

Abstract
Thrombospondin 1 (TSP) elicits potent antiinflammatory activities in vivo, as evidenced by persistent, multiorgan inflammation in TSP null mice. Herein, we report that DCs represent an abundant source of TSP at steady state and during activation. Human monocyte-derived immature dendritic cells (iDCs) spontaneously produce TSP, which is strongly enhanced by PGE2 and to a lesser extent by transforming growth factor (TGF) β, two soluble mediators secreted by macrophages after engulfment of damaged tissues. Shortly after activation via danger signals, DCs transiently produce interleukin (IL) 12 and tumor necrosis factor (TNF) α, thereby eliciting protective and inflammatory immune responses. Microbial stimuli increase TSP production, which is further enhanced by IL-10 or TGF-β. The endogenous TSP produced during early DC activation negatively regulates IL-12, TNF-α, and IL-10 release through its interactions with CD47 and CD36. After prolonged activation, DCs extinguish their cytokine synthesis and become refractory to subsequent stimulation, thereby favoring the return to steady state. Such “exhausted” DCs continue to release TSP but not IL-10. Disrupting TSP–CD47 interactions during their restimulation restores their cytokine production. We conclude that DC-derived TSP serves as a previously unappreciated negative regulator contributing to arrest of cytokine production, further supporting its fundamental role in vivo in the active resolution of inflammation and maintenance of steady state.
Keywords: PGE2, TGF-β, IL-12, CD47, CD36
Introduction
DCs play a central role in the induction of immunity and tolerance (1). In the absence of inflammation, immature DCs (iDCs) located in peripheral tissues are specialized in uptake of innocuous and cell-associated self Ag. They continuously capture Ag and migrate to the draining lymph node, where they can induce tolerance (1). Under the influence of danger signals (i.e., pathogens and necrotic cells), DCs undergo a process called maturation manifest by up-regulation of costimulatory molecules, secretion of pro- and antiinflammatory cytokines, and the ability to stimulate the differentiation of naive T cells into effector cells. Cytokine production by activated DCs (IL-12, TNF-α, and IL-10) is transient; after prolonged stimulation, DCs become refractory to subsequent activation signals (2). Although delayed production of IL-10 partially contributes to the arrest of cytokine production by activated DCs and the return to steady state, the immune system relies on additional negative feedback mechanisms to down-regulate proinflammatory cytokine release and/or terminate the response to Ag. These include soluble mediators such as PGE2, cyclopentenone PGs, and lipoxin A4 (3–5), or ligation of certain cell surface receptors (i.e., CD36, CD47, and CD51/CD61; references 6, 7). The latter receptors bind thrombospondin 1 (TSP), an extracellular matrix (ECM) protein predominantly secreted by platelets, monocytes, macrophages, and several nonhematopoietic cell types (8, 9). Also, TSP binds additional receptors as follows: α3β1, αIIbβ3, and cellular glycosaminoglycans. As a consequence of its binding to several cell surface receptors, TSP exerts myriad effects on cell function, which sometimes appear to be contradictory (10, 11). Some studies have shown that TSP promotes cell migration, cell adhesion, and T cell activation (for reviews see references 10 and 12). It inhibits angiogenesis, cell migration, and T cell activation, and promotes apoptosis (for reviews see references 10 and 12). Notably, TSP is composed of several distinct structural domains that specifically interact with a range of cellular receptors differentially expressed by a variety of cells (12), which could explain its broad spectrum of biological activities. Nevertheless, TSP displays predominantly antiinflammatory activities in vivo as strongly suggested by the inflammatory phenotype of TSP null mice (13).
We reported previously that CD47 ligation by anti-CD47 monoclonal antibodies or by TSP selectively down-regulates IL-12 production by monocytes. Moreover, CD47 ligation inhibits DC phenotypic maturation and cytokine release while leaving TGFβ secretion intact (7, 14). Although TSP is a major activator of TGF-β, which itself prevents DC maturation, the TSP inhibitory effect on APCs was shown to be independent of both TGF-β and IL-10 (7, 13, 15). Here, we demonstrate that iDCs express and secrete TSP, and show that TSP production is regulated by soluble mediators and maturation signals. Our results also support the negative regulatory effects of DC-derived TSP on cytokine release (i.e., IL-12, TNF-α, and IL-10) through interactions with CD36 and CD47 during early DC activation. Interestingly, only TSP–CD47 interactions appear to contribute to refractoriness of fully mature DCs to additional activation signals, suggesting an active role for TSP in the resolution of the inflammatory response after an environmental insult.
Materials and Methods
Dendritic Cell Preparation and Culture Conditions.
Peripheral blood mononuclear cells were isolated by density gradient centrifugation of heparinized blood from healthy volunteers with Lymphoprep (Nycomed). Monocytes were enriched by cold aggregation, followed by T and NK depletion as reported previously (14). Monocyte purity was shown to be >95% CD14+ cells by flow cytometry. Human monocyte-derived iDCs were prepared exactly as described previously (16). Every other day, two thirds of culture medium was replaced by fresh medium (RPMI 1640 10% FCS supplemented with 2 mM glutamine, 1 mM sodium pyruvate, 10 mM hepes, 100 IU penicillin, and 100 μg/ml streptomycin, containing GM-CSF and IL-4); nonadherent cells were harvested at day 5 to obtain iDCs. Mature DCs (mDCs) were generated after stimulation of iDCs (0.5 × 106/ml) in complete HB101 medium (Irvine Scientific) or complete medium (RPMI 1640 10% FCS containing low TSP levels [<30 ng/ml]) for the indicated periods of time. DCs were activated by Staphylococcus aureus Cowan I strain (SAC; 0.0025%) (wt/vol; Pansorbin; Calbiochem), 1 μg/ml LPS or CD40-L L-transfectants (or 1 μg/ml sCD40-L and 500 U/ml IFN-γ). IL-10 and TGF-β were used at 10 ng/ml and purchased from R&D Systems, and PGE2 was used at 100 nM (Sigma-Aldrich). Neutralizing anti-TSP mAbs III (clone 6.7) and I (clone A4.1) obtained from Lab Vision and control mAbs (IgG1 and IgM; Sigma-Aldrich) were used at 10 μg/ml. To examine DC exhaustion, mDCs were washed, counted, and restimulated overnight with L-transfectants CD40-L or sCD40-L and IFN-γ in RPMI 1640 10% FCS.
GeneChip Expression Analysis.
Human genome-wide gene expression was examined with the Human Genome U133A probe array (GeneChip; Affymetrix, Inc.), which contains the oligonucleotide probe set for ∼22,000 full-length genes, according to the manufacturer's protocol. Total RNA was extracted from ∼5 × 106 cells. Double-stranded cDNA was synthesized by means of Superscript Choice system (Life Technologies) and a T7-(dt)24 primer (Amersham Biosciences). The cDNA was subjected to the in vitro transcription in the presence of biotinylated nucleoside [Q]triophosphate by means of a BioArray High Yield RNA Transcript labeling kit (Enzo Diagnostics). The biotinylated cRNA was hybridized with a probe. After washing, the hybridized biotinylated cRNA was stained with PE-streptavidin (Molecular Probes) and scanned with an HP Gene Array Scanner (Affymetrix, Inc.). The fluorescence intensity of each probe was quantified with a computer program (Suite 4.0; Affymetrix, Inc.).
Flow Cytometric Analysis.
The phenotypes of iDCs and mDCs were determined by direct staining with PE-CD86 (clone BU63) and FITC-CD83 (clone HB15e; Becton Dickinson). A three-step procedure was used for intracytoplasmic detection of TSP. In brief, brefeldin A was added for the last 5 h of the culture; DCs were permeabilized with 0.5% saponin and fixed for 5 min with 0.1% paraformaldehyde. Cells were incubated with 2 μg/ml anti-TSP mAb (clone 6.7 or A3; Lab Vision) for 30 min at 4°C, followed by biotinylated goat anti–mouse IgG + IgM (1/500; Biosource International) for 30 min and stained with streptavidin-PE (Becton Dickinson). Stained cells were analyzed with a FACScalibur™ (Becton Dickinson).
Cytokine Measurement.
IL-12p70, TNF-α, and IL-10 release were assessed by two-site sandwich ELISA as described previously (7). The sensitivity of the assays was 6 pg/ml for IL-12 and 50 pg/ml for the other cytokines. For TSP measurement, plates were coated overnight with 0.5 μg/ml anti-TSP mAb (clone P10; Chemicon) and blocked with PBS containing 2% BSA and 0.1% Tween 20 for 2 h. After washing, 100-μl samples diluted in HB101 medium were added overnight. TSP purified from platelets (Calbiochem) was used as a standard. Plates were washed and incubated with detection polyclonal rabbit anti-TSP Ab (Calbiochem) and developed by HRPO-labeled mouse anti–rabbit IgG and substrate. The sensitivity of the assay was 10 ng/ml.
Results
Immature and Mature DCs Synthesize TSP.
TSP produced by macrophages facilitates the phagocytosis of apoptotic cells by establishing a molecular bridge between the phagocyte and the dying cell (17, 18). The iDC is another Ag-presenting cell specialized in the uptake of apoptotic cells, and is the only cell type capable of cell-associated Ag cross-presentation to naive T cells. Therefore, we postulated that iDCs could represent an additional source of TSP. Our initial microarray analysis performed on iDCs isolated from five donors revealed the constitutive presence of TSP transcript and its up-regulation by SAC (∼11-fold) and LPS (∼8-fold) stimulation. By using two complementary experimental approaches (intracellular staining and ELISA), we demonstrated that TSP is secreted spontaneously by iDCs and that its production is significantly increased during DC maturation. First, we confirmed that DCs express TSP by intracellular staining (Fig. 1) . Indeed, the staining reveals that TSP is readily detected in the whole population of unstimulated iDCs. Engagement of toll-like receptors (TLR) 4 or 2, by LPS and SAC respectively, significantly increases TSP expression (Fig. 1 A) with maximum levels observed after 48 h of stimulation (Fig. 1 B, P < 0.05, n = 5). TSP is hardly detectable in nonpermeabilized cell preparations, indicating that the vast majority of TSP is located intracellularly and not on the cell surface (unpublished data). Second, the measurement of TSP in the culture supernatant of unstimulated and maturing DCs confirmed that TSP is indeed secreted by DCs (Fig. 2) . TSP production is strongly increased during DC maturation in response to T cell–dependent or TLR-mediated activation signals (Fig. 2 A, P < 0.05, n = 9). The levels of spontaneous TSP production plateau between 48 h and 72 h (Fig. 2 B). Kinetic studies indicate that TSP production by maturing DCs is delayed when compared with that of IL-12 and TNF-α, but simultaneous with that of IL-10 (Fig. 2, B and C). These data establish that DCs are an abundant source of TSP in the steady state and that production can be enhanced by inflammatory signals.
Figure 1.

Immature and mature DCs express TSP. 0.5 × 106/ml monocyte-derived iDCs were cultured for 48 h (A) or the indicated time (B) in the absence or presence of 1 μg/ml LPS or 0.0025% SAC. Cells were permeabilized, fixed, and stained for intracellular TSP as described in Materials and Methods. (A) TSP staining is depicted as follows: iDCs (left, thick line); iDCs (middle, thin line) versus iDCs + LPS (thick line); and iDCs (right, thin line) versus iDCs + SAC (thick line). (dotted lines) Isotype control mAb. Data are from one representative experiment out of three (A) and out of five (B).
Figure 2.
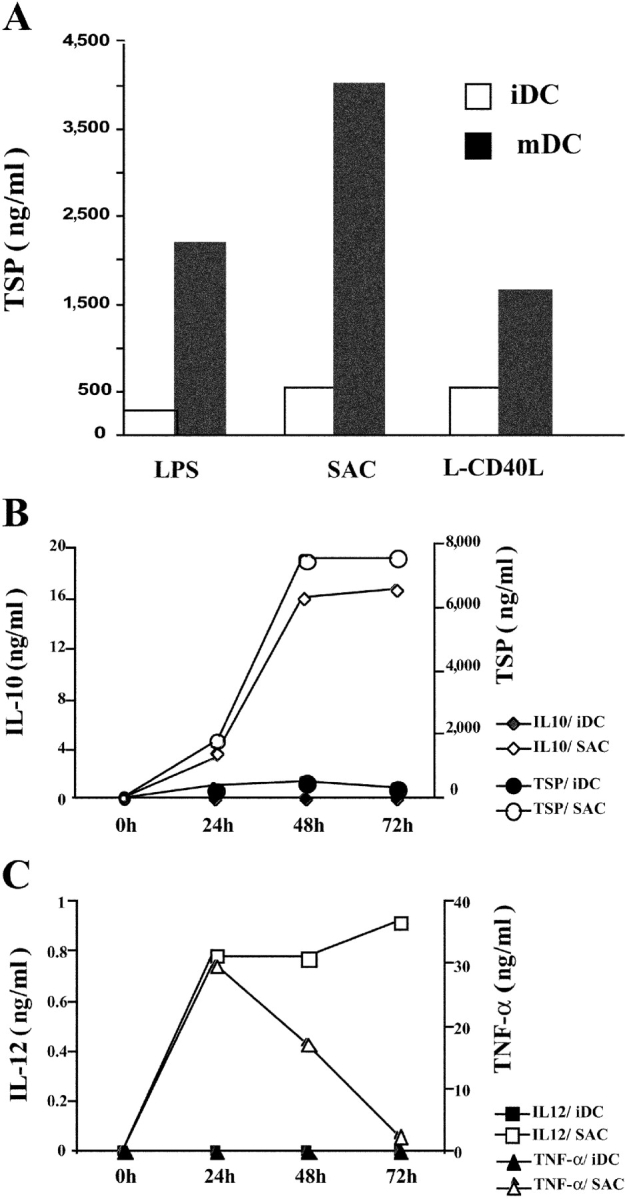
TSP secretion by activated DCs. (A) Monocyte-derived iDCs were cultured for 24 h in the absence or presence of LPS, SAC, or L-CD40L + IFN-γ. TSP production was determined in the culture supernatants by ELISA (sensitivity, 10 ng/ml). Data are from one representative experiment out of three. (B and C) Kinetics of cytokine production by SAC-activated DCs. Monocyte-derived iDCs were cultured for the indicated periods of time in the absence or presence of SAC. IL-12, TNF-α, IL-10, and TSP production was determined by ELISA. Data are from one representative experiment out of three.
Soluble Mediators Regulate TSP Production by Immature and Activated DCs.
TSP potentiates the phagocytosis of apoptotic cells by macrophages, leading to the up-regulation of TGF-β and PGE2 production by these cells (17, 18). Therefore, we examined whether PGE2, TGF-β, or IL-10, each of which exhibits well-established antiinflammatory properties, would in turn modulate TSP production by immature and maturing DCs. As shown in Fig. 3 , TGF-β but not IL-10 significantly increases TSP production by iDCs (Fig. 3 A). In contrast, both cytokines both significantly potentiate TSP production after 24 h of stimulation by SAC (Fig. 3 B). This enhancing effect is no longer observed after 48 h, when TSP levels reach a plateau (unpublished data). Strikingly, PGE2 increases the release of TSP by iDCs by >50-fold (Fig. 3 C) and 1-nM physiological concentrations of PGE2 are sufficient to increase TSP production (Fig. 3 E); this enhancing effect is confirmed by intracytoplasmic staining (Fig. 3 F). PGE2 further potentiates TSP secretion by activated DCs (Fig. 3 C), whereas it significantly down-regulates IL-12 and TNF-α release (Fig. 3 D).
Figure 3.

IL-10, TGF-β, and PGE2 enhance TSP secretion by DCs. (A and B) Monocyte-derived iDCs were cultured for 24 h in the absence or presence of SAC with or without IL-10 or TGF-β. TSP production was determined by ELISA and TSP levels expressed in fold increase compared with iDCs. Mean ± SEM of five experiments (paired Student's t test; *, P < 0.05). (C and D) Monocyte-derived iDCs were cultured for 24 h in the absence or presence of SAC with or without 100 nM PGE2. TSP and IL-12, TNF-α production was determined by ELISA. (C) TSP levels expressed in fold increase as in A and B. (D) IL-12 and TNF-α levels expressed as percentage of control response (100%, cytokine production by activated DCs). Mean ± SEM of five experiments (paired Student's t test; **, P < 0.01). (E) Dose–response curve of PGE2 effect on TSP secretion by iDCs. (F) Effect of 100 nM PGE2 on intracytoplasmic TSP expression by iDCs. (thin line) TSP staining of iDCs. (thick line) iDCs + PGE2. (dotted line) Control mAb. Data are from one representative experiment out of three for E and F.
We conclude that antiinflammatory molecules, including PGE2, IL-10, and TGF-β, provide a positive signal for TSP secretion by both iDCs and activated DCs. Because exogenous TSP reportedly down-modulates DC maturation (7), next we examined the role of endogenous TSP in the regulation of cytokine production.
Endogenous TSP Secretion Renders Fully Mature DCs Refractory to Further Activation.
TSP can potentially use two pathways to decrease proinflammatory cytokine release by maturing DCs. Ligation of CD36 and CD47, two TSP receptors on DCs, by their respective mAbs reportedly impairs IL-12 secretion (6, 7). Therefore, we attempted to block TSP binding to CD36 or CD47 at an early stage of DC activation by adding neutralizing anti-TSP mAbs together with a TLR activation signal. We found that anti-TSP mAb III and I, which interfere with the binding of TSP to CD47 and CD36 respectively, enhance TNF-α, IL-12, and IL-10 synthesis in response to SAC or in response to CD40L plus IFN-γ stimulation (Fig. 4 A and not depicted). It is worth noting that these mAbs do not by themselves modulate the expression of costimulatory molecules or affect the ability of DCs to stimulate the proliferation of allogeneic naive T cells (unpublished data). This indicates that endogenous TSP negatively regulates pro- and antiinflammatory cytokine release during DC maturation.
Figure 4.

Endogenous TSP negatively regulates cytokine release and mediates DC exhaustion. (A) Monocyte-derived iDCs were activated with SAC for 48 h in the presence of neutralizing anti-TSP mAbs or respective control mAbs (10 μg/ml). Anti-TSP mAb III selectively interferes with TSP–CD47 interactions and anti-TSP mAb I with TSP–CD36 interactions. Data are from one representative experiment out of six. (B) Monocyte-derived iDCs were cultured for 48 h with SAC, washed, and restimulated for 24 h with L-CD40L transfectants and IFN-γ (to mimic T cell interactions) in the presence or absence of the two different neutralizing anti-TSP mAbs. Data are from one representative experiment out of four. IL-12, TNF-α, and IL-10 production were determined by ELISA.
Next, we examined the possible role of endogenous TSP in DC exhaustion. DCs were cultured for 48 h with SAC (or LPS plus IFN-γ), washed, and restimulated for 24 h with L-CD40L transfectants and IFN-γ (to mimic T cell interactions) in the presence or absence of the two different neutralizing anti-TSP mAbs (Fig. 4 B). As reported previously by Langenkamp et al. (2), DCs have almost extinguished their cytokine release after 48-h maturation in response to secondary stimulation, resulting in the state referred to as exhaustion. Interestingly, we found that exhausted DCs still produced large amounts of TSP (ranging from 1,320 to 3,890 ng/ml; n = 5). Interruption of TSP–CD47 interactions (by anti-TSP mAb III) allows DCs to recover their ability to secrete cytokines. This is not the case with anti-TSP mAb I (i.e., inhibiting binding of TSP to CD36) nor with neutralizing anti–IL-10 mAb (unpublished data). The differential effect of the two anti-TSP mAbs can be partly explained by the loss of CD36 contrasting with the stability in CD47 expression during DC maturation (7, 19).
These data suggest that TSP interactions with CD47 and CD36 inhibit cytokine production at the early stages of DC activation by TLR ligands, whereas only TSP–CD47 is involved in the negative regulation of cytokine release at the final stage of DC maturation, when DCs encounter T cells. In conclusion, we define DC-derived TSP production as a negative feedback mechanism that is actively involved in the arrest of cytokine production and the loss of DC responsiveness to further stimulation.
Discussion
The present findings demonstrate that DCs represent a previously unknown and abundant source of TSP, both in the steady state and during activation by danger signals. Additionally, we present evidence that endogenous TSP actively renders DCs refractory to subsequent stimulation and, thus, contributes to the arrest of the inflammatory response. In fact, TSP is rapidly expressed at high levels in inflamed and damaged tissues (20). Resolution of inflammation is now considered to be an active process tightly regulated by stop signals (21), including antiinflammatory molecules such as lipid mediators (lipoxins and prostaglandin derivatives), TGF-β, IL-10, and TSP. Our data are consistent with the known predominant antiinflammatory functions of TSP in vitro and in vivo (10). TSP null mice have persistent inflammation in multiple organs, including the pancreas and the lung, and display a similar phenotype as TGF-β null mice (11, 13). TSP is reported to be a major activator of TGF-β in vivo because a TSP peptide-activating TGF-β reverses the TSP null phenotype toward wild type. Here, we show that TGF-β further increases TSP secretion by DCs. These results are in agreement with recent findings that TGF-β augments TSP expression by macrophages, and TGF-β–treated APCs promote the generation of regulatory T cells (22, 23). This positive feedback loop creates an environment favorable for the induction of anterior chamber-associated immune deviation, a form of tolerance ensuring the absence of inflammatory reaction in immune privileged sites.
IL-10 is another major inducer of tolerance and is presently considered to be the most potent down-regulator of proinflammatory cytokine secretion by DCs (24). IL-10–treated DCs promote the differentiation of naive T cells into regulatory T cells, which in turn secrete IL-10. Our data indicate that IL-10 increases TSP secretion by activated DCs. Kinetics of IL-10 and TSP release are very similar during DC maturation. However, TSP inhibits IL-10 production. Thus, fully mDCs continue secreting TSP whereas they stop producing IL-10 upon secondary stimulation. Interestingly, interruption of TSP–CD47 by anti-TSP mAb added at restimulation allows recovery of DC cytokine production (including IL-10), suggesting the existence of alternate pathways to highly regulate cytokine release when DCs encounter T cells.
Furthermore, our findings show that DC-derived TSP negatively regulates pro- and antiinflammatory cytokine secretion during early activation through interactions with CD47 and CD36. These results support our previous paper that exogenous TSP and a peptide derived from the C domain of TSP, which selectively binds to CD47, down-regulate DC cytokine release, with the exception of TGF-β (7). They are also consistent with a recent report indicating that anti-CD36 mAb delivers a negative signal to DCs, resulting in decreased IL-12 production and phenotypic maturation (6)
Most interestingly, we observed that physiologic concentrations of PGE2, and to a lesser extent TGF-β, enhance TSP secretion by steady-state iDCs in the absence of inflammatory signals. Both PGE2 and TGF-β are reportedly produced by macrophages after engulfment of apoptotic cells and the latter secrete TGF-β (25, 26). Thus, iDC-derived TSP may facilitate the phagocytosis of apoptotic cells by at least two mechanisms. First, TSP promotes CD47-mediated caspase-independent cell death, characterized by increased phosphatidylserine exposure on target cells (27). Second, it establishes a molecular bridge between αvβ3/CD36 on phagocytes and an unidentified receptor, possibly CD47, on dying cells (17). In that context, we postulate that iDC-derived TSP is likely to be involved in phagocytosis of damaged tissues by steady-state DCs that continuously tolerize lymph node T cells. Furthermore, TSP inhibits early T cell activation and promotes naive T cell anergy (28, 29), and active TGF-β displays potent immunoregulatory functions, including prevention of DC maturation and activation of regulatory T cells (15, 30). Altogether, it is possible that PGE2 and TGF-β further amplify a TSP/TGF-β/TSP-positive immunoregulatory loop established to ensure tolerance in the steady state. Consistent with numerous analyses, we observed that exogenous PGE2 strongly inhibits cytokine release by activated DCs (3). Under these inflammatory conditions, PGE2, like TGF-β and IL-10, markedly increases TSP.
Herein, we provide evidence that DC-derived TSP mediates DC exhaustion. After prolonged stimulation, DCs secrete TSP and TGF-β and promote the generation of nonpolarized T cells, a subset of central memory T cells (2). We propose that TSP exerts its antiinflammatory activity both in the steady state, contributing to the tolerogenic function of DCs, and at the end of the inflammatory response, allowing the rapid and active resolution of inflammation. The in vivo biological relevance of the present in vitro findings is supported strongly by the inflammatory phenotype of TSP null mice.
Acknowledgments
We are grateful to Dr. C. Maliszweski for his critical review of the manuscript and thank Ms. N. Hashimoto and Ms. H. Wakita for skillful technical assistance.
The project was supported by the Canadian Institute for Health Research (grant MOP-44090) and D. Braun was sponsored by the Association pour la Recherche contre le Cancer.
References
- 1.Steinman, R.M., D. Hawiger, and M.C. Nussenzweig. 2003. Tolerogenic dendritic cells. Annu. Rev. Immunol. 21:685–711. [DOI] [PubMed] [Google Scholar]
- 2.Langenkamp, A., M. Messi, A. Lanzavecchia, and F. Sallusto. 2000. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 1:311–316. [DOI] [PubMed] [Google Scholar]
- 3.Harizi, H., M. Juzan, V. Pitard, J.F. Moreau, and N. Gualde. 2002. Cyclooxygenase-2-issued prostaglandin e(2) enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J. Immunol. 168:2255–2563. [DOI] [PubMed] [Google Scholar]
- 4.Morelli, A.E., and A.W. Thomson. 2003. Dendritic cells under the spell of prostaglandins. Trends Immunol. 24:108–111. [DOI] [PubMed] [Google Scholar]
- 5.Aliberti, J., S. Hieny, C. Reis e Sousa, C.N. Serhan, and A. Sher. 2002. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat. Immunol. 3:76–82. [DOI] [PubMed] [Google Scholar]
- 6.Urban, B.C., N. Willcox, and D.J. Roberts. 2001. A role for CD36 in the regulation of dendritic cell function. Proc. Natl. Acad. Sci. USA. 98:8750–8755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demeure, C.E., H. Tanaka, V. Mateo, M. Rubio, G. Delespesse, and M. Sarfati. 2000. CD47 engagement inhibits cytokine production and maturation of human dendritic cells. J. Immunol. 164:2193–2199. [DOI] [PubMed] [Google Scholar]
- 8.Jaffe, E.A., J.T. Ruggiero, and D.J. Falcone. 1985. Monocytes and macrophages synthesize and secrete thrombospondin. Blood. 65:79–84. [PubMed] [Google Scholar]
- 9.Brown, E.J., and W.A. Frazier. 2001. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 11:130–135. [DOI] [PubMed] [Google Scholar]
- 10.Bornstein, P. 2001. Thrombospondins as matricellular modulators of cell function. J. Clin. Invest. 107:929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawler, J., M. Sunday, V. Thibert, M. Duquette, E.L. George, H. Rayburn, and R.O. Hynes. 1998. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J. Clin. Invest. 101:982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silverstein, R.L. 2002. The face of TSR revealed: an extracellular signaling domain is exposed. J. Cell Biol. 159:203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford, S.E., V. Stellmach, J.E. Murphy-Ullrich, S.M. Ribeiro, J. Lawler, R.O. Hynes, G.P. Boivin, and N. Bouck. 1998. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 93:1159–1170. [DOI] [PubMed] [Google Scholar]
- 14.Armant, M., M.-N. Avice, P. Hermann, M. Rubio, M. Kiniwa, G. Delespesse, and M. Sarfati. 1999. CD47 ligation selectively downregulates human interleukin 12 production. J. Exp. Med. 190:1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geissmann, F., P. Revy, A. Regnault, Y. Lepelletier, M. Dy, N. Brousse, S. Amigorena, O. Hermine, and A. Durandy. 1999. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J. Immunol. 162:4567–4675. [PubMed] [Google Scholar]
- 16.Tanaka, H., C.E. Demeure, M. Rubio, G. Delespesse, and M. Sarfati. 2000. Human monocyte-derived dendritic cells induce naive T cell differentiation into T helper cell type 2 (Th2) or Th1/Th2 effectors. Role of stimulator/responder ratio. J. Exp. Med. 192:405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savill, J., I. Dransfield, C. Gregory, and C. Haslett. 2002. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2:965–975. [DOI] [PubMed] [Google Scholar]
- 18.Fadok, V.A., D.L. Bratton, S.C. Frasch, M.L. Warner, and P.M. Henson. 1998. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 5:551–562. [DOI] [PubMed] [Google Scholar]
- 19.Albert, M.L., S.F. Pearce, L.M. Francisco, B. Sauter, P. Roy, R.L. Silverstein, and N. Bhardwaj. 1998. Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 188:1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raugi, G.J., J.E. Olerud, and A.M. Gown. 1987. Thrombospondin in early human wound tissue. J. Invest. Dermatol. 89:551–554. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence, T., D.A. Willoughby, and D.W. Gilroy. 2002. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2:787–795. [DOI] [PubMed] [Google Scholar]
- 22.Masli, S., B. Turpie, K.H. Hecker, and J.W. Streilein. 2002. Expression of thrombospondin in TGFbeta-treated APCs and its relevance to their immune deviation-promoting properties. J. Immunol. 168:2264–2273. [DOI] [PubMed] [Google Scholar]
- 23.Streilein, J.W., S. Masli, M. Takeuchi, and T. Kezuka. 2002. The eye's view of antigen presentation. Human Immunol. 63:435–443. [DOI] [PubMed] [Google Scholar]
- 24.Steinbrink, K., M. Wolfl, H. Jonuleit, J. Knop, and A.H. Enk. 1997. Induction of tolerance by IL-10-treated dendritic cells. J. Immunol. 159:4772–4780. [PubMed] [Google Scholar]
- 25.Fadok, V.A., D.L. Bratton, A. Konowal, P.W. Freed, J.Y. Westcott, and P.M. Henson. 1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101:890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorelik, L., and R.A. Flavell. 2002. Transforming growth factor-beta in T-cell biology. Nat. Rev. Immunol. 2:46–53. [DOI] [PubMed] [Google Scholar]
- 27.Mateo, V., L. Lagneaux, D. Bron, G. Biron, M. Armant, G. Delespesse, and M. Sarfati. 1999. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat. Med. 5:1277–1284. [DOI] [PubMed] [Google Scholar]
- 28.Li, Z., L. He, K. Wilson, and D. Roberts. 2001. Thrombospondin-1 inhibits TCR-mediated T lymphocyte early activation. J. Immunol. 166:2427–2436. [DOI] [PubMed] [Google Scholar]
- 29.Avice, M.-N., M. Rubio, M. Sergerie, G. Delespesse, and M. Sarfati. 2001. Role of CD47 in the induction of human naive T cell anergy. J. Immunol. 167:2459–2468. [DOI] [PubMed] [Google Scholar]
- 30.Yamagiwa, S., J.D. Gray, S. Hashimoto, and D.A. Horwitz. 2001. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J. Immunol. 166:7282–7289. [DOI] [PubMed] [Google Scholar]