

Abstract
gp49B1 is expressed on mast cells and inhibits immunoglobulin E–dependent activation and inflammation in vivo. We now show that gp49B1 is expressed on neutrophils and prevents neutrophil-dependent vascular injury in response to lipopolysaccharide (LPS). The intradermal (i.d.) injection of LPS into gp49B1-null (gp49B − / −) but not gp49B1-sufficient (gp49B + / +) mice elicited macroscopic hemorrhages by 24 h, which were preceded on microscopic analyses by significantly more intravascular thrombi (consisting of neutrophils, platelets, and fibrin) that occluded venules and by more tissue neutrophils than in gp49B + / + mice. However, there were no differences in the number of intact (nondegranulating) mast cells or the tissue levels of mediators that promote neutrophil recruitment. Hemorrhage was prevented by depleting neutrophils, blocking β2 integrin–intercellular adhesion molecule 1 interactions, or inhibiting coagulation. These characteristics indicate that gp49B − / − mice are exquisitely sensitive to a local Shwartzman reaction (LSR) after a single i.d. injection of LPS, whereas in the classic LSR, a second exposure is required for increased β2 integrin function, intravascular neutrophil aggregation, formation of occlusive thrombi, and hemorrhage. Moreover, LPS increased gp49B1 expression on neutrophils in vivo. The results suggest that gp49B1 suppresses the LPS-induced increase in intravascular neutrophil adhesion, thereby providing critical innate protection against a pathologic response to a bacterial component.
Keywords: Shwartzman reaction, thrombosis, hemorrhage, cell adhesion molecules, innate immunity
Introduction
A rapid inflammatory response to pathogenic microbes and their products is essential for the survival of all higher organisms. This response is mediated largely by the innate immune system through cells that reside constitutively in tissues, such as macrophages and mast cells, as well as by neutrophils that migrate from the blood to a site of invasion within hours (1–3). These cells are activated by pattern recognition receptors, such as the Toll-like receptors (TLRs), and by receptors that bind endogenous products formed in response to infection, such as receptors for certain complement fragments (4, 5). These innate receptors are encoded in the germline and hence are either expressed constitutively or induced rapidly in response to microbes or microbial products. This characteristic delineates the innate receptors from the elements of adaptive immunity, in which humoral and cellular defenses are generated over days to weeks through antigen-induced recombination of T cell receptors and B cell receptors/Ig genes followed by clonal expansion of the selected cells.
Despite the essential nature of innate inflammation, an overly vigorous response can be detrimental (6) as, for example, when toxic levels of cytokines such as TNF-α and IL-1β are produced and cause septic shock leading to multiple organ failure (7). Therefore, it seemed reasonable to speculate that the cells that mediate innate inflammation might express inhibitory receptors that can counterregulate the activation responses. We have previously shown that gp49B1, which is expressed constitutively on the surface of mouse mast cells and macrophages (8, 9), inhibits the high affinity receptor for IgE (FcɛRI)-dependent activation of mast cells in vitro through recruitment of the src homology type 2 domain-containing phosphatase 1 to the immunoreceptor tyrosine-based inhibitory motifs (ITIMs) located in the cytoplasmic domain of gp49B1 (10). Moreover, we determined that gp49B1 dampens FcɛRI-dependent mast cell activation and the resulting immediate inflammatory response in vivo in gp49B1-sufficient (gp49B + / +) mice as compared with gp49B1-null (gp49B − / −) mice (11). We subsequently found that mast cells in gp49B − / − mice are also more sensitive to activation induced by the cytokine stem cell factor (SCF; reference 12), which signals through its receptor c-kit (13). This finding revealed that the inhibitory capacity of gp49B1 extends beyond receptors of adaptive immunity, and it raised the possibility that the germline-encoded gp49B1 also suppresses inflammation induced by ligands that stimulate innate immune responses.
To address this issue, we compared the responses of gp49B − / − and gp49B + / + mice to LPS derived from Escherichia coli, which activates cells through TLR4 (4). We found that the intradermal (i.d.) injection of LPS in gp49B − / − mice elicited the formation of thrombi and hemorrhages in a manner fully dependent on neutrophils, β2 integrins, intercellular adhesion molecule (ICAM)-1, and coagulation. This response was suppressed in gp49B + / + mice. The thrombohemorrhagic response of gp49B − / − mice to LPS is reminiscent of the classic local Shwartzman reaction (LSR), in which an initial dermal exposure to LPS primes the skin for a thrombohemorrhagic vasculopathy in response to systemic challenge with LPS 18–24 h later (14). The first, local exposure increases expression of ICAM-1 on endothelial cells, and the second, systemic dose increases and prolongs β2 integrin expression and avidity on neutrophils (15). This sequence leads to pathologic adhesion of neutrophils to each other and to the endothelium, resulting in thrombus formation and hemorrhage (15). Here we show that neutrophils constitutively express gp49B1 and that in gp49B + / + but not in gp49B − / − mice the expression increases in response to LPS over a time course that precedes the development of the thrombohemorrhagic response in gp49B − / − mice. Our findings reveal that in the absence of gp49B1 on neutrophils, an increase in sensitivity to LPS makes a single exposure sufficient to elicit an LSR, possibly at the β2 integrin–ICAM-1 interaction step. Thus, gp49B1 constitutively inhibits a pathologic response to a microbial product that stimulates the innate immune system.
Materials and Methods
Mice.
Male gp49B + / + and gp49B − / − mice (N9 backcrossed to the BALB/c strain) were produced as previously described (11) and were 6–11-wk old when used for experiments. Mice were maintained in a specific pathogen-free barrier facility at the Dana-Farber Cancer Institute, and the studies were approved by the Animal Care and Use Committee.
Preparation of LPS.
LPS purified from E. coli 0111:B4 by phenol extraction and ion exchange chromatography (<1% protein; Sigma Ultra L-3024, lot 111k4046) was purchased from Sigma-Aldrich. 25 mg LPS was suspended in 10 ml endotoxin-free saline (Sigma S8776, lot 111K2351). After gentle mixing, the LPS solution was warmed to 37°C in a water bath for 30 min and then sonicated for 2 min in an ultrasonic water bath (Branson Sonifier 450; VWR Scientific) to increase the solubility. This LPS stock solution was then aliquoted into 0.5-ml Eppendorf tubes and stored at −20°C. For use, the LPS solution was warmed to 37°C for 30 min and sonicated for 2 min.
Antibodies.
Unlabeled and PE-labeled rat IgG2b anti–mouse Gr-1 mAb (clone RB6-8C5), rat IgG2a anti–mouse CD18 mAb (clone M18/2), rat IgG2a negative control mAb (clone eBR2a), rat IgG2b anti–mouse CD54 mAb (clone YN1/1.7.4), and rat IgG2b negative control mAb (clone KLH/G2b-1-2) were obtained from eBioscience (all non–PE-labeled Abs were functional grade). PE-labeled rat IgG2b negative control mAb (clone A95-1; BD Biosciences) and rat IgM negative control mAb (clone IR202; Zymed Laboratories) were obtained as noted. The rat IgM anti–mouse gp49B1 mAb B23.1 (8, 16) was produced in the ascites fluid of nu/nu mice and purified with the ImmunoPure IgM purification kit (Pierce Chemical Co.). Purified mAb B23.1 and rat IgM negative control were labeled with the Alexa Fluor 488 (Alexa) protein labeling kit (Molecular Probes, Inc.) according to the product instructions.
Tissue Thickness, Histologic, and Cytokine/Chemokine Analyses of LPS-induced Inflammation.
50 μg LPS in 20 μl saline was injected i.d. in the right ear of each mouse with a 25-μl Hamilton syringe (Hamilton Company) fitted with a 30G 1/2 gauge needle (Becton Dickinson). 20 μl saline was injected in the left ear as a negative control. At various times after the injection, ear thickness was measured with a caliper as previously described (12). Additional mice were killed by CO2 inhalation, and to record hemorrhages macroscopically, photographs of the entire bodies or ears of mice were taken with a digital camera (Coolpix 5000; Nikon). The ears were then excised, fixed in 4% paraformaldehyde for at least 8 h, and embedded in JB-4 glycolmethacrylate. 2-μm-thick full-length cross sections of ears were cut, placed on microscope slides, air dried, and stained with Diff-Quik to count mast cells (12) or for chloroacetate esterase activity to count neutrophils and thrombi. The number of cells and thrombi in each full-length cross section was divided by the number of lengths in the section to obtain the number of cells or thrombi per unit length. Occluding thrombi were defined as those blocking 100% of the diameter of blood vessels.
For the measurement of cytokines and chemokines 2 h after LPS injection (the time of maximal generation in pilot time course experiments), the ears were excised, weighed, and placed in a buffer consisting of calcium- and magnesium-free HBSS containing 1% NP-40, 10 mM Hepes, pH 7.2, 5 μM leupeptin, 5 μM pepstatin A, 200 μM 4-(2-aminoethyl)-benzenesulfonyl fluoride, and 20 μM aprotinin (all reagents were obtained from Sigma-Aldrich except NP-40, which was obtained from BDH Limited). The tissue was homogenized for 2 min at 4°C in a Minibeadbeater-8 Cell Disruptor (BioSpec Products, Inc.) at setting 2. The homogenates were centrifuged at 14,000 rpm for 30 min at 4°C in a Hermle Labnet Z233M microfuge. The supernatants were aspirated and additional insoluble material was removed by centrifugation of the supernatants at 4,000 rpm for 10 min at 4°C through 0.22 μm cellulose acetate filters in SPIN-X® centrifuge tubes (Costar). The amounts of IL-1β, KC, monocyte chemotactic protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, MIP-2, and TNF-α in the filtered extracts were measured with Quantikine® M colorimetric sandwich ELISA kits (R&D Systems) according to the manufacturer's instructions. Data were calculated as picograms of mediator per milligram of ear tissue.
Collection of Blood Samples and Analysis of Peripheral Blood Leukocytes.
Mice were injected with either LPS or saline as described above. At the times indicated below, mice were killed by CO2 inhalation, and blood was obtained by cardiac puncture with a 1-ml syringe that had been washed with 0.1 ml of 0.5 M EDTA solution (E-7889; Sigma-Aldrich). The blood was placed in a 2-ml Vacutainer® tube containing 3.6 mg dry dipotassium EDTA (Becton Dickinson), and within 30 min, samples (≥500 μl) were analyzed for leukocyte count and differential on an automatic cell counter (ADVIA® 120A; Bayer Diagnostics) in the Hematology Laboratory of Children's Hospital Boston.
Neutrophil Depletion and Blocking of Adhesion Molecules.
To deplete neutrophils, 100 μg anti–Gr-1 was injected i.p. 24 h before the i.d. injection of LPS (50 μg/ear) and saline in the right and left ears, respectively. The injection of mice with anti–Gr-1 does not deplete T or B lymphocytes, mononuclear phagocytes, or NK cells (17). To block the β2 chain of integrins (CD18) or ICAM-1 (CD54), 100 μg anti-CD18 or 100 μg anti-CD54 was injected i.v. 2 h before the i.d. injection with LPS (35 μg/ear) and saline in separate ears. Control animals were injected i.v. with 100 μg isotype-matched negative control mAb, or saline before the i.d. injection of LPS or saline.
Anticoagulant Treatment.
gp49B − / − mice received 2 mg (in 200 μl) warfarin [3-(α-acetonylbenzyl)-4-hydroxycoumarin] (Sigma-Aldrich) or vehicle control (15% acetone in pyrogen-free water; Sigma-Aldrich) by stomach tube 1 h before and 2 h after the i.d. injection of LPS (25 μg/ear) and saline in the right and left ears, respectively. The LPS dose was reduced somewhat for the studies with anti-adhesion molecule antibodies and the anticoagulant to illustrate better the effects of these interventions on macroscopic hemorrhage.
Flow Cytometric Analyses.
Samples of whole blood (400 μl) collected as described above were mixed with 7 ml ACK lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 111 μM disodium EDTA in Milli-Q water; all reagents from Sigma-Aldrich) and incubated at room temperature for 10 min. 7 ml HBA buffer (calcium- and magnesium-free HBSS containing 0.1% [wt/vol] BSA and 0.02% [wt/vol] sodium azide) was added, and samples were mixed well and centrifuged for 5 min at 1,000 rpm (Beckman GPR Centrifuge) at 4°C. The pelleted leukocytes were washed once by centrifugation in 10 ml HBA buffer, resuspended in 500 μl HBA buffer, and adjusted to 106 cells/ml. Bone marrow cells were isolated by flushing the femurs and tibias of mice with RPMI 1640 medium, and cells were resuspended as described for blood leukocytes. 100-μl portions of cells were distributed into the wells of a 96-well round-bottom cell culture plate (Costar 3799), and incubated for 30 min at 4°C with saturating concentrations of Alexa-labeled mAb B23.1, PE-labeled anti–Gr-1, or equal concentrations of isotype-matched negative control mAb. The cells were then washed in HBA buffer twice and analyzed on a FACSort™ with CELLQuest™ software (Becton Dickinson). The neutrophil population was identified on the basis of the anti–Gr-1 staining, and the mAb B23.1 positivity was measured on that cell population.
Statistical Analyses.
Data are expressed as mean ± SEM unless otherwise noted. Differences between groups of mice were assessed with Student's unpaired, two-tailed t test. P values <0.05 were defined as statistically significant.
Results
LPS-induced Hemorrhage, Thrombosis, and Tissue Neutrophilia in gp49B−/− Mice.
The right ears of gp49B + / + and gp49B − / − mice were injected i.d. with 50 μg LPS in 20 μl endotoxin-free sterile saline, and the left ears were injected with an equal volume of saline alone. 24 h (but not 12 h) after injection of LPS, gp49B − / − mice exhibited moderate to severe macroscopic hemorrhage in ears injected with LPS, whereas gp49B + / + mice did not (illustrated for three mice from a representative experiment in Fig. 1 A). In a series of experiments, 26 of 31 (84%) gp49B − / − mice exhibited a macroscopic hemorrhagic response 24 h after the injection of LPS, compared with only 1 of 15 (7%) gp49B + / + mice. Histologic analysis confirmed that no hemorrhage had occurred 12 h after the injection of LPS (Fig. 1 B, a and e), but hemorrhage was substantial by 24 h in gp49B − / − mice (Fig. 1 C, a and e). 12 h after the injection of LPS, microscopic examination revealed that fully occlusive venular thrombi, consisting of neutrophils, platelets, and fibrin (Fig. 1 B, b and f), were present in a significantly greater percentage of tissue lengths in gp49B − / − mice compared with gp49B + / + mice (6.8 ± 1.6% vs. 1.4 ± 0.65%, respectively; n = 7; P = 0.01). 24 h after the injection of LPS, the increase in occlusive thrombi in gp49B − / − mice compared with gp49B + / + mice (Fig. 1 C, b and f) was not quantitated because of the confounding effect of the intense hemorrhage in gp49B − / − mice.
Figure 1.




LPS-induced hemorrhage, thrombosis, and tissue neutrophilia in gp49B +/+ and gp49B −/− mice. gp49B + / + and gp49B − / − mice were injected i.d. with 20 μl saline alone (left ears) or containing 50 μg LPS (right ears). At the times indicated below, the mice were killed and their ears were removed, photographed, placed in fixative, embedded, and sectioned for staining with chloroacetate esterase. (A) The macroscopic appearance of the ears of three mice of each genotype 24 h after the injection of saline or LPS is shown from one experiment, representative of five experiments with a total of 15 gp49B + / + and 31 gp49B − / − mice. (B and C) Microscopic fields of tissue sections 12 (B) and 24 h (C) after the injection of LPS (a–c and e–g) or saline (d and h) show the extent of hemorrhage (a and e), thrombi (b and f), and tissue neutrophilia (c and g). In g, an area with minimal hemorrhage is shown to permit visualization of the neutrophils, which stain red with chloroacetate esterase. The data are representative of two (12 h) and three (24 h) experiments with 7 and 10 mice, respectively, of each genotype per time point. (D) The number of neutrophils in full-length tissue cross sections was enumerated and is expressed per microscopic unit length (140 μm) as mean ± SEM, n = 3 (2 and 6 h) and n = 7 (12 and 24 h). *, significant difference between gp49B + / + and gp49B − / − mice.
The prominence of neutrophils in the thrombohemorrhagic response (Fig. 1, B and C, c and g) led to an enumeration of the number of neutrophils in tissue sections. 2 and 6 h after the injection of LPS, few neutrophils had extravasated from the blood into the tissue in either gp49B + / + or gp49B − / − mice (Fig. 1 D). However, after 12 h, there were significant 5- and 12-fold increases in neutrophils in the ears of both gp49B + / + and gp49B − / − mice, respectively, compared with 6 h, and the number of neutrophils in gp49B − / − mice after the injection of LPS was significantly greater than that in gp49B + / + mice. 24 h after the injection of LPS, the number of neutrophils in the ears of gp49B + / + and gp49B − / − mice increased compared with 12 h, and the number of neutrophils continued to be significantly higher in gp49B − / − mice compared with gp49B + / + mice. In contrast, the numbers of neutrophils in the peripheral blood of gp49B + / + and gp49B − / − mice 24 h after i.d. injection of LPS were not significantly different (3,331 ± 332 vs. 3,484 ± 480 per μl, respectively; n = 8; P = 0.80).
Because earlier studies revealed that mast cells in the ears of gp49B − / − mice were hyperresponsive to degranulation by IgE-dependent passive cutaneous anaphylaxis or SCF as assessed by ear swelling and tissue histology (11, 12), these assays were also performed in response to LPS. Histologic analyses showed no significant difference in the number of intact (nondegranulating) mast cells in gp49B + / + and gp49B − / − mice 2 h after LPS injection (3.9 ± 0.3 and 3.7 ± 0.03 intact mast cells/unit length, respectively; n = 3 in one experiment; P = 0.4), or at 6 (n = 3 in one experiment), 12 (n = 7 in two experiments), or 24 (n = 10 in three experiments) h after injection. Early net tissue swelling as assessed with calipers was not significantly different in gp49B + / + and gp49B − / − mice 1 h after injection with LPS (6.0 ± 1.6 and 5.2 ± 2.4 × 10−2 mm, respectively; n = 6 gp49B + / + and n = 5 gp49B − / − mice in two experiments; P = 0.8), and there were no significant differences at 2, 4, or 8 h (all P > 0.3) after injection in the same two experiments.
Because mast cells as well as other gp49B1-expressing cell types elaborate cytokines and chemokines, the amounts of several of these mediators were assessed in extracts prepared from the ears of mice 2 h after the injection of LPS, the time point of their maximal values. There were no significant differences between gp49B + / + and gp49B − / − mice in the respective levels of IL-1β (12 ± 2 vs. 14 ± 4 pg/mg; n = 3 from one experiment), KC (31 ± 2 vs. 32 ± 1 pg/mg; n = 6 from two experiments; P = 0.7), MCP-1 (21 ± 0.9 vs. 22 ± 2 pg/mg; n = 6 from two experiments; P = 0.6), MIP-1α (39 ± 0.4 vs. 39 ± 0.4 pg/mg; n = 6 from two experiments; P = 0.9), MIP-2 (16 ± 1 vs. 17 ± 1 pg/mg; n = 6 from two experiments; P = 0.5), or TNF-α (1.7 ± 0.1 vs. 1.8 ± 0.1 pg/mg; n = 8 from three experiments; P = 0.3).
Role of Neutrophils in LPS-induced Hemorrhage in gp49B−/− Mice.
gp49B − / − mice were depleted of neutrophils by an i.p. injection of 100 μg anti–Gr-1 24 h before the i.d. injection of 50 μg LPS. Anti–Gr-1 elicited a 97% decrease in the number of peripheral blood neutrophils measured 24 h after the injection of LPS compared with mice injected i.p. with saline (Fig. 2 A). Moreover, treatment of gp49B − / − mice with anti–Gr-1 caused a >95% decrease in the number of neutrophils in the ears 24 h after the injection of LPS compared with mice treated i.p. with saline (Fig. 2 B). Treatment of mice with an i.p. injection of isotype-matched negative control Ig instead of anti–Gr-1 before LPS gave results comparable to those with the injection of saline (unpublished data). Importantly, as assessed both macroscopically (Fig. 2 C) and microscopically (unpublished data), the hemorrhagic response to LPS did not occur in the absence of neutrophils (0/11 vs. 11/11 for gp49B − / − mice treated or not treated with anti–Gr-1, respectively).
Figure 2.


Effects of neutrophil depletion on LPS-induced hemorrhage in gp49B −/− mice. gp49B − / − mice were injected i.p. with saline or 100 μg anti–Gr-1 24 h before the i.d. injection of 50 μg LPS. 24 h later, peripheral blood was collected and the neutrophil count was determined with an automated cell counter (A). The number of neutrophils in tissue cross sections (B) was determined as described for Fig. 1 D. Data are expressed as mean ± SEM, n = 8 (A) or n = 4 (B). Macroscopic hemorrhage is illustrated for four mice from one experiment (C), representative of three experiments with a total of 11 mice in each treatment group.
Role of Adhesion Molecules.
gp49B − / − mice were injected i.v. with 100 μg anti-CD18 or anti-CD54 2 h before the i.d. injection of LPS. Neither treatment reduced significantly the number of neutrophils in the peripheral blood 24 h after LPS injection compared with mice that received isotype-matched negative control Ab (Fig. 3 A). The numbers of neutrophils in ear tissue sections from mice treated with anti-CD18 or anti-CD54 were both decreased by ∼45% compared with those from mice that received negative control Ab (Fig. 3 B). Nevertheless, macroscopic hemorrhage was substantially inhibited in five of five gp49B − / − mice treated with anti-CD18 and in three of three mice treated with anti-CD54 (Fig. 3 C). Macroscopic hemorrhage was not inhibited in eight control mice treated with rat IgG2a (isotype-matched negative control for anti-CD18), rat IgG2b (isotype-matched negative control for anti-CD54), or saline.
Figure 3.
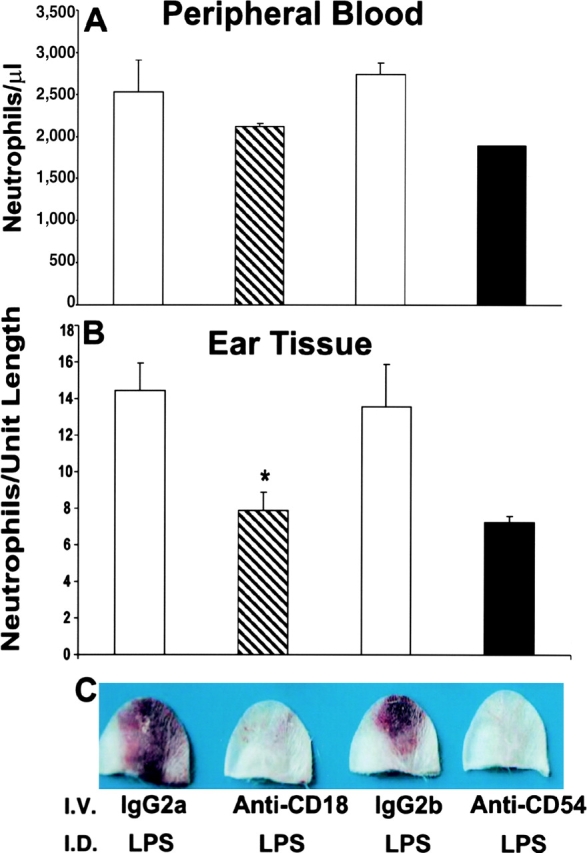
Effects of blocking adhesion molecules on LPS-induced hemorrhage in gp49B −/− mice. gp49B − / − mice were injected i.v. with 100 μg anti-CD18, anti-CD54, or isotype-matched negative control Ig (rat IgG2a and rat IgG2b, respectively) 2 h before the i.d. injection of 35 μg LPS. 24 h later, peripheral blood (A) and ear tissue (B) samples were analyzed for neutrophil numbers and macroscopic hemorrhage (C) as described for Fig. 2. Data are expressed as mean ± SEM, n = 3 (A and B). Hemorrhage illustrated in C is representative of two experiments with a total of five and three mice treated with anti-CD18 and anti-CD54, respectively. *, significant difference between mice injected i.p. with IgG2a versus anti-CD18.
Expression of gp49B1 on Neutrophils and Up-regulation by LPS.
The constitutive and LPS-induced expression of gp49B1 on both peripheral blood and bone marrow neutrophils was assessed by cytofluorographic analysis with the gp49B1-specific mAb B23.1 (16). Blood leukocytes were stained with Alexa-labeled mAb B23.1 and neutrophils were identified by simultaneous staining with PE-labeled anti–Gr-1. Essentially, all anti–Gr-1+ cells in the peripheral blood (Fig. 4 A) and bone marrow (Fig. 4 B) of naive gp49B + / + mice were also gp49B1+. Furthermore, an increase in the intensity of staining was apparent 1 h after the i.d. injection of LPS, reached a plateau by 8 h (unpublished data), and was maintained at 24 h (Fig. 4, A and B). 24 h after the injection of LPS, the mean fluorescence intensities of peripheral blood and bone marrow neutrophils from gp49B + / + mice had each increased approximately fourfold compared with mice injected with saline. No mAb B23.1-mediated staining was detected on peripheral blood or bone marrow neutrophils from gp49B − / − mice (unpublished data), implicating the absence of gp49B1 in the neutrophil-dependent thrombohemorrhagic response to LPS in gp49B − / − mice.
Figure 4.

Flow cytometric analysis of the expression of gp49B1 on peripheral blood and bone marrow neutrophils. gp49B + / + mice were injected i.d. with saline (dotted line) or with 50 μg LPS (filled curve and solid line). 24 h later, peripheral blood (A) and bone marrow (B) cells were stained with PE-labeled anti–Gr-1 and either Alexa-labeled mAb B23.1 anti-gp49B1 (dotted and solid lines) or Alexa-labeled isotype-matched negative control IgM (filled curve). Data are presented as histograms of the Alexa fluorescence of anti–Gr-1+ cells and are representative of 5 experiments with a total of 10 mice in each group.
Role of the Coagulation Pathway.
gp49B − / − mice were given 2 mg of the anticoagulant warfarin intragastrically 1 h before and 2 h after the LPS injection. Warfarin completely inhibited the macroscopic and microscopic hemorrhagic response in five of six gp49B − / − mice, whereas vehicle control did not inhibit hemorrhage in three of three mice. The effect of warfarin was not the result of neutrophil depletion because the numbers of neutrophils in the peripheral blood (3,660 ± 900 vs. 2,833 ± 450 per μl; n = 5 and 8, respectively; P = 0.38) and ears (10.2 ± 1.5 vs. 14.7 ± 1.5 per unit length; n = 5 and 3, respectively; P = 0.10) of LPS-treated gp49B − / − mice were not significantly different with or without warfarin treatment, respectively. Thus, the coagulation pathway is critical to the LPS-induced microangiopathy in the injected ear of gp49B − / − mice.
Discussion
We have previously shown that gp49B1 constitutively inhibits adaptive inflammation elicited by Ig-dependent mast cell activation in vivo (11). Here we report that gp49B1 is also expressed on neutrophils and suggest that it inhibits neutrophil-dependent innate inflammation induced by LPS. A single i.d. injection of LPS in the gp49B − / − mice resulted in a macroscopic thrombohemorrhagic response that was essentially absent from gp49B + / + mice (Fig. 1). Assessment of the requirements for the LPS-elicited lesion identified a role for the neutrophil (Fig. 2) and its integrin-mediated endothelial cell interactions (Fig. 3), as well as for the blood coagulation system. These are the essential components of the classic LSR, which in normal mice requires an i.d. priming dose of 7.5–50 μg LPS followed by a systemic challenge dose of 30–150 μg LPS 18–24 h later (18, 19). Because we found that the neutrophils of normal mice constitutively express gp49B1 and that its expression on neutrophils is up-regulated by the i.d. injection of LPS (Fig. 4), the exaggerated microangiopathy of gp49B − / − mice to i.d. LPS revealed gp49B1 as a negative regulator of LPS-mediated, neutrophil-dependent tissue injury.
gp49B1 is also expressed on mast cells (8) and macrophages (9) but, in contrast with our previous findings for mast cell activation-dependent inflammation elicited by IgE-dependent passive cutaneous anaphylaxis or SCF (11, 12), gp49B − / − mice did not exhibit more mast cell degranulation and early tissue swelling in response to LPS. Furthermore, the tissue levels of cytokines and chemokines produced by and/or active on gp49B1-expressing cells (IL-1β, KC, MCP-1, MIP-1α, MIP-2, and TNF-α; references 20–25) were not significantly different in extracts prepared from the ears of gp49B + / + and gp49B − / − mice 2 h after LPS injection, the time of maximal production of these mediators. Taken together, the data suggest that the LSR exhibited by gp49B − / − mice is not a result of increased exocytosis of tissue-resident mast cells or cytokine/chemokine generation by macrophages or other cell types, but rather, a heightened activation response of neutrophils. Nevertheless, in the absence of a direct demonstration of increased neutrophil activation in situ, a contribution from the absence of gp49B1 on cells other than neutrophils in the generation of the LSR after a single LPS injection cannot be entirely ruled out.
The essential role of intravascular neutrophils in the classic LSR (18) has been defined in terms of the adhesive functions of β2 integrins and ICAM-1 (15). Neutrophils contribute to the pathology of the classic LSR by enlarging the size of thrombi when aggregated with each other and enmeshed with platelets and fibrin, and they provide firm adhesion of the thrombi to the endothelium via a β2 integrin–ICAM-1 interaction. The resulting stasis in blood flow leads to hypoxia that causes the disintegration of the blood vessel endothelium and ensuing hemorrhage. Furthermore, LPS-activated neutrophils that are immobilized in blood vessels release mediators such as elastase and superoxide that are toxic to endothelial cells. Indeed, the classic LSR is inhibited by a protease inhibitor and vascular damage similar to the LSR is induced by the injection of lysosomal extracts from neutrophils (26, 27).
The induction of hemorrhage in gp49B − / − mice 24 h after a single i.d. injection of LPS (Fig. 1 C) was preceded at 12 h by the occlusion of significantly more venules with thrombi consisting of neutrophils, platelets, and fibrin compared with gp49B + / + mice (Fig. 1 B). In addition, there were significantly more neutrophils in the tissue of gp49B − / − mice both 12 and 24 h after injection of LPS (Fig. 1 D). The greater amount of tissue neutrophilia did not simply represent the availability of more neutrophils for diapedesis through blood vessels, because the numbers of peripheral blood neutrophils in gp49B + / + and gp49B − / − mice were not significantly different 24 h after injection of the animals with LPS. We also found that mouse peripheral blood neutrophils express CD18, CD11b, and CD54 (unpublished data). The levels of these molecules were up-regulated after the i.d. injection of LPS, but there were no differences in the levels of their expression in the gp49B + / + and gp49B − / − mice. Nevertheless, that neutrophils were required for the hemorrhagic response to i.d. LPS in gp49B − / − mice was established by attenuation of the lesion with their depletion (Fig. 2) and was related to their adhesion function. Intravenous provision of the anti-CD18 or anti-CD54 2 h before the i.d. injection of LPS inhibited the hemorrhagic response at 24 h (Fig. 3 C) without depleting blood or ear neutrophils (Fig. 3, A and B), suggesting that increased avidity of one or more β2 integrins for ICAM-1 in gp49B − / − mice is a key mechanistic step. We consider the greater tissue neutrophilia in gp49B − / − mice to be a secondary manifestation of this effect. Anti-CD11a did not inhibit hemorrhage in gp49B − / − mice, even though CD11a is expressed on essentially all mouse leukocytes except mast cells. It was not possible to assess the role of CD11b because the injection of anti-CD11b depleted peripheral blood neutrophils and reiterated the inhibitory effect of anti–Gr-1. In rabbits, the classic LSR is inhibited by anti-CD54, anti-CD11b, and anti-CD18, but not by anti-CD11a, under conditions in which neutrophils were still present in the vasculature as determined histologically (15).
gp49B1 is expressed on mast cells, macrophages, NK cells, and T cells (8, 28–31), and mouse bone marrow and splenic neutrophils bind a mAb directed to an epitope shared by gp49A and gp49B1 (32). Using the gp49B1-specific mAb B23.1 (11, 16), we now show that peripheral blood and bone marrow neutrophils not only constitutively express this inhibitory receptor but also up-regulate its expression after the i.d. injection of LPS (Fig. 4). The molecule that bound mAb B23.1 on the surface of peripheral blood neutrophils from LPS-treated mice was full-length gp49B1 as assessed by immunoprecipitation with mAb B23.1 followed by deglycosylation, SDS-PAGE, and immunoblotting with anti-gp49B302–313 (8, 16, and unpublished data).
The presence of fibrin and platelets in the occluding thrombi of LPS-treated gp49B − / − mice suggested a contribution of the blood coagulation pathway, as noted by others for the classic LSR (33). That the administration of the anticoagulant warfarin inhibited the hemorrhagic response in gp49B − / − mice to i.d. LPS without decreasing the number of peripheral blood or ear tissue neutrophils indicates that the coagulation pathway is essential to the vasculopathy. The characteristic involvement of the coagulation cascade in the classic LSR may reflect LPS-mediated activation of the pathway through the production of tissue factor by endothelial cells and mononuclear phagocytes (34). The formation of a complex between tissue factor and factor VII initiates fibrin formation, and administration of anti–factor VII before the systemic dose of LPS inhibits the classic LSR (33). Hence, for both the single-dose LSR in gp49B − / − mice and the classic LSR, neutrophils and the coagulation system are both necessary, but neither alone is sufficient.
The LSR is a model for thrombohemorrhagic vasculopathy that can occur in response to infection with Gram-negative bacteria, such as during meningococcal sepsis (35, 36). Furthermore, some of the characteristics of the LSR described above occur in vasculitic syndromes initiated by immune complexes and complement or through unknown etiologies (36, 37). In addition, the LSR resembles in many respects the generalized Shwartzman reaction, in which both doses of LPS are given systemically, resulting in a β2 integrin–dependent, lethal thrombohemorrhagic syndrome that is a model for the disseminated intravascular coagulation that may occur during septic shock (37). Our findings provide the first presumptive evidence of counterregulation of the response to a TLR agonist by an ITIM-bearing receptor in vivo. This conceivably reflects the recruitment of src homology type 2 domain-containing phosphatase 1 by the ITIMs of gp49B1, leading to reversal of LPS-induced tyrosine phosphorylation (38–40). Because gp49B1 is the closest mouse analogue of the human ITIM-bearing receptor termed leukocyte Ig-like receptor 5 (or Ig-like transcript 3; references 41–43), our results suggest that the expression of leukocyte Ig-like receptor 5 and/or other inhibitory receptors on myeloid cells may contribute to the outcome of innate immune responses in human inflammatory conditions. Moreover, the findings presented here when coupled with our previous studies establish that gp49B1 inhibits a functionally diverse group of activating receptors involved in both adaptive and innate immune responses.
Acknowledgments
The authors thank Li Fen Chen and Weili Chang for technical assistance.
Supported by grants AI-31599, AI-41144, and HL-36110 from the National Institutes of Health and by grants from GlaxoSmithKline and the Hyde and Watson Foundation.
Abbreviations used in this paper: Alexa, Alexa Fluor 488; ICAM, intercellular adhesion molecule; i.d., intradermal; ITIM, immunoreceptor tyrosine-based inhibitory motif; LSR, local Shwartzman reaction; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein; SCF, stem cell factor; TLR, Toll-like receptor.
References
- 1.Aderem, A., and D.M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593–623. [DOI] [PubMed] [Google Scholar]
- 2.Galli, S.J., M. Maurer, and C.S. Lantz. 1999. Mast cells as sentinels of innate immunity. Curr. Opin. Immunol. 11:53–59. [DOI] [PubMed] [Google Scholar]
- 3.Burg, N.D., and M.H. Pillinger. 2001. The neutrophil: function and regulation in innate and humoral immunity. Clin. Immunol. 99:7–17. [DOI] [PubMed] [Google Scholar]
- 4.Janeway, C.A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. [DOI] [PubMed] [Google Scholar]
- 5.Fujita, T. 2002. Evolution of the lectin-complement pathway and its role in innate immunity. Nat. Rev. Immunol. 2:346–353. [DOI] [PubMed] [Google Scholar]
- 6.Nathan, C. 2002. Points of control in inflammation. Nature. 420:846–852. [DOI] [PubMed] [Google Scholar]
- 7.Dinarello, C.A. 2000. Proinflammatory cytokines. Chest. 118: 503–508. [DOI] [PubMed] [Google Scholar]
- 8.Katz, H.R., A.C. Benson, and K.F. Austen. 1989. Activation- and phorbol ester-stimulated phosphorylation of a plasma membrane glycoprotein antigen expressed on mouse IL-3-dependent mast cells and serosal mast cells. J. Immunol. 142:919–926. [PubMed] [Google Scholar]
- 9.LeBlanc, P.A., S.W. Russell, and S.-M.T. Chang. 1982. Mouse mononuclear phagocyte heterogeneity detected by monoclonal antibodies. J. Reticuloendothelial Soc. 32:219–231. [PubMed] [Google Scholar]
- 10.Lu-Kuo, J.M., D.M. Joyal, K.F. Austen, and H.R. Katz. 1999. gp49B1 inhibits IgE-initiated mast cell activation through both immunoreceptor tyrosine-based inhibitory motifs, recruitment of the src homology 2 domain-containing phosphatase-1, and suppression of early and late calcium mobilization. J. Biol. Chem. 274:5791–5796. [DOI] [PubMed] [Google Scholar]
- 11.Daheshia, M., D.S. Friend, M.J. Grusby, K.F. Austen, and H.R. Katz. 2001. Increased severity of local and systemic anaphylactic reactions in gp49B1-deficient mice. J. Exp. Med. 194:227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feldweg, A.F., D.S. Friend, J.S. Zhou, Y. Kanaoka, M. Daheshia, L. Li, K.F. Austen, and H.R. Katz. 2003. gp49B1 suppresses stem cell factor-induced mast cell activation-secretion and attendant inflammation in vivo. Eur. J. Immunol. 33:2262–2268. [DOI] [PubMed] [Google Scholar]
- 13.Zsebo, K.M., D.A. Williams, E.N. Geissler, V.C. Broudy, F.H. Martin, H.L. Atkins, R.-Y. Hsu, N.C. Birkett, K.H. Okino, D.C. Murdock, et al. 1990. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 63:213–224. [DOI] [PubMed] [Google Scholar]
- 14.Stetson, C.A. 1951. Studies on the mechanism of the Shwartzman phenomenon. Certain factors involved in the production of the local hemorrhagic necrosis. J. Exp. Med. 93:489–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Argenbright, L.W., and R.W. Barton. 1992. Interactions of leukocyte integrins with intercellular adhesion molecule 1 in the production of inflammatory vascular injury in vivo. The Shwartzman reaction revisited. J. Clin. Invest. 89:259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katz, H.R., E. Vivier, M.C. Castells, M.J. McCormick, J.M. Chambers, and K.F. Austen. 1996. Mouse mast cell gp49B1 contains two immunoreceptor tyrosine-based inhibition motifs and suppresses mast cell activation when coligated with the high-affinity Fc receptor for IgE. Proc. Natl. Acad. Sci. USA. 93:10809–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wipke, B.T., and P.M. Allen. 2001. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 167:1601–1608. [DOI] [PubMed] [Google Scholar]
- 18.Stetson, C.A., and R.A. Good. 1951. Shwartzman phenomenon-participation of polymorphonuclear leukocytes. J. Exp. Med. 93:49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scholzen, T.E., C. Sunderkotter, D.H. Kalden, T. Brzoska, M. Fastrich, T. Fisbeck, C.A. Armstrong, J.C. Ansel, and T.A. Luger. 2003. α-melanocyte stimulating hormone prevents lipopolysaccharide-induced vasculitis by down-regulating endothelial cell adhesion molecule expression. Endocrinology. 144:360–370. [DOI] [PubMed] [Google Scholar]
- 20.Burd, P.R., H.W. Rogers, J.R. Gordon, C.A. Martin, S. Jayaraman, S.D. Wilson, A.M. Dvorak, S.J. Galli, and M.E. Dorf. 1989. Interleukin 3–dependent and –independent mast cells stimulated with IgE and antigen express multiple cytokines. J. Exp. Med. 170:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon, J.R., and S.J. Galli. 1991. Release of both preformed and newly synthesized tumor necrosis factor α (TNF-α)/cachectin by mouse mast cells stimulated via the FcɛRI. A mechanism for the sustained action of mast cell–derived TNF-α during IgE-dependent biological responses. J. Exp. Med. 174:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beutler, B., N. Krochin, I.W. Milsark, C. Luedke, and A. Cerami. 1986. Control of cachectin (tumor necrosis factor) synthesis: mechanisms of endotoxin resistance. Science. 232:977–980. [DOI] [PubMed] [Google Scholar]
- 23.Echtenacher, B., D.N. Männel, and L. Hültner. 1996. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 381:75–77. [DOI] [PubMed] [Google Scholar]
- 24.Driscoll, K.E. 1994. Macrophage inflammatory proteins: biology and role in pulmonary inflammation. Exp. Lung Res. 20:473–490. [DOI] [PubMed] [Google Scholar]
- 25.Introna, M., R.C.J. Bast, C.S. Tannenbaum, T.A. Hamilton, and D.O. Adams. 1987. The effect of LPS on expression of the early “competence” genes JE and KC in murine peritoneal macrophages. J. Immunol. 138:3891–3896. [PubMed] [Google Scholar]
- 26.Halpern, B.N. 1963. Inhibition of the local hemorrhagic Shwartzman reaction by a polypeptide possessing potent antiprotease activity. Proc. Soc. Exp. Biol. Med. 115:273–276. [DOI] [PubMed] [Google Scholar]
- 27.Movat, H.Z., and S. Wasi. 1985. Severe microvascular injury induced by lysosomal releasates of human polymorphonuclear leukocytes. Increase in vasopermeability, hemorrhage, and microthrombosis due to degradation of subendothelial and perivascular matrices. Am. J. Pathol. 121:404–417. [PMC free article] [PubMed] [Google Scholar]
- 28.LeBlanc, P.A., and C.A. Biron. 1984. Mononuclear phagocyte maturation: a cytotoxic monoclonal antibody reactive with postmonoblast stages. Cell. Immunol. 83:242–254. [DOI] [PubMed] [Google Scholar]
- 29.Wang, L.L., I.K. Mehta, P.A. LeBlanc, and W.M. Yokoyama. 1997. Mouse natural killer cells express gp49B1, a structural homolog of human killer inhibitory receptors. J. Immunol. 158:13–17. [PubMed] [Google Scholar]
- 30.Rojo, S., D.N. Burshtyn, E.O. Long, and N. Wagtmann. 1997. Type I transmembrane receptor with inhibitory function in mouse mast cells and NK cells. J. Immunol. 158:9–12. [PubMed] [Google Scholar]
- 31.Gu, X., A. Laouar, J. Wan, M. Daheshia, J. Lieberman, W.M. Yokoyama, H.R. Katz, and N. Manjunath. 2003. The gp49B1 inhibitory receptor regulates IFN-γ responses of T cells and NK cells. J. Immunol. 170:4095–4101. [DOI] [PubMed] [Google Scholar]
- 32.Matsumoto, Y., L.L. Wang, W.M. Yokoyama, and T. Aso. 2001. Uterine macrophages express the gp49B inhibitory receptor in midgestation. J. Immunol. 166:781–786. [DOI] [PubMed] [Google Scholar]
- 33.Zivelin, A., L.V. Rao, and S.I. Rapaport. 1995. Evidence for an essential role of tissue factor dependent blood coagulation in the pathogenesis of the local Shwartzman reaction. Blood Cells Mol. Dis. 21:9–19. [DOI] [PubMed] [Google Scholar]
- 34.Bokarewa, M.I., J.H. Morrissey, and A. Tarkowski. 2002. Tissue factor as a proinflammatory agent. Arthritis Res. 4:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dahle, J.S. 1983. Pathogenesis of hemorrhagic skin lesions in meningococcal disease. NIPH Ann. 6:49–53. [PubMed] [Google Scholar]
- 36.Hess, D.C. 1997. Cerebral lupus vasculopathy. Mechanisms and clinical relevance. Ann. N. Y. Acad. Sci. 823:154–168. [DOI] [PubMed] [Google Scholar]
- 37.Belmont, H.M., J. Buyon, R. Giorno, and S. Abramson. 1994. Up-regulation of endothelial cell adhesion molecules characterizes disease activity in systemic lupus erythematosus. The Shwartzman phenomenon revisited. Arthritis Rheum. 37:376–383. [DOI] [PubMed] [Google Scholar]
- 38.Beaty, C.D., T.L. Franklin, Y. Uehara, and C.B. Wilson. 1994. Lipopolysaccharide-induced cytokine production in human monocytes: role of tyrosine phosphorylation in transmembrane signal transduction. Eur. J. Immunol. 24:1278–1284. [DOI] [PubMed] [Google Scholar]
- 39.Williams, L.M., and A.J. Ridley. 2000. Lipopolysaccharide induces actin reorganization and tyrosine phosphorylation of Pyk2 and paxillin in monocytes and macrophages. J. Immunol. 164:2028–2036. [DOI] [PubMed] [Google Scholar]
- 40.Okugawa, S., Y. Ota, T. Kitazawa, K. Nakayama, S. Yanagimoto, K. Tsukada, M. Kawada, and S. Kimura. 2003. Janus kinase 2 is involved in lipopolysaccharide-induced activation of macrophages. Am. J. Physiol. Cell Physiol. 285:C399–C408. [DOI] [PubMed] [Google Scholar]
- 41.Arm, J.P., C. Nwankwo, and K.F. Austen. 1997. Molecular identification of a novel family of human immunoglobulin superfamily members that possess immunoreceptor tyrosine-based inhibition motifs and homology to the mouse gp49B1 inhibitory receptor. J. Immunol. 159:2342–2349. [PubMed] [Google Scholar]
- 42.Borges, L., M.-L. Hsu, N. Fanger, M. Kubin, and D. Cosman. 1997. A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules. J. Immunol. 159:5192–5196. [PubMed] [Google Scholar]
- 43.Cella, M., C. Dohring, J. Samaridis, M. Dessing, M. Brockhaus, A. Lanzavecchia, and M. Colonna. 1997. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J. Exp. Med. 185:1743–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]