macrophages. First, there might be substances present in cultures of resting macrophages that can activate NF-κB through signal transduction pathways that are dependent on MyD88. Stimuli of endogenous origin that signal through MyD88 include IL-1α, IL-1β, IL-18, β-defensin 2 (38), minimally modified low density lipoproteins (39), necrotic cells (40, 41), and heat shock proteins (42). For example, monocytes and macrophages express mRNA for IL-1β and TNF as they adhere to extracellular matrix (43, 44). Second, there might be substances that activate NF-κB through pathways that do not involve MyD88. However, macrophages may require MyD88 to respond to signals that elicit the production of these substances. For example, under resting conditions, WT macrophages secreted TNF and SAA3, but MyD88−/− macrophages released less of these proteins. TNF activates NF-κB via the TNF receptor, whereas SAA3 activates NF-κB through the receptor for advanced glycation end-products (45). Neither the TNF receptor nor receptor for advanced glycation end-products depend on MyD88, but MyD88 appears to have helped mediate the signals that led to secretion of TNF and SAA3 by resting macrophages. Finally, MyD88 may regulate additional pathways besides NF-κB that can also modulate the response to IFN-γ (Sun, D. and Ding, A., personal communication).
Active self-priming of macrophages for subsequent activation is fundamentally different from an earlier concept of macrophage activation, in which the resting cell was passive and the exogenous-activating stimuli microbial products and lymphocyte-derived mediators were called “priming” and “activating” agents, depending on the order of their addition (3, 8, 46, 47).
Despite its marked effect on the macrophage response to IFN-γ, subnormal basal NF-κB activity had little impact on the ability of MyD88−/− macrophages to respond to Mtb. Most macrophage genes that were regulated by Mtb responded independently of MyD88, even genes that were subnormally expressed in resting MyD88−/− macrophages. Unlike the response to IFN-γ on the part of MyD88−/− macrophages, wherein many genes failed to reach expression levels comparable to that of the WT cells, expression of Mtb-induced genes in MyD88−/− macrophages was often quantitatively similar to that of the WT cells.
The MyD88-independent pathways by which Mtb activates the macrophage remain to be identified. Mycobacterial products such as lipoproteins, LAM, and phosphatidylinositol stimulate macrophages via TLR2 (25, 26). However, a TLR2-dependent, MyD88-independent signal transduction pathway has not been described. Live Mtb activates macrophages not only through TLR2 but also through TLR4 (48). Some of the genes (e.g., IRG1, IP10, GARG16) that were regulated by Mtb in an MyD88-independent manner in our study were also induced by LPS via an MyD88-independent pathway (16, 49), and it is possible that Mtb also activates this pathway via TLR4. NO production and anti-mycobacterial activity were retained in activated, Mtb-infected MyD88−/− cells. This is in agreement with MyD88-independent activation of the iNOS promoter in Mtb-stimulated macrophage-like cells (50). Many receptors besides TLRs have been shown to mediate interactions of Mtb with macrophages. The mannose receptor, complement receptors, scavenger receptors, CD14, CD44, and Fcγ receptors may all participate in phagocytosis of Mtb by macrophages (51, 52). Engagement of the mannose receptor triggers production of reactive oxygen intermediates by macrophages as well as induction of matrix metalloproteinase-9 in response to Mtb (53, 54). CD14 is involved in LAM-stimulated release of TNF, IL-1, and IL-8 (55, 56). Dendritic cell–specific ICAM-3–grabbing nonintegrin mediates uptake of Mtb by human monocyte-derived dendritic cells and transmits signals that interfere with their maturation (57, 58). Dendritic cell–specific ICAM-3–grabbing nonintegrin has also been found on macrophages (59) and thus may transmit Mtb-induced signals in macrophages as well. By whichever receptors, it is clear that live, intact Mtb can activate macrophages in large part independently of MyD88.
On the other hand, the MyD88-dependent component of the macrophage response to Mtb included induction of IL-1β and IL-6, which play critical roles in the immune response to and control of Mtb in vivo. IL-6–deficient mice displayed enhanced susceptibility to infection with Mtb (32), which might be related to impaired production of IFN-γ during the early phase of infection, before adaptive T cell immunity has fully developed (35). IL-1α and IL-1β double knockout mice and IL-1R type I–deficient mice showed increased bacterial growth and defective granuloma formation after infection with Mtb (33, 34). Thus, our results with cultured macrophages do not predict the course of experimental tuberculosis in MyD88−/− mice, where some critical cells will respond to Mtb before IFN-γ is produced, some will encounter IFN-γ but not Mtb, and some will react to both.
In conclusion, these results demonstrate that resting primary mouse macrophages prime themselves in an MyD88-dependent manner via autocrine factors, including TNF, to respond to a classical activating signal, IFN-γ. MyD88-dependent basal activation of NF-κB appears to be one of the mechanisms by which MyD88 scales the macrophage response to IFN-γ. Finally, it appears that live, virulent Mtb activates macrophages in large part via TLR adaptors other than MyD88 or via receptors other than TLRs.
Acknowledgments
We thank L. Lu, J. Nezezon, and C. Hickey for excellent technical assistance, and S. Bekiranov and T. Gaasterland for help analyzing data from pilot experiments not included in this report.
Supported by National Institutes of Health grants HL61241 (to C. Nathan), HL68525 (to S. Ehrt), AI30165 (to A. Ding), and a Cancer Research Institute Predoctoral Fellowship Training Grant (to S. Shi). The Department of Microbiology and Immunology acknowledges the support of the William Randolph Hearst Foundation.
Abbreviations used in this paper: BMMφ, bone marrow–derived macrophages; EMSA, electrophoretic mobility shift assay; FPR, formyl peptide receptor; iNOS, inducible nitric oxide synthase; IRG1, immune responsive gene 1; LAM, lipoarabinomannan; MARCO, macrophage receptor with collagenous domain; MOI, multiplicity of infection; Mtb, Mycobacterium tuberculosis; NF, nuclear factor; NO, nitric oxide; qRT-PCR, quantitative RT-PCR; SAA3, serum amyloid A; TLR, Toll-like receptor.
The online version of this article contains supplemental material.
References
- 1.Mackaness, G.B. 1969. The influence of immunologically committed lymphoid cells on macrophage activity in vivo. J. Exp. Med. 129:973–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nathan, C.F., M.L. Karnovsky, and J.R. David. 1971. Alterations of macrophage functions by mediators from lymphocytes. J. Exp. Med. 133:1356–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams, D.O., and T.A. Hamilton. 1984. The cell biology of macrophage activation. Annu. Rev. Immunol. 2:283–318. [DOI] [PubMed] [Google Scholar]
- 4.Mosser, D.M. 2003. The many faces of macrophage activation. J. Leukoc. Biol. 73:209–212. [DOI] [PubMed] [Google Scholar]
- 5.Gordon, S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3:23–35. [DOI] [PubMed] [Google Scholar]
- 6.Nathan, C.F., T.J. Prendergast, M.E. Wiebe, E.R. Stanley, E. Platzer, H.G. Remold, K. Welte, B.Y. Rubin, and H.W. Murray. 1984. Activation of human macrophages. Comparison of other cytokines with interferon-γ. J. Exp. Med. 160:600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flynn, J.L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93–129. [DOI] [PubMed] [Google Scholar]
- 8.Pace, J.L., and S.W. Russell. 1981. Activation of mouse macrophages for tumor cell killing. I. Quantitative analysis of interactions between lymphokine and lipopolysaccharide. J. Immunol. 126:1863–1867. [PubMed] [Google Scholar]
- 9.Ding, A.H., C.F. Nathan, and D.J. Stuehr. 1988. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J. Immunol. 141:2407–2412. [PubMed] [Google Scholar]
- 10.Munoz-Fernandez, M.A., M.A. Fernandez, and M. Fresno. 1992. Synergism between tumor necrosis factor-alpha and interferon-gamma on macrophage activation for the killing of intracellular Trypanosoma cruzi through a nitric oxide-dependent mechanism. Eur. J. Immunol. 22:301–307. [DOI] [PubMed] [Google Scholar]
- 11.Sypek, J.P., and D.J. Wyler. 1991. Antileishmanial defense in macrophages triggered by tumor necrosis factor expressed on CD4+ T lymphocyte plasma membrane. J. Exp. Med. 174:755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alderson, M.R., R.J. Armitage, T.W. Tough, L. Strockbine, W.C. Fanslow, and M.K. Spriggs. 1993. CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J. Exp. Med. 178:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2:675–680. [DOI] [PubMed] [Google Scholar]
- 14.Medzhitov, R., P. Preston-Hurlburt, E. Kopp, A. Stadlen, C. Chen, S. Ghosh, and C.A. Janeway, Jr. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell. 2:253–258. [DOI] [PubMed] [Google Scholar]
- 15.Jones, B.W., K.A. Heldwein, T.K. Means, J.J. Saukkonen, and M.J. Fenton. 2001. Differential roles of Toll-like receptors in the elicitation of proinflammatory responses by macrophages. Ann. Rheum. Dis. 60:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto, M., S. Sato, H. Hemmi, H. Sanjo, S. Uematsu, T. Kaisho, K. Hoshino, O. Takeuchi, M. Kobayashi, T. Fujita, et al. 2002. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 420:324–329. [DOI] [PubMed] [Google Scholar]
- 17.Horng, T., G.M. Barton, R.A. Flavell, and R. Medzhitov. 2002. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 420:329–333. [DOI] [PubMed] [Google Scholar]
- 18.Oshiumi, H., M. Matsumoto, K. Funami, T. Akazawa, and T. Seya. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 4:161–167. [DOI] [PubMed] [Google Scholar]
- 19.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 11:115–122. [DOI] [PubMed] [Google Scholar]
- 20.Scanga, C.A., J. Aliberti, D. Jankovic, F. Tilloy, S. Bennouna, E.Y. Denkers, R. Medzhitov, and A. Sher. 2002. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 168:5997–6001. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi, O., K. Hoshino, and S. Akira. 2000. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 165:5392–5396. [DOI] [PubMed] [Google Scholar]
- 22.Seki, E., H. Tsutsui, N.M. Tsuji, N. Hayashi, K. Adachi, H. Nakano, S. Futatsugi-Yumikura, O. Takeuchi, K. Hoshino, S. Akira, et al. 2002. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 169:3863–3868. [DOI] [PubMed] [Google Scholar]
- 23.Edelson, B.T., and E.R. Unanue. 2002. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J. Immunol. 169:3869–3875. [DOI] [PubMed] [Google Scholar]
- 24.Weighardt, H., S. Kaiser-Moore, R.M. Vabulas, C.J. Kirschning, H. Wagner, and B. Holzmann. 2002. Cutting edge: myeloid differentiation factor 88 deficiency improves resistance against sepsis caused by polymicrobial infection. J. Immunol. 169:2823–2827. [DOI] [PubMed] [Google Scholar]
- 25.Heldwein, K.A., and M.J. Fenton. 2002. The role of Toll-like receptors in immunity against mycobacterial infection. Microbes Infect. 4:937–944. [DOI] [PubMed] [Google Scholar]
- 26.Stenger, S., and R.L. Modlin. 2002. Control of Mycobacterium tuberculosis through mammalian Toll-like receptors. Curr. Opin. Immunol. 14:452–457. [DOI] [PubMed] [Google Scholar]
- 27.Ehrt, S., D. Schnappinger, S. Bekiranov, J. Drenkow, S. Shi, T.R. Gingeras, T. Gaasterland, G. Schoolnik, and C. Nathan. 2001. Reprogramming of the macrophage transcriptome in response to interferon-γ and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp. Med. 194:1123–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie, Q.W., H.J. Cho, J. Calaycay, R.A. Mumford, K.M. Swiderek, T.D. Lee, A. Ding, T. Troso, and C. Nathan. 1992. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science. 256:225–228. [DOI] [PubMed] [Google Scholar]
- 29.Keller, S.A., E.J. Schattner, and E. Cesarman. 2000. Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood. 96:2537–2542. [PubMed] [Google Scholar]
- 30.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1– and IL-18–mediated function. Immunity. 9:143–150. [DOI] [PubMed] [Google Scholar]
- 31.Inoue, M., F.P. Ross, J.M. Erdmann, Y. Abu-Amer, S. Wei, and S.L. Teitelbaum. 2000. Tumor necrosis factor alpha regulates alpha(v)beta5 integrin expression by osteoclast precursors in vitro and in vivo. Endocrinology. 141:284–290. [DOI] [PubMed] [Google Scholar]
- 32.Ladel, C.H., C. Blum, A. Dreher, K. Reifenberg, M. Kopf, and S.H. Kaufmann. 1997. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 65:4843–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juffermans, N.P., S. Florquin, L. Camoglio, A. Verbon, A.H. Kolk, P. Speelman, S.J. van Deventer, and T. van Der Poll. 2000. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J. Infect. Dis. 182:902–908. [DOI] [PubMed] [Google Scholar]
- 34.Yamada, H., S. Mizumo, R. Horai, Y. Iwakura, and I. Sugawara. 2000. Protective role of interleukin-1 in mycobacterial infection in IL-1 alpha/beta double-knockout mice. Lab. Invest. 80:759–767. [DOI] [PubMed] [Google Scholar]
- 35.Saunders, B.M., A.A. Frank, I.M. Orme, and A.M. Cooper. 2000. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect. Immun. 68:3322–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conti, L., J. Hiscott, M. Papacchini, A. Roulston, M.A. Wainberg, F. Belardelli, and S. Gessani. 1997. Induction of relA(p65) and I kappa B alpha subunit expression during differentiation of human peripheral blood monocytes to macrophages. Cell Growth Differ. 8:435–442. [PubMed] [Google Scholar]
- 37.Riches, D.W., and G.A. Underwood. 1991. Expression of interferon-beta during the triggering phase of macrophage cytocidal activation. Evidence for an autocrine/paracrine role in the regulation of this state. J. Biol. Chem. 266:24785–24792. [PubMed] [Google Scholar]
- 38.Biragyn, A., P.A. Ruffini, C.A. Leifer, E. Klyushnenkova, A. Shakhov, O. Chertov, A.K. Shirakawa, J.M. Farber, D.M. Segal, J.J. Oppenheim, and L.W. Kwak. 2002. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 298:1025–1029. [DOI] [PubMed] [Google Scholar]
- 39.Miller, Y.I., S. Viriyakosol, C.J. Binder, J.R. Feramisco, T.N. Kirkland, and J.L. Witztum. 2003. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J. Biol. Chem. 278:1561–1568. [DOI] [PubMed] [Google Scholar]
- 40.Li, M., D.F. Carpio, Y. Zheng, P. Bruzzo, V. Singh, F. Ouaaz, R.M. Medzhitov, and A.A. Beg. 2001. An essential role of the NF-kappa B/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J. Immunol. 166:7128–7135. [DOI] [PubMed] [Google Scholar]
- 41.Basu, S., R.J. Binder, R. Suto, K.M. Anderson, and P.K. Srivastava. 2000. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 12:1539–1546. [DOI] [PubMed] [Google Scholar]
- 42.Vabulas, R.M., H. Wagner, and H. Schild. 2002. Heat shock proteins as ligands of toll-like receptors. Curr. Top. Microbiol. Immunol. 270:169–184. [DOI] [PubMed] [Google Scholar]
- 43.Haskill, S., C. Johnson, D. Eierman, S. Becker, and K. Warren. 1988. Adherence induces selective mRNA expression of monocyte mediators and proto-oncogenes. J. Immunol. 140:1690–1694. [PubMed] [Google Scholar]
- 44.Fuhlbrigge, R.C., D.D. Chaplin, J.M. Kiely, and E.R. Unanue. 1987. Regulation of interleukin 1 gene expression by adherence and lipopolysaccharide. J. Immunol. 138:3799–3802. [PubMed] [Google Scholar]
- 45.Miyata, T., O. Hori, J. Zhang, S.D. Yan, L. Ferran, Y. Iida, and A.M. Schmidt. 1996. The receptor for advanced glycation end products (RAGE) is a central mediator of the interaction of AGE-beta2microglobulin with human mononuclear phagocytes via an oxidant-sensitive pathway. Implications for the pathogenesis of dialysis-related amyloidosis. J. Clin. Invest. 98:1088–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meltzer, M.S. 1981. Macrophage activation for tumor cytotoxicity: characterization of priming and trigger signals during lymphokine activation. J. Immunol. 127:179–183. [PubMed] [Google Scholar]
- 47.Hibbs, J.B., J.B. Weinberg, and H.A. Chapman. 1979. Modulation of the tumoricidal function of activated macrophages by bacterial endotoxin and mammalian macrophage activation factor(s). Adv. Exp. Med. Biol. 121B:433–453. [DOI] [PubMed] [Google Scholar]
- 48.Means, T.K., S. Wang, E. Lien, A. Yoshimura, D.T. Golenbock, and M.J. Fenton. 1999. Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 163:3920–3927. [PubMed] [Google Scholar]
- 49.Kawai, T., O. Takeuchi, T. Fujita, J. Inoue, P.F. Muhlradt, S. Sato, K. Hoshino, and S. Akira. 2001. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167:5887–5894. [DOI] [PubMed] [Google Scholar]
- 50.Means, T.K., B.W. Jones, A.B. Schromm, B.A. Shurtleff, J.A. Smith, J. Keane, D.T. Golenbock, S.N. Vogel, and M.J. Fenton. 2001. Differential effects of a Toll-like receptor antagonist on Mycobacterium tuberculosis-induced macrophage responses. J. Immunol. 166:4074–4082. [DOI] [PubMed] [Google Scholar]
- 51.Ernst, J.D. 1998. Macrophage receptors for Mycobacterium tuberculosis. Infect. Immun. 66:1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leemans, J.C., S. Florquin, M. Heikens, S.T. Pals, R. van der Neut, and T. Van Der Poll. 2003. CD44 is a macrophage binding site for Mycobacterium tuberculosis that mediates macrophage recruitment and protective immunity against tuberculosis. J. Clin. Invest. 111:681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rivera-Marrero, C.A., W. Schuyler, S. Roser, J.D. Ritzenthaler, S.A. Newburn, and J. Roman. 2002. M. tuberculosis induction of matrix metalloproteinase-9: the role of mannose and receptor-mediated mechanisms. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L546–L555. [DOI] [PubMed] [Google Scholar]
- 54.Ezekowitz, R.A., K. Sastry, P. Bailly, and A. Warner. 1990. Molecular characterization of the human macrophage mannose receptor: demonstration of multiple carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1 cells. J. Exp. Med. 172:1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, Y., M. Doerfler, T.C. Lee, B. Guillemin, and W.N. Rom. 1993. Mechanisms of stimulation of interleukin-1 beta and tumor necrosis factor-alpha by Mycobacterium tuberculosis components. J. Clin. Invest. 91:2076–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pugin, J., I.D. Heumann, A. Tomasz, V.V. Kravchenko, Y. Akamatsu, M. Nishijima, M.P. Glauser, P.S. Tobias, and R.J. Ulevitch. 1994. CD14 is a pattern recognition receptor. Immunity. 1:509–516. [DOI] [PubMed] [Google Scholar]
- 57.Tailleux, L., O. Schwartz, J.L. Herrmann, E. Pivert, M. Jackson, A. Amara, L. Legres, D. Dreher, L.P. Nicod, J.C. Gluckman, et al. 2003. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 197:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geijtenbeek, T.B., S.J. Van Vliet, E.A. Koppel, M. Sanchez-Hernandez, C.M. Vandenbroucke-Grauls, B. Appelmelk, and Y. Van Kooyk. 2003. Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Lent, P.L., C.G. Figdor, P. Barrera, K. Van Ginkel, A. Sloetjes, W.B. Van Den Berg, and R. Torensma. 2003. Expression of the dendritic cell-associated C-type lectin DC-SIGN by inflammatory matrix metalloproteinase-producing macrophages in rheumatoid arthritis synovium and interaction with intercellular adhesion molecule 3-positive T cells. Arthritis Rheum. 48:360–369. [DOI] [PubMed] [Google Scholar]