

Abstract
Relatively little is known about the pathway leading to the presentation of glycolipids by CD1 molecules. Here we show that the adaptor protein complex 3 (AP-3) is required for the efficient presentation of glycolipid antigens that require internalization and processing. AP-3 interacts with mouse CD1d, and cells from mice deficient for AP-3 have increased cell surface levels of CD1d and decreased expression in late endosomes. Spleen cells from AP-3–deficient mice have a reduced ability to present glycolipids to natural killer T (NKT) cells. Furthermore, AP-3–deficient mice have a significantly reduced NKT cell population, although this is not caused by self-tolerance that might result from increased CD1d surface levels. These data suggest that the generation of the endogenous ligand that selects NKT cells may also be AP-3 dependent. However, the function of MHC class II–reactive CD4+ T lymphocytes is not altered by AP-3 deficiency. Consistent with this divergence from the class II pathway, NKT cell development and antigen presentation by CD1d are not reduced by invariant chain deficiency. These data demonstrate that the AP-3 requirement is a particular attribute of the CD1d pathway in mice and that, although MHC class II molecules and CD1d are both found in late endosomes or lysosomes, different pathways mediate their intracellular trafficking.
Keywords: intracellular localization, trafficking, lipid antigen, lymphocyte selection, MHC II trafficking pathway
Introduction
CD1 molecules are a third family of antigen-presenting molecules. CD1a, CD1b, and CD1c present cell wall glycolipids from mycobacteria for host defense (1, 2) and brain-derived glycolipids (3). CD1d presents glycolipids to natural killer T (NKT) cells, a population of T lymphocytes that may have important immune regulatory functions (4). NKT cells typically have been defined as lymphocytes that express both TCR and NK1.1, but much attention has been focused on the NKT cell subset that in mice expresses a TCR α chain encoded by a rearrangement of Vα14 to Jα18 (formerly Jα281). This rearrangement forms an invariant CDR3α region (Vα14i) in these lymphocytes (5), and here we refer to them as Vα14i NKT cells in order to designate this subset more precisely. Many Vα14i NKT cells are self-reactive to APC-expressing mouse CD1d (mCD1d) (6–8); this presumably requires presentation by mCD1d of an endogenous, self-glycolipid ligand. Furthermore, nearly all of them show enhanced responses when mCD1d presents the synthetic phytosphingolipid, α–d-galactosyl ceramide (αGalCer) (9, 10). αGalCer was obtained from an extract of a marine sponge as a result of a screen for compounds that could prevent the metastases of transplanted tumors to the livers of mice (11). This glycolipid belongs to the category of glycosphingolipids that include the gangliosides, but the α anomeric linkage of the galactose to the lipid is unusual, since nearly all glycosphingolipids have a β linkage of the sugar to the lipid. Therefore, it is generally assumed that αGalCer is not a natural ligand for Vα14i NKT cells, but the structure of this natural ligand remains to be determined. The pathway leading to the presentation of glycolipid antigens by CD1d molecules is not well defined. Furthermore, although both CD1 molecules and MHC class II molecules are found in endosomes, features required for proper CD1 localization and antigen presentation that distinguish it from MHC class II molecules have not been identified. CD1b, CD1c, and CD1d all have a tyrosine-containing sequence in their cytoplasmic tails that mediates their localization to various types of endosomal compartments (12). Mice bearing a germ line deletion of the cytoplasmic tail of mCD1d have been generated, and APC from these mice have a defect in glycolipid antigen presentation (13). These mice also have a greatly reduced number of Vα14i NKT lymphocytes, despite greater than normal levels of surface mCD1d expression, strongly suggesting that the natural ligand required for the selection or homeostasis of these cells depends on the normal endosomal trafficking of mCD1d. These findings do not, however, identify a particular role for early or late endosomes in mCD1d antigen presentation.
Four AP complexes are known to bind to the tyrosine or dileucine-containing sequence motifs in transmembrane proteins in order to direct their selective localization to subsets of endosomal compartments (14, 15). This suggests that one or more of these could be involved in mCD1d trafficking, but the role of these adaptors in antigen presentation remains to be defined. AP-1 is important for the trafficking of proteins from the trans-Golgi network to endosomes (16), and AP-2 is involved in the internalization of membrane proteins to recycling compartments (17). Adaptor protein complex 3 (AP-3), by contrast, is involved in the localization of membrane proteins to lysosomes, platelet-dense granules, and melanosomes (18). Each AP complex is a heterotetramer. For example, the AP-3 complex found in most cells is composed of β3A, δ, μ3A, and σ3 subunits (19). Spontaneous mutants of the AP-3 β3A and δ subunits in mice were uncovered because of their effects on coat color, although these animals also have defects in platelets and other systems (15, 20). The β3A mutants, known as pearl mice, are considered a model for the Hermansky-Pudlak syndrome, which is characterized by misregulation of the biogenesis/function of melanosomes, lysosomes, and bleeding diathesis in these patients due to defects in platelet dense granules (21). We have shown that disaccharide analogs of αGalCer, such as Gal(α1→2)αGalCer, require processing of their carbohydrate portion to generate the antigenic monosaccharide αGalCer. Lysosomal enzymes mediate this antigen-processing event, and mCD1d is found in lysosomes (22), but it is not known if the mCD1d molecules in lysosomes are functional in antigen presentation. To address this question, we have analyzed mCD1d mutants having alterations in the cytoplasmic tail, and additionally, we have focused on mice bearing mutations in AP-3. The data define a role for AP-3 in mCD1d antigen presentation, but not for MHC class II presentation, and they strongly support the hypothesis that mCD1d localization to late endosomes or lysosomes is required for both antigen presentation and development of Vα14i NKT cells.
Materials and Methods
Mice.
Pearl (pe/pe) mutant and heterozygous pe/+ mice on the C57BL/6 background were a gift from Dr. Richard Swank (Roswell Park Memorial Institute, Buffalo, NY). These mice have a deletion in the COOH terminal region of the β3A subunit of the AP-3 adaptor, thereby creating a hypomorphic allele (20). The β3B protein, the neuronal form of β3, is not affected by this mutation (23). Pearl mice were bred in the vivarium of the La Jolla Institute for Allergy and Immunology by mating heterozygous females (black coat) to homozygous males (gray coat). The offspring were typed as pe/pe based on their gray coat color. Mocha (mh/mh) mutant and heterozygous (mh/+) mice are of a mixed genetic background and were obtained from the Jackson Laboratory. C57BL/6, C57BL/6 RAG1−/−, Ii − / − (B6.129S6.litm1liz), and B6 CD45.1(B6.SJL-Ptprca Pep3b/BoyJ) congenic mice were obtained from the Jackson Laboratory. Jα18−/− (Jα281−/−) mice were a gift from Dr. M. Taniguchi (Chiba University, Chiba, Japan).
Cell Lines.
B cell lymphoma A20 cells and Vα14i NKT cell hybridomas 1.2, 1.4, 3C3, and 2C12 were described previously (10, 24). The Vα14i NKT cell hybridomas share the Vα14i α chain, express different TCR β chains, and all have reactivity for αGalCer presented by mCD1d. The fetal skin dendritic cell (FSDC) line was a gift from Dr. Paula Ricciardi-Castagnoli (Instituto Dermopatico dell'Immacolata, Istituto di Ricovero e Cura a Carattere Scientifico, Rome, Italy) (25). Whole splenocytes and/or DCs were prepared from pe/+, pe/pe, B6, and Ii − / − mice according to procedures reported previously (10).
Generation of Cell Lines Expressing WT or Mutant mCD1d.
Primers were designed to contain 5′ BamHI or 3′ SalI sites to facilitate subcloning. To generate mouse CD1d (mCD1d)/a chimeras, in which the cytoplasmic tail and transmembrane region are derived from CD1a, a chimeric oligonucleotide encoding aa G271 to W279 of the mCD1d α3 domain and aa V274 to S281 of the human CD1a transmembrane domain, 5′-GGAGGACAGGATATCATCCTCTACTGGGAGCATCACAGTTCC-3′, paired with an oligonucleotide from aa F307 to the stop codon in the hCD1a cytoplasmic domain, 5′-GCGTCGACTTAACAGAAACAGCGTTTCCTGAACC-3′, were used to amplify the transmembrane and cytoplasmic domains of hCD1a from the plasmid πH3M-CD1a (26, 27). The amplified fragment was then paired with an oligonucleotide encompassing the NH2 terminus of mCD1d, 5′-GCGGATCCTGTGTAGAACTCTGGCGCTATGCGG-3′, to amply the ectodomain of mCD1d using pHβANeo-CD1.1 (28) as the PCR template. The mCD1d/b construct was made in a similar fashion except that a PCR product containing the transmembrane and cytoplasmic domains of human CD1b was first amplified using an oligonucleotide encoding aa G271 to W279 of the mCD1d α3 domain and aa I273 to P281 of the transmembrane domain of human CD1b, 5′-GGAGGACAGGATATCATCCTCTACTGGAGAAACCCCACC-3′, paired with an oligonucleotide from aa R312 to the stop codon in the hCD1b cytoplasmic domain, 5′-GCGTCGACTCATGGGATATTCTGATATGACCGGCG-3′, using the plasmid πH3M-CD1b (26, 27) as the template. To make a chimera containing the mCD1d ectodomain and transmembrane region fused to the cytoplasmic tail of the low density lipoprotein receptor (LDLr) chimera, called mCD1d/ldlr, total RNA from a C57BL/6 mouse liver was extracted using a High Pure RNA kit (Roche), and first strand cDNA was synthesized using a First-Strand cDNA Synthesis kit (Amersham Biosciences) according the manufacturers' instructions. RT-PCR was then performed to obtain the cytoplasmic domain of the mouse LDLr using a pair of oligonucleotides encoding aa V299 to W308 of the transmembrane domain of mCD1d and the 5′ portion of the cytoplasmic domain of mLDLr, 5′-GTGGTGGGTGCTGTAGTCTACTATATCTGGAGGAACTGGCGGCTGAAGAAC-3′, and the 3′ end of the mLDLr cytoplasmic domain, 5′-GCGTCGACTCATGCCACATCGTCCTCCAGGC-3′. The amplified cytoplasmic fragment was then used as a 3′ primer paired with the 5′ mCD1d primer to amplify the ectodomain and the transmembrane domain of mCD1d. These PCR products were cloned into TOPO-pBlunt (Invitrogen), and all vector sequences were verified by dideoxynucleotide sequencing using an Applied Biosystems Prism 310 genetic analyzer (PerkinElmer). The WT and mutant mCD1d constructs were then digested with BamHI and SalI and subcloned into pBabe/puro vector for retrovirus production and transduction into A20 B lymphoma cells and FSDC cells as described previously (29). After selection, the high-expressing cells were selected by fluorescence-activated cell sorting using the 1B1 anti-mCD1d mAb (BD Biosciences).
Yeast Two-hybrid Assays.
The constructs Gal4AD-μ3A in the pACTII (LEU2) plasmid and Gal4BD-TGN38 in the pGBT9 (TRP1) plasmid have been described previously (30, 31) MATCHMAKER GAL4 Two-Hybrid System 3 (CLONTECH Laboratories, Inc.) was used for the generation of GAL4BD-mCD1d and yeast transformation. GAL4BD-mCD1d was generated by ligation of pGBKT7, the GAL4BD vector, with synthetic double-strand DNA encoding the entire 10 aa cytoplasmic domain of mCD1d. The construct was confirmed by sequencing. The Saccharomyces cerevisiae strain Y187 was transformed according the manufacturer's instruction with GAL4AD-μ3A together with GAL4BD-mCD1d, GAL4BD-TGN38, or the pGBKT7 vector, and selected in synthetic-defined medium without leucine and tryptophan. β-galactosidase activities of Y187 transformants in liquid culture were assayed using o-nitrophenyl-β-d-galactopyranoside according the manufacturer's instruction. Results are normalized by growth and cell numbers used and expressed as the means ± SD of three independent determinations.
Surface Plasmon Resonance Analysis of AP-3 Binding to mCD1d Cytoplasmic Tail Peptides.
The interaction between mCD1d cytoplasmic tail peptides and the cytoplasmic adaptor complex AP-3 was determined in real time by surface plasmon resonance using a BIAcore 3000 biosensor (Biacore AB). Two peptides corresponding to WT mCD1d (CIWRRRSAYQDIR) and a mutant tail peptide (CIWRRRSAAQDIR) in which the tyrosine residue was substituted for alanine (in bold) were synthesized with an NH2-terminal cysteine residue to allow their directed immobilization via the Thiol group to a CM5 biosensor surface (Biacore AB). The AP-3 complex was purified from pig brain cytosol using chromatography through DEAE sepharose, hydroxyapatite, Mono-Q, and a final size-exclusion chromatography step (unpublished data). The interaction experiments were performed in buffer A (20 mM Hepes, 150 mM NaCl, 10 mM KCl, 2 mM MgCl2, 0.2 mM DTT, pH 7.0) at a flow rate of 20 μl/min. The association phase, involving perfusion of AP-3 over the biosensor, was recorded for 2 min, followed by dissociation for 2 min with buffer A. A short pulse injection (15 s) of 20 mM NaOH/0.5% SDS was used to regenerate the sensor chip surface after each experimental cycle. AP-3 was used at protein concentrations ranging from 50 to 750 nM. To exclude distortions due to injection and mixing, segments of the sensograms recorded 15–20 s after switching from buffer flow to adaptor solution or 5–10 s after switching back to running buffer, were used for the calculations of association and dissociation rate, respectively.
Kinetic parameters and equilibrium dissociation constants were determined from sensograms recorded at different adaptor concentrations. The association constant ka, the dissociation constant kd and the equilibrium constant KD= kd/ka were calculated using Biacore kinetic evaluation software, assuming pseudo-first order kinetics A + B = AB. The model calculates the association rate constant, ka, and the steady-state response level, Req, by fitting data to the equation R = Req(1-e−[ ka Cn + kd ][t − t0]), where t is the time in seconds, Req is the steady-state response level, and C the molar concentration of adaptors in the injection solution. The steric interference factor n, which describes the valency of the interaction between μA and VSR-PS1, was set to 1. The dissociation rate constant, kd, was determined by fitting the data to the equation R = R0e− kd (t − t0), where R0 is the response level at the beginning of the dissociation phase. This model has been applied recently to describe the interactions of adaptors with cytosolic domains (32–34) and is described in more detailed elsewhere (35, 36).
Immunofluorescent Labeling and Confocal Microscopy.
Splenic DCs were enriched by differential adherence and Percoll gradient centrifugation as described previously (10). FSDC transfectants, or spleen cells, were fixed, permeabilized, and blocked before the addition of the antibodies as previously described (10, 22). mCD1d molecules were labeled with biotinylated mAb 1B1 followed by Cy3-conjugated streptavidin (Jackson ImmunoResearch Laboratories). For the colocalization experiments, cells were stained with FITC-conjugated anti–mouse Lamp-2 mAb (BD Biosciences). The fluorescently labeled cells were analyzed with a Bio–Rad Laboratories MRC 1024 ES laser scanning confocal microscope.
Flow Cytometry.
Cells were washed once in PBS with 10% FCS (staining buffer) and blocked in staining buffer containing anti-Fc receptor antibody 2.4G2 (1:50) for 15 min at 4°C. Cells were stained in 100 μl vol with mAb to mCD1d, CD4, CD8α, NK1.1, TCR β, and CD11c, used at 1:100 in staining buffer at 4°C for 20 min. All primary antibodies were directly conjugated to fluorophores and obtained from BD Biosciences. Cells were analyzed on a FACScalibur (Becton Dickinson) cytometer. Vα14i NKT cells were stained with the mCD1d-αGalCer tetramers as described earlier (37).
Antigen Presentation Assays.
Glycosphingolipid antigens, αGalCer, Galα(1→2)αGalCer, and Galβ(1→3)αGalCer have been described previously (22) and were a gift from the Kirin Pharmaceutical Research Corporation, Gunma, Japan. Antigen presentation assays have been described previously (10, 22, 24, 38). APC used were either A20 cells transduced with WT mCD1d or mutant constructs or whole splenocytes. Pearl and heterozygous pe/+ littermates were immunized subcutaneously with 100 μg of hen egg lysozyme (HEL) emulsified 1:1 in complete Freund's adjuvant (CFA; Difco Laboratories). After 10 d, mice were killed and the draining LNs were removed. Cells were resuspended in HL-1 medium (BioWhittaker) supplemented with 100 μg/ml streptomycin, 100 U/ml penicillin G (Life Technologies), and cultured in 96-well plates at 4 × 105 cells per well with the various concentrations of the immunodominant peptide of HEL, p74–96. Proliferation was measured by addition of 1 μCi of [3H]-thymidine (ICN, Inc.) and measurement of uptake for the last 18 h of a 4-d culture.
BM Chimeras.
BM cells were isolated from the femurs of C57BL/6, B6 CD45.1 congenic, pe/+, and pe/pe mice, washed, and magnetically depleted of Thy1.2-positive cells. 10 million purified BM cells were coinjected into each recipient. Recipients were either irradiated Jα18−/− (1030 rads) or RAG1−/− (750 rads) mice. Chimeric animals were analyzed 10 wk after adoptive transfer.
Results
mCD1d Localization to Lysosomes Is Required for Efficient Glycolipid Presentation.
To determine how an altered distribution in the endosomal pathway might affect antigen presentation, transfectants expressing chimeric molecules with the mCD1d ectodomain were generated. Chimeric molecules containing the transmembrane and cytoplasmic domains of human CD1a (mCD1d/a) or the transmembrane domain of the mouse LDLr (mCD1d/ ldlr) (Fig. 1 a) were predicted to be found predominantly in recycling compartments or early endosomes. In the case of the LDL receptor fusion, internalization should be mediated by clathrin-dependent mechanisms (39, 40), whereas a different mechanism governs internalization mediated by the CD1a cytoplasmic tail (41). By contrast, CD1b is found predominantly in late endosomes and lysosomes, and therefore fusion of the mCD1d ectodomain to the transmembrane and cytoplasmic regions of human CD1b (mCD1d/b) (Fig. 1 a) should cause expression of the chimeric protein in late endosomes or lysosomes and the cell surface (12).
Figure 1.

Chimeric mCD1d molecules are expressed on the cell surface. (a) Constructs encoding chimeric mCD1d molecules with the transmembrane regions and cytoplasmic tails from either human CD1a (mCD1d/a) or human CD1b (mCD1d/b) or the cytoplasmic tail of the mouse LDLr (mCD1d/ldlr) are depicted. WT mCD1d sequences are indicated in white rectangles and donor sequences for making the mCD1d chimeras are indicated in stippled rectangles. The entire cytoplasmic tail amino acid sequences are shown, with potential adaptor binding motifs in bold type, except for the mCD1d/ldlr chimera, which has a longer cytoplasmic tail. In this case, only the sequence motif important for endosomal localization is indicated. (b) A20 transfectants with the indicated chimeric constructs were stained with PE-conjugated rat anti-mCD1d mAb, and the expression level of surface mCD1d molecules was analyzed by flow cytometry. Isotype control stainings are shown with shaded fill under the curve.
Fig. 1 b shows that A20 transfectants expressing high levels of WT mCD1d and each of the three chimeric mCD1d molecules were generated, although slightly higher surface levels were obtained for cells expressing the WT and the mCD1d/a chimera. The results from analysis of intracellular distribution of these mCD1d molecules by confocal microscopy were consistent with the expected expression patterns. WT mCD1d colocalized extensively with Lamp-2, as did the mCD1d/b chimera (Fig. 2) . By contrast, transfectants expressing the mCD1d/a fusion exhibited a very predominant surface expression but relatively little colocalization with Lamp-2. The mCD1d/ldlr chimeric protein was found in intracellular vesicles that are presumably recycling compartments, but like the mCD1d/a fusion, it also had little colocalization with Lamp-2 (Fig. 2). The antigen-presenting ability of the chimeric mCD1d molecules was tested using Vα14i NKT cell hybridomas. Analogs of αGalCer that have additional sugars linked to the 2′ and/or 3′ carbon of the galactose cannot be recognized without internalization and carbohydrate processing, which are required to generate the monosaccharide αGalCer (22). mCD1d can bind to the lipid portions of these analogs (22), but removal of the outer sugar is required to allow for TCR recognition. All of the mCD1d mutants could present αGalCer (Fig. 3) , which does not require internalization and processing. Their ability to do so, however, partially reflected the level of surface expression, with the WT and mCD1d/a chimera stimulating slightly higher levels of IL-2 release when pulsed with this compound. However, the mCD1d/1a and mCD1d/ldlr chimeras had a greatly reduced capacity to present Galα(1→2)αGalCer. This was not observed for the mCD1d/b chimera. Although it has a reduced level of surface mCD1d expression compared with the mCD1d/a chimera (Fig. 1 b), mCD1d/b was much better at presenting Gal(α1→2)αGalCer (Fig. 3). The data are consistent with the hypothesis that the localization of mCD1d to late endosomes and/or lysosomes is required for the efficient presentation of those glycosphingolipids that require internalization and carbohydrate processing in lysosomes.
Figure 2.
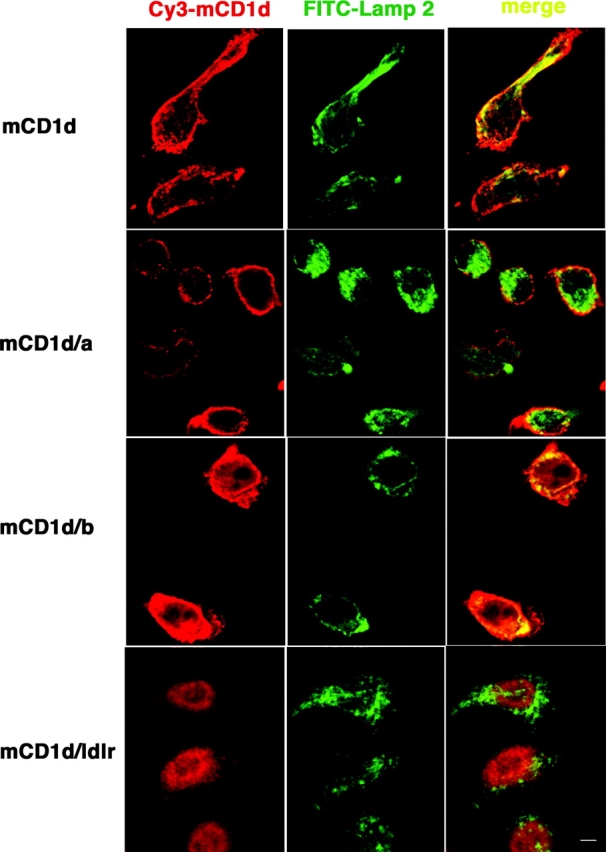
Chimeric mCD1d molecules display differential intracellular localization. FSDC transfectants expressing chimeric mCD1d molecules were double labeled with biotinylated anti-mCD1d followed by Cy3-conjugated streptavidin and FITC-conjugated Lamp-2 and analyzed by confocal microscopy. The colocalization of the mCD1d (red) and Lamp-2 (green) channels is depicted with a yellow signal. Bars, 10 μm.
Figure 3.

Chimeric mCD1d molecules not found in late endosomes/lysosomes show reduced antigen-presenting capacity. A20 cells expressing WT or mutant mCD1d molecules were pulsed with the indicated amounts of antigens, either αGalCer or Galα(1→2)GalCer, washed, and cultured with the 1.2 Vα14i NKT cell hybridoma. Cell-free supernatants were tested by ELISA for IL-2 production. The error bars indicate the SEM of triplicate measurements. Similar results were obtained in four independent experiments and also with the 1.4 and 3C3 Vα14i NKT cell hybridomas (not depicted).
The Pearl Mutation Affects Antigen Presentation by mCD1d.
Data from the cells that express chimeric mCD1d molecules described above demonstrated the importance of mCD1d localization to late endosomes/lysosomes. Because AP-3 is involved in the lysosomal localization of some proteins, we tested the ability of APC from AP-3βA mutant pearl (pe/pe) mice to present the same glycolipid antigens. As shown in Fig. 4 , spleen cells from pe/pe mice had a reduced ability to present two oligosaccharide analogs of αGalCer that require internalization and processing in lysosomes. Several findings indicate that this reduction was not due to differences in the cell populations found in the spleen of pe/pe mice or to an inability to express mCD1d. First, we did not find decreased numbers of DCs and B cells in the spleen of pe/pe mice (unpublished data). Second, αGalCer could be presented by pe/pe APC, indicative of a significant level of surface mCD1d expression (Fig. 4). There was a partial reduction in the immune response to αGalCer mediated by pearl APC, but this was less pronounced than the response to compounds that are dependent on internalization and processing (Fig. 4).
Figure 4.

The AP-3 adaptor protein complex is important for antigen presentation by mCD1d. Total spleen cells from pearl mice or heterozygous littermates were pulsed with either Galα(1→2)GalCer, Galβ(1→3)GalCer, or αGalCer. The 1.2 Vα14i NKT cell hybridoma was cultured with the pulsed APC, and IL-2 release was measured. The response of the 3C3 hybridoma was similar (not depicted). The experiments were repeated with APC from three different sets of mice.
Intracellular Distribution of mCD1d Is Altered in Pearl Mice.
We tested for an alteration in surface expression of mCD1d in cells from pearl mice using flow cytometry. Surprisingly, despite decreased antigen-presenting ability, increased levels of surface mCD1d were detected in several cell types tested, including whole thymocytes (unpublished data) and DCs from thymus and spleen (Fig. 5 a). The findings of decreased antigen-presenting ability coupled with increased surface mCD1d expression are reminiscent of those obtained from mice that express a form of mCD1d missing its cytoplasmic tail due to a germ line mutation (13). Therefore, we analyzed the intracellular distribution of mCD1d in splenocytes from C57BL/6 and pearl mice by confocal microscopy. Immunofluorescent labeling revealed extensive colocalization of mCD1d with Lamp-2 in cells from WT mice (Fig. 5 b). In splenocytes from pearl mice, however, surface labeling with anti-mCD1d mAb was prominent but there was considerably less labeling of intracellular vesicles and reduced colocalization with Lamp-2.
Figure 5.



The AP-3 adaptor protein complex is important for trafficking of mCD1d. (a) Increased levels of mCD1d were detected by flow cytometry on the surface of DCs from the thymus (left) and spleen (right) from pearl mice. Representative data from four experiments. (b) Confocal microscopy on spleen cells from pearl mice. Spleen cells from B6 or pearl enriched for DCs were double labeled with anti-mCD1d and Lamp-2 as described above and analyzed by confocal microscopy. Magnification is 144×. (c) Binding of mCD1d to the μ subunit of the adaptor complex AP-3. Yeast cells were transformed with the GAL4AD-μ3A along with GAL4BD-mCD1d, GAL4BD-TGN38 (as a positive control), or pGBKT7 vector only (as a negative control). β-Galactosidase activities were assayed to quantify specific interactions. Results are representative of four independent experiments and expressed as the mean of β-galactosidase units ± SD of three independent determinations.
The Cytoplasmic Tail of mCD1d Interacts with the μ Subunit of AP-3.
Lysosomal targeting of several proteins is accomplished by direct interaction of the tyrosine-based motif located at their cytoplasmic tails with the μ subunit of the AP-3 complex (15). To determine if mCD1d employs the same mechanism for its lysosomal trafficking, we used a yeast two-hybrid system to examine if the tyrosine motif in the cytoplasmic tail of mCD1d interacts with the AP-3 μ subunit. As shown in Fig. 5 c, the interaction is evident by the expression of β-galactosidase activity of yeast transformants containing both the AP-3 μ subunit and the mCD1d cytoplasmic tail. This interaction appears to be even stronger than that between the AP-3 μ subunit with the TGN38 cytoplasmic tail construct, the positive control.
We also used surface plasmon resonance to measure the interaction of synthetic peptides corresponding to the cytoplasmic tail of WT mCD1d and a variant in which the tyrosine residue was substituted for alanine, to purified AP-3. AP-3 bound to the WT tail with an equilibrium-binding constant of 540 nM (Table I). Binding was decreased by almost fivefold when AP-3 was passed over the mutant CD1d peptide (Y > A), due to a decreased on rate. This demonstrates that optimal AP-3 binding to CD1d requires a functional tyrosine motif (YQDIR). The specificity of the AP-3 binding to the mouse CD1d cytoplasmic tail was further corroborated by competition experiments in which AP-3 was preincubated with a 10- to 50-fold molar excess of soluble peptides corresponding to the mouse and human CD1d tyrosine motifs. Whereas preincubation of AP-3 with the soluble mouse CD1d peptide completely inhibited subsequent binding to the immobilized mouse CD1d cytoplasmic tail sequence on the biosensor surface, we could not detect any effect by preincubating AP-3 with a soluble human CD1d peptide (unpublished data). This is consistent with previous data indicating that human CD1d does not interact with AP-3 (34), and it highlights both the specificity of this interaction and a major difference between human and mouse CD1d.
Table I.
Biophysical Parameters of AP-3 Complex Binding to WT and Tyrosine Mutant Peptides from the mCD1d Cytoplasmic Tail
| ka(1/M × s) | kd (1/s) | KD = kd/ka (μM) | |
|---|---|---|---|
| WT | 8.9 × 103 | 4.8 × 10−3 | 0.54 |
| Y > A | 1.9 × 103 | 5.0 × 10−3 | 2.63 |
Vα14i NKT Cells Are Decreased in Pearl Mice.
In mice that have a germline deletion in the cytoplasmic tail of mCD1d, the number of Vα14i T cells is greatly diminished (13). We therefore determined if there was a similar decrease in AP-3–deficient pearl mice. There was a marked reduction in the percentage (Fig. 6 a) and absolute number (Fig. 6 b) of Vα14i NKT cells in thymus, spleen, and liver, ranging up to approximately fivefold. Consistent with this decreased cell number, the in vivo response to αGalCer was also strongly reduced (unpublished data). The results from intracellular cytokine staining, however, indicated that the residual Vα14i NKT cell population was capable of responding to αGalCer by producing cytokines (unpublished data).
Figure 6.

Pearl mice have reduced Vα14i T cells. (a) The liver and thymus of pearl and heterozygous control mice was measured by flow cytometry on cells stained with the αGalCer-mCD1d tetramer and a mAb to the TCR β chain. The percentages indicated are the averages obtained from all the mice tested. (b) Compilation of the absolute numbers of Vα14i NKT cells, detected as αGalCer/mCD1d tetramer+ and TCR β+ cells. n, number of mice tested. The horizontal bar indicates the mean value of the cell number.
The decreased numbers of Vα14i NKT cells in pearl mice could be caused by enhanced negative selection due to the increased levels of surface mCD1d expression (Fig. 5 a). We tested this possibility by analyzing BM chimeras that received T cell–depleted BM from CD45.1 congenic C57BL/6 mice along with marrow from either pearl mice or their heterozygous littermates. The recipients were either irradiated Jα18−/− mice or RAG-1−/− mice, both of which do not have cells that stain with the αGalCer/mCD1d tetramer. Fig. 7 a shows that BM cells from both donors contributed to the pool of Vα14i NKT cells detected in the liver of the recipient mice 10 wk after transfer. Although cells from the CD45.1 congenic C57BL/6 donor always contributed the majority of the Vα14i NKT cell population, there was no difference in the percentage of tetramer+ cells originating from pearl homozygous and heterozygous donors. Based on this result, we conclude that the requirement for AP-3 expression is not cell autonomous, i.e., AP-3 expression is not required in either the Vα14i NKT cell itself or its precursor. A detailed comparison of the total number of Vα14i NKT cells (including both donors) in different organs of the chimeric recipients is compiled in Fig. 7 b. There is no decrease in the Vα14i NKT cell number when the cotransferred BM cells were from pearl mice. This suggests that the increased level of surface mCD1d expression on thymocytes and other cell types derived from the pearl donor did not cause negative selection of Vα14i NKT cells.
Figure 7.

Increased surface mCD1d expression does not cause deletion of Vα14i NKT cells in pearl mice. (a) The percent tetramer+ cells in the livers of Jα18−/− recipients of the indicated types of BM are depicted. Tetramer+ cells were gated and analyzed for expression of CD45.1, indicating derivation from the C57BL/6 donor. Cotransferred CD45.1-negative (CD45.2+) cells were derived from either the pe/+ donors (left) or the pe/pe donors (right). Representative data from one of two experiments are shown. (b) The total number of donor-derived Vα14i NKT cells in the liver (L), spleen (S), and thymus (T) of the indicated recipients.
The Phenotype of Mocha Mice Is Similar to Pearl Mice.
We also analyzed the mocha (mh/mh) strain of mice, which lack AP-3 function due to a mutation in a different subunit, AP-3δ, which results in the complete loss of this subunit (23). As shown in Fig. 8 a, similar to pe/pe mice, there were increased surface levels of mCD1d expression on thymocytes from mocha mice compared with the heterozygous controls. Spleen cell levels of mCD1d were heterogeneous, but an increase in surface mCD1d expression also was observed (Fig. 8 a). Furthermore, mocha mice had a greatly reduced number of Vα14i NKT cells (Fig. 8 b), whereas CD4+ T cells numbers were not altered (unpublished data). Additionally, presentation of glycolipid antigens that require internalization and processing to Vα14i NKT cell hybridomas is reduced using APC from mocha mice (unpublished data). Overall, the data demonstrate that mice with defects in either AP-3β3A or AP-3δ have a similar phenotype with respect to mCD1d-mediated antigen presentation and Vα14i NKT cell development.
Figure 8.

Mocha mice have elevated surface mCD1d levels and reduced Vα14i NKT cells. (a) Measurement of surface levels of mCD1d by flow cytometry on whole thymocytes and total splenocytes from mocha mice and control heterozygous mice. (b) The liver, spleen, and thymus of mocha mice have significantly reduced percentages (not depicted) and absolute numbers of Vα14i NKT cells. Comparison of the absolute numbers of Vα14i NKT cells from mh/mh and mh/+ control mice. n, number of mice tested. The horizontal bar indicates the mean value of the cell number.
The CD1d and MHC Class II Trafficking Pathways Are Different.
The defect in AP-3–defective pearl mice might lead to a disruption of vesicular traffic that could alter MHC class II trafficking, which might diminish the function of conventional CD4+ and Vα14i NKT cells. However, flow cytometry analysis indicated that the number of CD4+ T cells was not decreased in the spleen and thymus of pearl mice (Fig. 9 a and unpublished data). Also, the proportion of CD4+ T cells in the spleen with a phenotype typical of recently activated/memory cells was not different when pearl mice and pe/+ controls were compared (Fig. 9 a).
Figure 9.

Conventional CD4+ T cells are normal in pearl mice. (a) Comparison of the number of naive and memory CD4+ T cells in the spleen of pearl and heterozygous control mice as measured by flow cytometry. (b) Pearl and control mice were primed with HEL and the response of CD4+ T cells from draining LNs to the indicated concentrations of the immunodominant peptide of HEL were measured. The data are the average of five sets of mice.
To assess antigen presentation function, mice were primed with HEL, and the recall proliferative response of T cells in the draining LN to the dominant HEL peptide (p74–96) presented by the I-Ab class II molecule was measured. As shown in Fig. 9 b, T cells from pearl mice were capable of making a normal CD4 T cell proliferative response, the difference between pearl and pe/+ mice was not statistically significant (P > 0.13). Therefore, there is no evidence for a global or severe deficiency in conventional MHC class II–reactive, CD4+T cells in the absence of AP-3 function.
The invariant chain (Ii) is important for the endosomal localization of MHC class II molecules (42), and a biochemical association between Ii and CD1d has been reported (43). To further compare the pathways leading to MHC II and CD1d antigen presentation, we analyzed the development of Vα14i NKT cells and APC function in invariant chain knockout (Ii − / −) mice. As reported previously (44), mCD1d surface expression in the thymus of Ii − / − mice is similar to C57BL/6J control mice, whereas mCD1d surface expression in the spleen of Ii − / − mice is increased (unpublished data). The lack of Ii, however, did not alter the percentage or the total number of Vα14i NKT cells in thymus and liver (Fig. 10 a). Furthermore, as shown in Fig. 10 b, splenocytes from Ii − / − mice showed no reduction in their ability to present the glycolipid antigens αGalCer and Galα(1→2)αGalCer to Vα14i NKT cell hybridomas. The greater response mediated by Ii − / − splenic APC could reflect the higher surface levels of mCD1d or perhaps a positive effect of the absence of Ii on glycosphingolipid antigen presentation.
Figure 10.


Vα14i NKT cell number and mCD1d antigen presentation are normal in Ii− /2 mice. (a) The thymus, spleen, and liver of Ii − / − mice were analyzed by flow cytometry on cells stained with the αGalCer-mCD1d tetramer and a mAb to the TCR β chain. The percentages indicated are the averages from the mice (n = 4) tested. (b) The 1.2 hybridoma was cultured with total spleen cells from C56BL/6J and Ii − / − mice in the presence of the indicated antigens, and IL-2 release was measured as described in experimental procedures. The error bars indicate the SEM of triplicate measurements. Similar results were obtained in three different sets of mice in independent experiments and also with the 1.4 and 3C3 hybridomas (not depicted).
Discussion
The pathway leading to the presentation of glycolipid antigens by CD1 molecules is just beginning to be elucidated. Although the importance of the endosomal localization of different CD1 molecules is now well established (41), the relative importance of different types of endosomes for CD1d presentation has not been determined. Here, using chimeric molecules we showed that the type of endosomal compartment that mCD1d localizes to is critical for the effective presentation of some antigens. The mCD1d/b chimera was very efficient at presentation of glycolipid antigens that require processing, suggesting that the processing event and the loading of mCD1d both occur in late endosomal or lysosomal vesicles. The striking difference in the ability of the CD1d/b and CD1d/a chimeras to present these antigens is consistent with previous experiments on CD1a and CD1b (41, 45, 46) and with the hypothesis that the different human CD1 isoforms are adapted to survey different types of endosomes (41). Mice lack CD1a, CD1b, and CD1c homologues. As a result, it is possible that mCD1d may need to survey multiple vesicular compartments, and in fact, it is found in early and late endosomes (44).
Consistent with experiments implicating a requirement for mCD1d localization to late endosomes or lysosomes, we showed that the function of the adaptor protein AP-3 is required for the efficient presentation of glycolipid antigens that require internalization and processing. The analysis of strains of mice having spontaneous mutations for either the β3A or the δ subunits confirms that AP-3 deficiency is responsible for defects in mCD1d-mediated recognition and Vα14i NKT cell selection, as opposed to some other genetic change that might have occurred in these strains. The response to αGalCer also was decreased when AP-3–deficient APC were tested but to a lesser extent. This is consistent with the view that presentation of this antigen can occur after surface loading of mCD1d molecules at neutral pH, although optimal αGalCer presentation may occur only after internalization and loading of mCD1d molecules in endosomal compartments (9, 10, 22). It is important to note that the decrease in antigen presentation occurred despite increased levels of surface mCD1d expression by spleen cells, including DC, from the mutant mice.
While this article was in preparation, it was shown that human CD1b also requires AP-3 function for intracellular localization and for antigen presentation in vitro (47). However, human CD1d did not require AP-3 (47), a result we corroborated here in a competition-binding experiment. Although the two CD1d homologues both contain a YXXφ motif (where X is any amino acid and φ is one having a bulky, hydrophobic side chain), the cytoplasmic tail of human CD1d is significantly different from that of the mouse. Therefore, subtle changes in the sequence of the YXXφ motif are important, and the ability of human CD1d to bind adaptors and to localize to specific compartments is likely to be different from mCD1d.
Interestingly, AP-3–deficient mice also showed reduced numbers of Vα14i NKT cells. The results from analysis of mixed BM chimeras indicated that AP-3 expression is not required in the Vα14i NKT cell itself, and the reduced cell numbers are not due to negative selection. The data are consistent with the hypothesis that the intact mCD1d cytoplasmic tail must interact with AP-3 in order to efficiently load the natural glycolipid ligand that selects Vα14i NKT cells and, furthermore, that the types of events governing the processing and mCD1d loading of synthetic compounds, such as Galα(1→2)αGalCer, and the loading of the elusive natural ligand exhibit similar requirements. Although these are logical conclusions, they remain speculative inferences in the absence of knowledge about the nature of the endogenous ligand(s) driving the development and expansion of Vα14i NKT cells. Interestingly, the magnitude of the decrease in Vα14i NKT cell number is similar to the decrease in mice bearing a germ line mutation in mCD1d that removes the tyrosine-containing cytoplasmic tail (13). These data suggest that the essential in vivo function of the mCD1d cytoplasmic tail sequence is likely to be mediated in part by AP-3, although an in vivo role for other AP complexes in CD1d trafficking is not excluded. However, in both the AP-3 and cytoplasmic tail mutants the defect is not complete, and the residual cells are functional, suggesting a minor, AP-3– and mCD1d cytoplasmic tail–independent pathway for mCD1d to influence the differentiation of the Vα14i NKT cell population.
The results from previous pulse–chase labeling studies indicated that mCD1d arrives rapidly at the cell surface, is then internalized, and undergoes several rounds of subsequent recycling between the cell surface and endosomes (44). Human CD1b might follow a similar pathway (48). mCD1d molecules eventually accumulate in lysosomes, although this process takes several hours (44). Collectively, these findings suggest that the mCD1d molecules important for presentation of the synthetic glycosphingolipids, and perhaps the natural ligand as well, are relatively mature molecules that may have undergone several rounds of recycling, and potentially, several rounds of exchange of glycolipid antigens before localizing to lysosomes. However, it remains possible that a subpopulation of newly synthesized mCD1d molecules travels from the Golgi network directly to late endosomes or lysosomes.
CD1 proteins and MHC class II molecules are both found in endosomes, and most of the previously published work on CD1 antigen presentation has emphasized similarities to the MHC class II pathway. For example, CD1b molecules were reported to be present in the MIIC (12, 49), and human CD1d was reported to be associated with MHC class II (43). Deficiencies in the cathepsins responsible for Ii degradation have effects on Vα14i NKT cells and/or CD1d function. Cathepsin L is required for Vα14i NKT cell differentiation (50), and in one report cathepsin S was reported to alter both the intracellular trafficking of mCD1d and its ability to present synthetic glycolipid antigens (51). However, the authors considered it most likely that this phenotype in cathepsin S–deficient mice was an indirect effect on the biogenesis of endosomal compartments due to reduced catabolism of Ii. Furthermore, it was reported recently that mCD1d is associated with Ii during its biosynthesis and that the autoreactivity of Vα14i NKT cells for mCD1d is reduced when Ii-deficient APCs were used to stimulate them (44). Based on these results, it was suggested that Ii might play a role in blocking the mCD1d groove in the ER, similar to one of its roles in MHC class II presentation (42). However, our data indicate that the effects of Ii deficiency on Vα14i NKT cells are much less dramatic than the effects on conventional CD4+ T cells. The lack of Ii apparently does not prevent loading of the natural ligand that selects Vα14i NKT cells, nor does it prevent the loading of antigens that require internalization and processing in late endosomes.
The data presented here clearly highlight the differences in the pathways required for the localization to late endosomes/lysosomes of mCD1d and MHC class II and the accessory molecules required for their optimal antigen-presenting function. This is evidenced by the difference in magnitude of the influence of Ii and the requirement for AP-3 function. Here we showed that the number and phenotype of CD4+ T cells and the CD4+ T cell response to a model protein antigen are not affected by the absence of AP-3. Consistent with this, it was reported recently that the localization and catabolism of Ii and the route of MHC class II-Ii transport are not altered in the absence of AP-3 (52, 53), correlating with the lack of interaction between Ii and AP-3 (54). Therefore, although it remains possible that AP-3–directed trafficking is needed for some class II–dependent immune responses, this cannot be true in the majority of cases. However, the requirement for AP-3 function in mCD1d presentation suggests the intriguing possibility that there could be additional molecules that play a role specifically in CD1-dependent pathways of antigen presentation.
Acknowledgments
We thank Dr. Hilde Cheroutre for critical reading of the manuscript, Drs. Olga Naidenko and Stéphane Sidobre (La Jolla Institute for Allergy and Immunology) for the mCD1d tetramers, Dr. Juan S. Bonifacino (Cell Biology Metabolism Branch, The National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD) for providing Gal4AD-μ3A and Gal4BD-TGN38 constructs, and Donald Martin for technical assistance.
This work was supported by National Institutes of Health grant RO1 AI40617 (to M. Kronenberg), a grant from the Human Frontiers of Science Program (to M. Kronenberg), a grant from the Neose foundation (to T.I. Prigozy), and grants from the University of Oslo and the Research Council of Norway (to O. Bakke). D. Elewaut is the recipient of a Career Development award from the Crohn's and Colitis Foundation of America. A.P. Lawton is the recipient of a National Research Service award from the National Institutes of Health (AI52552). This is publication number 480 from the La Jolla Institute for Allergy and Immunology.
D. Elewaut, A.P. Lawton, and N.A. Nagarajan contributed equally to this work.
D. Elewaut's present address is Dept. of Rheumatology, University Hospital Gent, De Pintelaan 185, 9000 Gent, Belgium.
E. Maverakis' present address is Dept. of Pathology, Harvard Medical School, 25 Shattuck St., Boston, MA 02115.
E. Sercarz's present address is Torrey Pines Institute for Molecular Studies, 3550 General Atomics Ct., San Diego, CA 92121.
T.I. Prigozy's present address is Tampa Bay Research Institute, 10900 Roosevelt Blvd., St. Petersburg, FL 33712.
Abbreviations used in this paper: AP-3, adaptor protein complex 3; FSDC, fetal skin dendritic cell; αGalCer, α–d-galactosyl ceramide; HEL, hen egg lysozyme; LDLr, low density lipoprotein receptor; NKT, natural killer T.
References
- 1.Beckman, E.M., S.A. Porcelli, C.T. Morita, S.M. Behar, S.T. Furlong, and M.B. Brenner. 1994. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature. 372:691–694. [DOI] [PubMed] [Google Scholar]
- 2.Porcelli, S.A., and R.L. Modlin. 1999. The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu. Rev. Immunol. 17:297–329. [DOI] [PubMed] [Google Scholar]
- 3.Shamshiev, A., A. Donda, I. Carena, L. Mori, L. Kappos, and G. De Libero. 1999. Self glycolipids as T-cell autoantigens. Eur. J. Immunol. 29:1667–1675. [DOI] [PubMed] [Google Scholar]
- 4.Godfrey, D.I., K.J. Hammond, L.D. Poulton, M.J. Smyth, and A.G. Baxter. 2000. NKT cells: facts, functions and fallacies. Immunol. Today. 21:573–583. [DOI] [PubMed] [Google Scholar]
- 5.Lantz, O., and A. Bendelac. 1994. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8− T cells in mice and humans. J. Exp. Med. 180:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bendelac, A., O. Lantz, M.E. Quimby, J.W. Yewdell, J.R. Bennink, and R.R. Brutkiewicz. 1995. CD1 recognition by mouse NK1+ T lymphocytes. Science. 268:863–865. [DOI] [PubMed] [Google Scholar]
- 7.Chiu, Y.H., J. Jayawardena, A. Weiss, D. Lee, S.H. Park, A. Dautry-Varsat, and A. Bendelac. 1999. Distinct subsets of CD1d-restricted T cells recognize self-antigens loaded in different cellular compartments. J. Exp. Med. 189:103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gui, M., J. Li, L.J. Wen, R.R. Hardy, and K. Hayakawa. 2001. TCR beta chain influences but does not solely control autoreactivity of V alpha 14J281T cells. J. Immunol. 167:6239–6246. [DOI] [PubMed] [Google Scholar]
- 9.Kawano, T., J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno, R. Nakagawa, H. Sato, E. Kondo, et al. 1997. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 278:1626–1629. [DOI] [PubMed] [Google Scholar]
- 10.Burdin, N., L. Brossay, Y. Koezuka, S.T. Smiley, M.J. Grusby, M. Gui, M. Taniguchi, K. Hayakawa, and M. Kronenberg. 1998. Selective ability of mouse CD1 to present glycolipids: alpha-galactosylceramide specifically stimulates V alpha 14+ NK T lymphocytes. J. Immunol. 161:3271–3281. [PubMed] [Google Scholar]
- 11.Morita, M., K. Motoki, K. Akimoto, T. Natori, T. Sakai, E. Sawa, K. Yamaji, Y. Koezuka, E. Kobayashi, and H. Fukushima. 1995. Structure-activity relationship of alpha-galactosylceramides against B16-bearing mice. J. Med. Chem. 38:2176–2187. [DOI] [PubMed] [Google Scholar]
- 12.Sugita, M., R.M. Jackman, E. van Donselaar, S.M. Behar, R.A. Rogers, P.J. Peters, M.B. Brenner, and S.A. Porcelli. 1996. Cytoplasmic tail-dependent localization of CD1b antigen-presenting molecules to MIICs. Science. 273:349–352. [DOI] [PubMed] [Google Scholar]
- 13.Chiu, Y.H., S.H. Park, K. Benlagha, C. Forestier, J. Jayawardena-Wolf, P.B. Savage, L. Teyton, and A. Bendelac. 2002. Multiple defects in antigen presentation and T cell development by mice expressing cytoplasmic tail-truncated CD1d. Nat. Immunol. 3:55–60. [DOI] [PubMed] [Google Scholar]
- 14.Boehm, M., and J.S. Bonifacino. 2001. Adaptins: the final recount. Mol. Biol. Cell. 12:2907–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson, M.S., and J.S. Bonifacino. 2001. Adaptor-related proteins. Curr. Opin. Cell Biol. 13:444–453. [DOI] [PubMed] [Google Scholar]
- 16.Huang, F., A. Nesterov, R.E. Carter, and A. Sorkin. 2001. Trafficking of yellow-fluorescent-protein-tagged mu1 subunit of clathrin adaptor AP-1 complex in living cells. Traffic. 2:345–357. [DOI] [PubMed] [Google Scholar]
- 17.Rapoport, I., M. Miyazaki, W. Boll, B. Duckworth, L.C. Cantley, S. Shoelson, and T. Kirchhausen. 1997. Regulatory interactions in the recognition of endocytic sorting signals by AP-2 complexes. EMBO J. 16:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daugherty, B.L., K.S. Straley, J.M. Sanders, J.W. Phillips, M. Disdier, R.P. McEver, and S.A. Green. 2001. AP-3 adaptor functions in targeting P-selectin to secretory granules in endothelial cells. Traffic. 2:406–413. [DOI] [PubMed] [Google Scholar]
- 19.Simpson, F., A.A. Peden, L. Christopoulou, and M.S. Robinson. 1997. Characterization of the adaptor-related protein complex, AP-3. J. Cell Biol. 137:835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang, W., C. Li, D.M. Ward, J. Kaplan, and S.L. Mansour. 2000. Defective organellar membrane protein trafficking in Ap3b1-deficient cells. J. Cell Sci. 113:4077–4086. [DOI] [PubMed] [Google Scholar]
- 21.Huizing, M., Y. Anikster, and W.A. Gahl. 2000. Hermansky-Pudlak syndrome and related disorders of organelle formation. Traffic. 1:823–835. [DOI] [PubMed] [Google Scholar]
- 22.Prigozy, T.I., O. Naidenko, P. Qasba, D. Elewaut, L. Brossay, A. Khurana, T. Natori, Y. Koezuka, A. Kulkarni, and M. Kronenberg. 2001. Glycolipid antigen processing for presentation by CD1d molecules. Science. 291:664–667. [DOI] [PubMed] [Google Scholar]
- 23.Kantheti, P., X. Qiao, M.E. Diaz, A.A. Peden, G.E. Meyer, S.L. Carskadon, D. Kapfhamer, D. Sufalko, M.S. Robinson, J.L. Noebels, and M. Burmeister. 1998. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 21:111–122. [DOI] [PubMed] [Google Scholar]
- 24.Brossay, L., S. Tangri, M. Bix, S. Cardell, R. Locksley, and M. Kronenberg. 1998. Mouse CD1-autoreactive T cells have diverse patterns of reactivity to CD1+ targets. J. Immunol. 160:3681–3688. [PubMed] [Google Scholar]
- 25.Girolomoni, G., M.B. Lutz, S. Pastore, C.U. Assmann, A. Cavani, and P. Ricciardi-Castagnoli. 1995. Establishment of a cell line with features of early dendritic cell precursors from fetal mouse skin. Eur. J. Immunol. 25:2163–2169. [DOI] [PubMed] [Google Scholar]
- 26.Aruffo, A., and B. Seed. 1987. Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proc. Natl. Acad. Sci. USA. 84:8573–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aruffo, A., and B. Seed. 1989. Expression of cDNA clones encoding the thymocyte antigens CD1a, b, c demonstrates a hierarchy of exclusion in fibroblasts. J. Immunol. 143:1723–1730. [PubMed] [Google Scholar]
- 28.Teitell, M., H.R. Holcombe, L. Brossay, A. Hagenbaugh, M.J. Jackson, L. Pond, S.P. Balk, C. Terhorst, P.A. Peterson, and M. Kronenberg. 1997. Nonclassical behavior of the mouse CD1 class I-like molecule. J. Immunol. 158:2143–2149. [PubMed] [Google Scholar]
- 29.Shamshiev, A., A. Donda, T.I. Prigozy, L. Mori, V. Chigorno, C.A. Benedict, L. Kappos, S. Sonnino, M. Kronenberg, and G. De Libero. 2000. The alphabeta T cell response to self-glycolipids shows a novel mechanism of CD1b loading and a requirement for complex oligosaccharides. Immunity. 13:255–264. [DOI] [PubMed] [Google Scholar]
- 30.Ohno, H., J. Stewart, M.C. Fournier, H. Bosshart, I. Rhee, S. Miyatake, T. Saito, A. Gallusser, T. Kirchhausen, and J.S. Bonifacino. 1995. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science. 269:1872–1875. [DOI] [PubMed] [Google Scholar]
- 31.Ohno, H., M.C. Fournier, G. Poy, and J.S. Bonifacino. 1996. Structural determinants of interaction of tyrosine-based sorting signals with the adaptor medium chains. J. Biol. Chem. 271:29009–29015. [DOI] [PubMed] [Google Scholar]
- 32.Boll, M., H. Daniel, and B. Gasnier. 2003. The SLC36 family: proton-coupled transporters for the absorption of selected amino acids from extracellular and intracellular proteolysis. Pflugers Arch. 10.1007/S00424-003-1073-4. [DOI] [PubMed]
- 33.Heilker, R., U. Manning-Krieg, J.F. Zuber, and M. Spiess. 1996. In vitro binding of clathrin adaptors to sorting signals correlates with endocytosis and basolateral sorting. EMBO J. 15:2893–2899. [PMC free article] [PubMed] [Google Scholar]
- 34.Honing, S., M. Sosa, A. Hille-Rehfeld, and K. von Figura. 1997. The 46-kDa mannose 6-phosphate receptor contains multiple binding sites for clathrin adaptors. J. Biol. Chem. 272:19884–19890. [DOI] [PubMed] [Google Scholar]
- 35.Boll, W., I. Rapoport, C. Brunner, Y. Modis, S. Prehn, and T. Kirchhausen. 2002. The mu2 subunit of the clathrin adaptor AP-2 binds to FDNPVY and YppO sorting signals at distinct sites. Traffic. 3:590–600. [DOI] [PubMed] [Google Scholar]
- 36.Jonsson, U., L. Fagerstam, B. Ivarsson, B. Johnsson, R. Karlsson, K. Lundh, S. Lofas, B. Persson, H. Roos, I. Ronnberg, et al. 1991. Real-time biospecific interaction analysis using surface plasmon resonance and a sensor chip technology. Biotechniques. 11:620–627. [PubMed] [Google Scholar]
- 37.Matsuda, J.L., O.V. Naidenko, L. Gapin, T. Nakayama, M. Taniguchi, C.R. Wang, Y. Koezuka, and M. Kronenberg. 2000. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brossay, L., O. Naidenko, N. Burdin, J. Matsuda, T. Sakai, and M. Kronenberg. 1998. Structural requirements for galactosylceramide recognition by CD1-restricted NK T cells. J. Immunol. 161:5124–5128. [PubMed] [Google Scholar]
- 39.Anderson, R.G., J.L. Goldstein, and M.S. Brown. 1977. A mutation that impairs the ability of lipoprotein receptors to localise in coated pits on the cell surface of human fibroblasts. Nature. 270:695–699. [DOI] [PubMed] [Google Scholar]
- 40.Lehrman, M.A., J.L. Goldstein, M.S. Brown, D.W. Russell, and W.J. Schneider. 1985. Internalization-defective LDL receptors produced by genes with nonsense and frameshift mutations that truncate the cytoplasmic domain. Cell. 41:735–743. [DOI] [PubMed] [Google Scholar]
- 41.Sugita, M., E.P. Grant, E. van Donselaar, V.W. Hsu, R.A. Rogers, P.J. Peters, and M.B. Brenner. 1999. Separate pathways for antigen presentation by CD1 molecules. Immunity. 11:743–752. [DOI] [PubMed] [Google Scholar]
- 42.Cresswell, P. 1994. Assembly, transport, and function of MHC class II molecules. Annu. Rev. Immunol. 12:259–293. [DOI] [PubMed] [Google Scholar]
- 43.Kang, S.J., and P. Cresswell. 2002. Regulation of intracellular trafficking of human CD1d by association with MHC class II molecules. EMBO J. 21:1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jayawardena-Wolf, J., K. Benlagha, Y.H. Chiu, R. Mehr, and A. Bendelac. 2001. CD1d endosomal trafficking is independently regulated by an intrinsic CD1d-encoded tyrosine motif and by the invariant chain. Immunity. 15:897–908. [DOI] [PubMed] [Google Scholar]
- 45.Briken, V., R.M. Jackman, G.F. Watts, R.A. Rogers, and S.A. Porcelli. 2000. Human CD1b and CD1c isoforms survey different intracellular compartments for the presentation of microbial lipid antigens. J. Exp. Med. 192:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaible, U.E., K. Hagens, K. Fischer, H.L. Collins, and S.H. Kaufmann. 2000. Intersection of group I CD1 molecules and mycobacteria in different intracellular compartments of dendritic cells. J. Immunol. 164:4843–4852. [DOI] [PubMed] [Google Scholar]
- 47.Sugita, M., X. Cao, G.F. Watts, R.A. Rogers, J.S. Bonifacino, and M.B. Brenner. 2002. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity. 16:697–706. [DOI] [PubMed] [Google Scholar]
- 48.Briken, V., R.M. Jackman, S. Dasgupta, S. Hoening, and S.A. Porcelli. 2002. Intracellular trafficking pathway of newly synthesized CD1b molecules. EMBO J. 21:825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prigozy, T.I., P.A. Sieling, D. Clemens, P.L. Stewart, S.M. Behar, S.A. Porcelli, M.B. Brenner, R.L. Modlin, and M. Kronenberg. 1997. The mannose receptor delivers lipoglycan antigens to endosomes for presentation to T cells by CD1b molecules. Immunity. 6:187–197. [DOI] [PubMed] [Google Scholar]
- 50.Honey, K., K. Benlagha, C. Beers, K. Forbush, L. Teyton, M.J. Kleijmeer, A.Y. Rudensky, and A. Bendelac. 2002. Thymocyte expression of cathepsin L is essential for NKT cell development. Nat. Immunol. 3:1069–1074. [DOI] [PubMed] [Google Scholar]
- 51.Riese, R.J., G.P. Shi, J. Villadangos, D. Stetson, C. Driessen, A.M. Lennon-Dumenil, C.L. Chu, Y. Naumov, S.M. Behar, H. Ploegh, et al. 2001. Regulation of CD1 function and NK1.1(+) T cell selection and maturation by cathepsin S. Immunity. 15:909–919. [DOI] [PubMed] [Google Scholar]
- 52.Caplan, S., E.C. Dell'Angelica, W.A. Gahl, and J.S. Bonifacino. 2000. Trafficking of major histocompatibility complex class II molecules in human B-lymphoblasts deficient in the AP-3 adaptor complex. Immunol. Lett. 72:113–117. [DOI] [PubMed] [Google Scholar]
- 53.Sevilla, L.M., S.S. Richter, and J. Miller. 2001. Intracellular transport of MHC class II and associated invariant chain in antigen presenting cells from AP-3-deficient mocha mice. Cell. Immunol. 210:143–153. [DOI] [PubMed] [Google Scholar]
- 54.Hofmann, M.W., S. Honing, D. Rodionov, B. Dobberstein, K. von Figura, and O. Bakke. 1999. The leucine-based sorting motifs in the cytoplasmic domain of the invariant chain are recognized by the clathrin adaptors AP1 and AP2 and their medium chains. J. Biol. Chem. 274:36153–36158. [DOI] [PubMed] [Google Scholar]