

Abstract
Autoimmune gastritis and Helicobacter pylori–associated gastric atrophy develop through similar mechanisms involving the proton pump H+,K+–adenosine triphosphatase as autoantigen. Here, we report that H. pylori–infected patients with gastric autoimmunity harbor in vivo–activated gastric CD4+ T cells that recognize both H+,K+–adenosine triphosphatase and H. pylori antigens. We characterized the submolecular specificity of such gastric T cells and identified cross-reactive epitopes from nine H. pylori proteins. Cross-reactive H. pylori peptides induced T cell proliferation and expression of T helper type 1 functions. We suggest that in genetically susceptible individuals, H. pylori infection can activate cross-reactive gastric T cells leading to gastric autoimmunity via molecular mimicry.
Keywords: Helicobacter pylori, T cell epitopes, autoreactive T cells, mimicry, mucosal immunity
Introduction
Several mechanisms have been proposed for how pathogens might induce activation and critical expansion of autoreactive T cells and start autoimmune disease (1–6). Activation of resting autoreactive T cells may be achieved by viral and bacterial superantigens that bind a variety of MHC class II molecules and activate large numbers of T cells, irrespective of their specificity (7). Pathogen-induced tissue inflammation may result in local activation of APCs and enhanced processing/presentation of self antigens that causes T cell priming, followed by T cell activation and expansion of additional specificities (epitope spreading; references 8, 9). Another mechanism would imply that the inflammatory setting and the paracrine secretion of T cell growth factors induce the expansion of activated autoreactive T cells, whose small number was previously insufficient to drive an autoimmune disease. Such a mechanism is referred to as bystander activation (10). Moreover, a microbial antigen can include an epitope that is structurally similar to an autoantigen epitope, providing the basic element of the mechanism referred to as molecular mimicry (5, 6, 11–14).
Autoimmune chronic gastritis (AIG) is an organ-specific inflammatory disease leading to gastric atrophy, hypochloridria, and eventually to pernicious anemia. AIG is characterized by lymphocytic infiltrates in the gastric mucosa and by destruction of parietal cells, resulting in mucosal atrophy (15). In most AIG patients, serum anti-parietal cell autoantibodies (PCAs) are detectable. The autoantigen recognized is the gastric H+,K+–adenosine triphosphatase (ATPase), the proton pump, localized in the parietal cell canaliculi (16, 17). H+,K+-ATPase is also the target of autoreactive T cells that infiltrate the gastric mucosa of AIG patients (18).
Helicobacter pylori infection is one of the most common bacterial infections in humans and its clinical outcomes are highly variable, including chronic gastritis, duodenal or gastric ulcers, mucosal atrophy, gastric carcinoma, or gastric lymphoma (19). In H. pylori–infected patients who develop gastric corpus atrophy, an increased incidence of positive PCAs has been reported, which significantly decreases after H. pylori eradication (20, 21). Based on the similarities between H. pylori–induced corpus atrophy and classical AIG, we inferred that in some individuals genetically predisposed to organ-specific autoimmunity due to their MHC class II haplotype, H. pylori infection plays a role in induction or exacerbation of gastric autoimmunity (20).
To test this hypothesis, we selected four women with chronic AIG and current H. pylori infection. Biopsy specimens of their gastric mucosa were cultured in IL-2–conditioned medium to expand in vivo–activated gastric T cells. Specimens were disrupted, and single T cell blasts were cloned (18). Gastric T cell clones were screened for their ability to proliferate in response to H+,K+-ATPase and/or to a H. pylori lysate. In addition to CD4+ T cell clones that proliferated to H. pylori lysate or to H+,K+-ATPase, we found a remarkable number of gastric T cell clones that recognized both H+,K+-ATPase and H. pylori antigens, and we identified their cross-reactive epitopes at the molecular level.
Materials and Methods
Generation of Gastric T Cell Clones.
Four women (mean age, 45; range, 29–53 yr) with chronic AIG and thyroiditis provided their informed consent for this work. Their MHC haplotypes were as follows: HLA-A2, A11, B35, B41, DRB1*0701, and DRB1*1303 in patient 1; HLA-A1, A24, B15, B35, DRB1*0404, and DRB1*0803 in patient 2; HLA-A23, A32, B27, B35, DRB1*0403, and DRB1*1303 in patient 3; and HLA-A2, A3, B18, B50, DRB1*0301, and DRB1*1104 in patient 4. All patients had serum PCAs and thyroid peroxidase autoantibodies, but not intrinsic factor autoantibodies or hematologic abnormalities. All the patients were receiving levothyroxine for hypothyroidism. Patient 4 also suffered from vitiligo. At the beginning of this work, all patients had positive serology for H. pylori infection, positive [13C]urea breath test, and positive histology for H. pylori at gastroscopy. None of the patients had H. pylori eradication. At the end of a 4-yr follow-up, in all patients, the [13C]urea breath test was found consistently negative.
Biopsy specimens of gastric mucosa were cultured for 7 d in RPMI 1640 medium supplemented with 50 U/ml of human IL-2 to expand in vivo–activated T cells. Specimens were disrupted, and single T cell blasts were cloned (18). In brief, T cell blasts were seeded under limiting dilution conditions (0.3 cells/well) in round-bottomed microwells containing 6,000 rad 105 irradiated autologous mononuclear cells (as feeder cells) and 1% vol/vol PHA in a final volume of 0.2 ml of complete medium supplemented with 20 U/ml IL-2, 5% human serum, and 10% FCS. Growing microcultures were supplemented, at weekly intervals, with 20 U/ml IL-2 and 105 irradiated feeder cells. Clones were screened for responsiveness to medium, 5 μg/ml of porcine albumin, 0.5 μg/ml of porcine gastric H+,K+-ATPase (22, 23), and H. pylori lysate (10 μg/ml aqueous extract of NCTC11637 strain) by measuring [3H]TdR uptake after 60 h (18).
Analysis of TCR Vβ Chain Repertoire of Gastric T Cell Clones.
The repertoire of the TCR Vβ chain of H+,K+-ATPase–specific Th clones was analyzed with a panel of 22 mAbs specific to the following: Vβ1, Vβ2, Vβ4, Vβ7, Vβ9, Vβ11, Vβ14, Vβ16, Vβ18, Vβ20, Vβ21.3, Vβ22, and Vβ23 (Beckman Coulter); and Vβ3.1, Vβ5.1, Vβ5.2, Vβ5.3, Vβ6.7, Vβ8, Vβ12, Vβ13, and Vβ17 (AMS Biotechnology GmbH). Isotype-matched nonspecific Ig were used as negative control. Data acquisition was performed in a FACSCalibur™ flow cytometer using the CELLQuest™ software program (Becton Dickinson). From each T cell clone, mRNA was extracted by mRNA direct isolation kit (QIAGEN). For cDNA synthesis, the same amount of mRNA (50 ng) was used, and cDNA was synthesized by Moloney murine leukemia virus-reverse transcriptase (New England Biolabs, Inc.) and oligo-(dT) primers according to the manufacturer's protocol. cDNA mix of all samples was amplified under equal conditions by a 30-cycle PCR using Vβ T cell receptor typing amplimer kit for Vβ10, Vβ15, and Vβ19 (CLONTECH Laboratories, Inc.) according to the manufacturer's instructions.
Generation of H+,K+-ATPase Overlapping Peptides and Prediction of Candidate Cross-reactive H. pylori Peptides.
To span the 1,034–amino acid α chain and the 270–amino acid β chain of porcine H+,K+-ATPase, 205 and 56 overlapping 15-mer peptides with a 10–amino acid overlap, respectively, were prepared by automated simultaneous multiple peptide synthesis, as described previously (24). Amino acid sequences of H+,K+-ATPase epitopes recognized by the 13 cross-reactive T cell clones were used in silico to identify H. pylori peptides that might be candidates for cross-reactivity. Homologies between the 13 H+,K+-ATPase epitopes and peptides present in both genomes of H. pylori J99 and 26695 strains were screened by using the basic local alignment search tool (BLAST) server of the National Center for Biotechnology Information. Standard BLAST search parameters were used with the following adaptations: word size, 2; Expect 100000; Matrix, Blosum 45 with existence 19 and extension 1 penalty settings. Amino acid sequences in H. pylori J99 and 26695 open reading frames (ORFs) with sufficient homology (top 100 of the BLAST results) to the relevant H+,K+-ATPase epitopes and with a minimal length of nine amino acids were studied for the presence of motifs that would predict binding to patient MHC class II alleles by using the ProPred MHC class II Binding Peptide Prediction Server (25). The threshold of prediction was set at 3%. Identified H. pylori peptides were adjusted to a 15–amino acid length based on the appropriate H. pylori ORFs.
Submolecular Specificity of Gastric T Cell Clones Reactive to H+,K+-ATPase or to Both H+,K+-ATPase and H. pylori Lysate.
Equal amounts of each of the 261 overlapping peptides of H+,K+-ATPase were pooled to have 20 pools. 4 × 104 T cell blasts from each clone were cultured in triplicate for 3 d together with 1.5 × 105 irradiated autologous mononuclear cells in the presence of medium, 5 μg/ml of porcine albumin, 0.5 μg/ml H+,K+-ATPase, 10 μg/ml H. pylori lysate or equal aliquots from each of the 20 pools in which each peptide component was present at a 10 μg/ml final concentration. After 60 h, [3H]TdR uptake was measured. Mitogenic index (MI) was calculated as the ratio between counts in stimulated cultures and those in unstimulated cultures. Each clone was retested for proliferation to the individual peptide components of the pool that had induced a MI > 5. Finally, each clone was tested for proliferation to H+,K+-ATPase, H. pylori lysate, and the appropriate H+,K+-ATPase peptide (as positive controls); the couple of flanking H+,K+-ATPase peptides (negative controls) and the series of H. pylori peptides were identified as possible candidates for cross-reactivity to the appropriate H+,K+-ATPase epitope.
In some experiments, the effect of 5 μg/ml anti–HLA-DR (clone G46–6) or anti–HLA-DQ (clone TU169; BD Biosciences) monoclonal antibodies or their isotype control (mouse IgG2a) on T cell clone proliferation induced by the appropriate H+,K+-ATPase and H. pylori cross-reactive peptides was assessed.
Cytokine Production Induced by Peptides.
T cell blasts of each cross-reactive clone (5 × 105) were cocultured in triplicate tubes for 48 h in 0.5 ml medium with 5 × 105 irradiated autologous APCs in the presence of medium, 0.5 μg/ml H+,K+-ATPase, 10 μg/ml H. pylori lysate, the appropriate H+,K+-ATPase peptide, and the H. pylori cross-reactive peptide that induced proliferation (10 μg/ml), as well as control peptides that failed to induce proliferation. Duplicate samples of each supernatant were assayed for IL-4, IL-5, and IFN-γ content by ELISA assays (18).
Perforin-mediated Cytolytic Activity and Fas–Fas Ligand-mediated Apoptotic Killing.
The ability of T cell clones to express perforin-mediated cytotoxicity was assessed in a lectin-dependent assay against 51Cr-labeled P815 murine mastocytoma cells as described previously (26).
The ability of gastric T cell clones to induce Fas–Fas ligand-mediated apoptosis was assessed using Fas+ Jurkat cells as target and the anti-Fas antagonistic mAb M3 (Immunex), as described previously (18, 27).
Results
Submolecular Specificity of Gastric T Cell Clones Reactive to H+,K+-ATPase.
A total of 154 CD4+ and 49 CD8+ T cell clones were obtained from the gastric biopsies of four H. pylori–infected AIG patients. All gastric clones were screened for proliferation to H. pylori lysate, H+,K+-ATPase, or porcine albumin (control antigen). No proliferation was detected in any of the CD8+ clones and in 108 CD4+ T cell clones, although they all proliferated to IL-2. In contrast, 18 CD4+ gastric clones (donor 1, five; donor 2, two; donor 3, four; and donor 4, seven) showed significant proliferation (MI range, 38–212) to H. pylori lysate, but not to H+,K+-ATPase or porcine albumin, and 15 CD4+ clones (donor 1, three; donor 2, five; donor 3, two; and donor 4, five) proliferated to H+,K+-ATPase (MI range, 28–179), but not to H. pylori lysate or porcine albumin. Interestingly, a third group of 13 CD4+ clones was found that proliferated almost equally well to both H+,K+-ATPase and H. pylori lysate, but not to porcine albumin (Fig. 1) .
Figure 1.
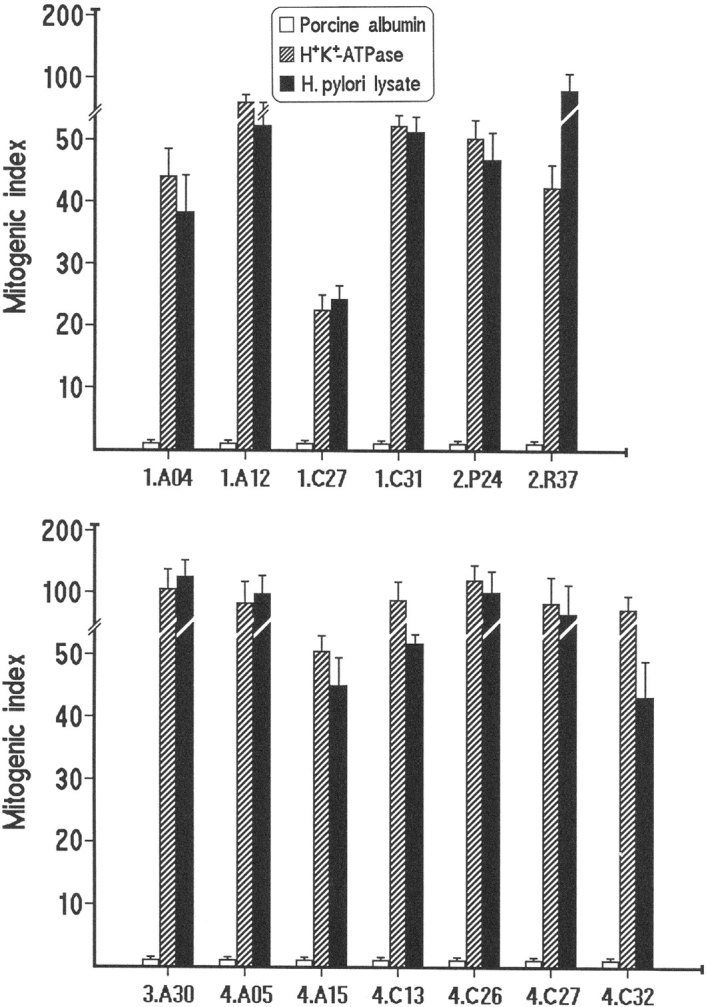
Proliferation to H+,K+-ATPase and H. pylori of gastric T cell clones. T cell blasts from 13 CD4+ clones derived from the gastric mucosa of H. pylori–infected patients with chronic autoimmune gastritis (four from patient 1, two from patient 2, one from patient 3, and six from patient 4) were cocultured with autologous irradiated APCs in the presence of optimal concentrations of H+,K+-ATPase, H. pylori lysate, or porcine albumin as control antigen. Results represent mean values (±SD) of MIs measured in five consecutive experiments.
T cell blasts from each of the 28 H+,K+-ATPase–reactive T cell clones were screened for proliferation in response to 205 overlapping peptides for the α chain and 56 peptides for the β chain of H+,K+-ATPase (Table I). In the series of 15 H+,K+-ATPase–specific clones that failed to proliferate to H. pylori lysate, 6 clones recognized an epitope in the α chain and 9 clones found their epitope in the β chain of the proton pump. Interestingly, the α881–895 epitope was recognized by a couple of clones from donor 2 (2.P02 and 2.P14), in spite of their different expression of TCR-Vβ. Likewise, different TCR-Vβ regions were expressed by the two clones from donor 3 and the three clones from donor 4 that recognized the same epitopes (β231-245 and β76-90, respectively).
Table I.
Epitope Specificity of Gastric T Cell Clones from H. pylori–infected AIG Patients
| T cell clones reactive to H+,K+-ATPase
|
T cell clones reactive to both H+,K+-ATPase and H. pylori lysate
|
|||||
|---|---|---|---|---|---|---|
| Patient | H+,K+-ATPase epitope recognized | Mean MI ± SD | H+,K+-ATPase epitope recognized | Mean MI ± SD | ||
| 1 | C11 (Vβ5.3) | α31-45 | 65 ± 9 | A12 (Vβ19) | α576-590 | 64 ± 10 |
| A01 (Vβ6.7) | β81-95 | 21 ± 2 | C31 (Vβ13) | α621-635 | 79 ± 9 | |
| C26 (Vβ13) | β166-180 | 22 ± 2 | A04 (Vβ13) | α781-795 | 194 ± 16 | |
| C27 (Vβ13) | β216-230 | 57 ± 12 | ||||
| 2 | Q08 (Vβ4) | α1-15 | 22 ± 1 | P24 (Vβ4) | α46-60 | 23 ± 3 |
| P34 (Vβ19) | α151-165 | 134 ± 11 | R37 (Vβ15) | α836-850 | 50 ± 7 | |
| P02 (Vβ8) | α881-895 | 23 ± 2 | ||||
| P14 (Vβ19) | α881-895 | 21 ± 2 | ||||
| R17 (Vβ4) | β111-125 | 29 ± 4 | ||||
| 3 | A42 (Vβ8) | β231-245 | 122 ± 36 | A30 (Vβ13) | α836-850 | 49 ± 6 |
| B46 (Vβ17) | β231-245 | 68 ± 19 | ||||
| 4 | C39 (Vβ9) | α351-365 | 53 ± 4 | A15 (Vβ18) | α181-195 | 39 ± 2 |
| A09 (Vβ23) | β76-90 | 97 ± 12 | C32 (Vβ16) | α241-255 | 87 ± 9 | |
| A33 (Vβ6.7) | β76-90 | 108 ± 21 | C27 (Vβ5.2) | α256-270 | 137 ± 17 | |
| C15 (Vβ22) | β76-90 | 72 ± 13 | C26 (Vβ21.3) | α516-530 | 104 ± 9 | |
| C03 (Vβ16) | β81-95 | 113 ± 19 | A05 (Vβ16) | α621-635 | 99 ± 12 | |
| C13 (Vβ14) | β11-25 | 91 ± 14 | ||||
In the series of 13 clones that proliferated to both H+,K+-ATPase and H. pylori lysate, 11 recognized their epitope in the α chain and 2 clones in the β chain. Two clones from different patients (2.R37 and 3.A30, bearing different TCR-Vβ) recognized the same α836-850 epitope, and two other clones in this series (1.C31 and 4.A05) recognized the same α621-635 epitope. No overlap was found between the H+,K+-ATPase epitopes recognized by clones reactive only to H+,K+-ATPase and the H+,K+-ATPase epitopes recognized by clones able to proliferate to both H+,K+-ATPase and H. pylori lysate (Table I).
Evidence for clonality of the cross-reactive CD4+ T cell clones was provided by the unique products of PCR analysis of TCR-Vβ mRNA expression in clones 1.A12 (Vβ19) and 2.R37 (Vβ15; unpublished data) or by the cytofluorimetric patterns of single TCR-Vβ expression shown by those clones. Each T cell clone was stained by only one of the TCR-Vβ chain–specific monoclonal antibodies, showing a single peak of fluorescence intensity (Fig. 2) .
Figure 2.

TCR Vβ chain repertoire of cross-reactive gastric T cell clones. The clonality of T cell clones reactive to both H+,K+-ATPase and H. pylori lysate was analyzed by a panel of 22 monoclonal antibodies specific for human TCR Vβ chain families, as detailed in Materials and Methods. T cell blasts from each clone were divided in aliquots and stained with each of the monoclonal antibody and the appropriate isotype controls.
Screening for Cross-reactive H. pylori Peptides with Homology to H+,K+-ATPase Epitopes.
Homologies between the 13 H+,K+-ATPase epitopes and peptides present in both genomes of H. pylori J99 and 26695 strains were screened by using a bioinformatic method. For clone 1.A12 reactive to the α576-590 epitope and clone 4.C133 reactive to β11-25, no cross-reactive candidate was predicted in H. pylori J99 or hp26695 ORFs. For clone 1.C27 that proliferated to the β216-230 H+,K+-ATPase epitope, the six predicted H. pylori peptides were synthesized, but none of them induced significant proliferation (MI > 5). It is of note that the H+,K+-ATPase epitopes recognized by the other 10 gastric clones reactive to both H+,K+-ATPase and H. pylori lysate were 100% identical in the pig and human molecules. For each of these 10 gastric clones, a cross-reactive peptide was found in the series (n = 73) of candidate H. pylori peptides we had synthesized (Table II). The two clones from different patients (2.R37 and 3.A30), that shared recognition of the α836–850 H+,K+-ATPase epitope, also shared the cross-reactive recognition of the 11-25 sequence of a lipopolysaccharide biosynthesis protein of H. pylori. The relative potency in inducing T cell clone proliferation of the self and of the corresponding microbial peptides was assessed by comparison of dose–response curves. At 10 nM, self- and cross-reactive peptides were almost equally potent in inducing T cell clone proliferation. At lower doses (such as 1 or 0.1 nM), the MI obtained with the appropriate self peptides was consistently higher (1.4–2-fold) than that obtained with the corresponding microbial peptides (Fig. 3) . However, at concentrations as low as 1 pM, both self- and cross-reactive microbial peptides were still stimulatory. To know the MHC restriction elements required for recognition of self- or cross-reactive epitopes, T cell clones were stimulated by the appropriate H+,K+-ATPase and H. pylori cross-reactive peptides in the presence of irradiated autologous APCs treated with anti–HLA-DR or anti–HLA-DQ monoclonal antibodies. Addition in culture of anti–HLA-DR consistently resulted in virtual abrogation of the proliferative response by T cell clones, whereas anti–HLA-DQ was unable to affect peptide-induced proliferation (Table III).
Table II.
Cross-reactive H+,K+-ATPase and H. pylori Peptides Recognized by Gastric T Cell Clones
| T cell clones (epitope) |
Amino acid sequence recognized
|
H. pylori protein including the cross-reactive peptide (position) |
|
|---|---|---|---|
| H+,K+-ATPase bacterial peptide | MI ± SD | ||
| 1.C31 (α621-635) | IRVIMVTGDHPITAK | 79 ± 9 | |
| VRVDVRRLDHLMNLI | 66 ± 5 | Histidine kinase (264-278) | |
| 1.A04 (α781-795) | NLKKSIAYTLTKNIP | 194 ± 16 | |
| ISNLPYYIATRLVLN | 108 ± 12 | Dimethyl adenosine transferase (99-113) | |
| 2.P24 (α46-60) | KKEMEINDHQLSVAE | 23 ± 3 | |
| LNNYQKENSLYNHNL | 27 ± 2 | Penicillin-binding protein 2 (104-118) | |
| 2.R37 (α836-850) | KAESDIMHLRPRNPK | 50 ± 7 | |
| NMRVFIIHLSPKTCK | 19 ± 2 | LPS biosynthesis protein (11-25) | |
| 3.A30 (α836-850) | KAESDIMHLRPRNPK | 49 ± 6 | |
| NMRVFIIHLSPKTCK | 16 ± 1 | LPS biosynthesis protein (11-25) | |
| 4.A15 (α181-195) | VIRDGDKFQINADQL | 39 ± 2 | |
| VVQGGDKFHAPVLVD | 20 ± 1 | Acetate kinase (93-107) | |
| 4.C32 (α241-255) | CTHESPLETRNIAFF | 87 ± 9 | |
| VIQIGPMPTPAIAFL | 75 ± 8 | Phosphoglucosamine mutase (70-84) | |
| 4.C27 (α256-270) | STMCLEGTAQGLVVN | 137 ± 17 | |
| ALDSLEKVVARLVVK | 34 ± 2 | VirB4 homologue (78-92) | |
| 4.C26 (α516-530) | VMKGAPERVLERCSS | 104 ± 9 | |
| VFKGIPGLSLEAVEK | 47 ± 4 | GidA (571-585) | |
| 4.A05 (α621-635) | IRVIMVTGDHPITAK | 99 ± 12 | |
| IRIVKTTGDKILDAP | 51 ± 6 | Porphobilinogen deaminase (35-49) | |
For each gastric T cell clone reactive to both H+,K+-ATPase and H. pylori, a single H. pylori cross-reactive peptide was identified. Identical amino acid residues in the recognized H+,K+-ATPase peptide and the cross-reactive H. pylori peptide are in boldface. H. pylori proteins containing a cross-reactive epitope recognized by one of the T cell clones in this study are as follows: histidine kinase [HP0392]; dimethyl adenosine transferase [HP1431]; penicillin-binding protein 2 [HP1565]; LPS (lipopolysaccharide) biosynthesis protein [HP0805]; acetate kinase [HP0903]; phosphoglucosamine mutase [HP0075]; VirB4 homologue [HP0017]; GidA, glucose-inhibited division protein A [HP0213]; and porphobilinogen deaminase [HP0237]. Codes in brackets indicate the annotated gene number of H. pylori 26695.
Figure 3.

Dose–response effect of graded concentrations of the self (closed symbols) and the corresponding bacterial peptides (open symbols) on the proliferative response of gastric T cell clones reactive to both H+,K+-ATPase and H. pylori lysate. Results represent mean values ± SD of 6 representative out of 10 T cell clones tested.
Table III.
Effect of Addition in Culture of Anti-DR or Anti-DQ Monoclonal Antibodies on the Proliferative Response to Self and Cross-reactive Peptides by Gastric T Cell Clones Reactive to both H+,K+-ATPase and H. pylori Lysate
| T cell clones |
Proliferative responsea in the presence of:
|
|||
|---|---|---|---|---|
| Peptide added in culture | Isotype control | Anti-DR | Anti-DQ | |
| 1.C31 | H+,K+-ATPase α621-635 | 110 ± 7 | 1.6 ± 0.1 | 115 ± 9 |
| Histidine kinase, 264-278 | 101 ± 5 | 1.5 ± 0.1 | 100 ± 7 | |
| 1.A04 | H+,K+-ATPase α781-795 | 173 ± 10 | 1.1 ± 0.1 | 160 ± 8 |
| Dimethyl adenosine transferase, 99-113 | 148 ± 11 | 1.0 ± 0.1 | 151 ± 10 | |
| 2.P24 | H+,K+-ATPase α46-60 | 31 ± 2 | 1.2 ± 0.1 | 30 ± 3 |
| Penicillin-binding protein 2, 104-118 | 38 ± 2 | 0.9 ± 0.1 | 38 ± 3 | |
| 2.R37 | H+,K+-ATPase α836-850 | 61 ± 4 | 1.1 ± 0.1 | 58 ± 5 |
| LPS biosynthesis protein, 11-25 | 32 ± 3 | 1.2 ± 0.1 | 35 ± 4 | |
| 3.A30 | H+,K+-ATPase α836-850 | 42 ± 4 | 1.0 ± 0.1 | 40 ± 5 |
| LPS biosynthesis protein, 11-25 | 22 ± 2 | 1.3 ± 0.1 | 23 ± 1 | |
| 4.A15 | H+,K+-ATPase α181-195 | 48 ± 3 | 1.4 ± 0.1 | 49 ± 4 |
| Acetate kinase, 93-107 | 29 ± 2 | 1.0 ± 0.1 | 30 ± 2 | |
| 4.C32 | H+,K+-ATPase α241-255 | 98 ± 9 | 1.1 ± 0.1 | 96 ± 8 |
| Phosphoglucosamine mutase, 70-84 | 89 ± 9 | 1.2 ± 0.1 | 90 ± 10 | |
| 4.C27 | H+,K+-ATPase α256-270 | 118 ± 10 | 1.9 ± 0.2 | 106 ± 11 |
| VirB4 homologue, 78-92 | 59 ± 4 | 0.9 ± 0.1 | 58 ± 5 | |
| 4.C26 | H+,K+-ATPase α516-530 | 127 ± 9 | 1.4 ± 0.1 | 126 ± 10 |
| GidA, 571-585 | 56 ± 6 | 1.3 ± 0.1 | 55 ± 7 | |
| 4.A05 | H+,K+-ATPase α621-635 | 97 ± 9 | 1.0 ± 0.1 | 96 ± 8 |
| Porphobilinogen deaminase, 35-49 | 60 ± 6 | 1.1 ± 0.1 | 62 ± 5 | |
Values are MI ± SD.
GidA, glucose-inhibited division protein A.
Cytokine Production Induced by H+,K+-ATPase and H. pylori Cross-reactive Peptides.
Mapping of the H+,K+-ATPase peptide specificity and the cross-reactive recognition of H. pylori peptides based on proliferative response was accomplished with the assessment of IFN-γ, IL-4, and IL-5 production induced by the relevant peptides. Upon appropriate stimulation, all gastric clones produced IFN-γ, but neither IL-4 nor IL-5, thus showing a Th1 profile (Fig. 4) . In contrast, either H+,K+-ATPase peptides or H. pylori cross-reactive peptides that failed to induce proliferation also failed to induce cytokine production. From these data, we concluded that the appropriate H. pylori cross-reactive peptides are as powerful as the specific H+,K+-ATPase peptides in inducing a number of gastric T cells to proliferate and to express their Th1 functional profile. All gastric clones reactive to both H+,K+-ATPase and H. pylori lysate expressed effector functions typical of Th1 cells, such as perforin-mediated cytotoxicity (range of specific 51Cr release, 36–67%) and Fas–Fas ligand-mediated proapoptotic activity (range of specific 51Cr release, 29–51%), which was substantially inhibited (range 37–69%) by an anti-Fas antagonistic antibody. Likewise, gastric clones reactive only to H+,K+-ATPase were able to express both perforin-mediated cytotoxicity (range of specific 51Cr release, 24–72%) and proapoptotic activity in the same target cells (range of specific 51Cr release, 22–59%).
Figure 4.

IFN-γ production by cross-reactive gastric T cell clones. Cytokine production was assessed in response to H. pylori lysate, H+,K+-ATPase, the H+,K+-ATPase epitope and the cross-reactive H. pylori epitope that induced T cell clone proliferation. For each clone, controls included medium alone, a couple of H+,K+-ATPase peptides flanking the stimulatory epitope and the series of candidate H. pylori epitopes that failed to induce T cell clone proliferation. Numbers in parentheses indicate the location of cross-reactive epitopes within the indicated H. pylori proteins. Results represent mean values ± SD of 4 representative out of 10 T cell clones tested.
Discussion
The presence in H. pylori–infected AIG patients of gastric T cells reactive to H. pylori antigens is in agreement with earlier demonstration of H. pylori–specific T cells in the gastric mucosa of H. pylori–infected individuals without AIG (28). Likewise, the detection of H+,K+-ATPase-specific T cells in the gastric mucosa of AIG patients confirms previous observation in H. pylori noninfected patients with AIG (18). The important point of the present paper is the demonstration that all four H. pylori–infected AIG patients harbored in their gastric mucosa in vivo–activated T cells that reacted to both H+,K+-ATPase and H. pylori. The analysis of the submolecular specificity of T cell clones reactive only to H+,K+-ATPase and of clones reactive to both H+,K+-ATPase and H. pylori showed that most of the former (9 out of 15) recognized their epitope in the β chain of the enzyme, whereas most of the latter (11 out of 13) found their epitope in the α chain of H+,K+-ATPase. Therefore, a number of H+,K+-ATPase epitopes are “private,” whereas other epitopes, mainly in the α chain, are similar to, or cross-reactive with, epitopes of H. pylori antigens.
For each of 10 cross-reactive gastric clones, bioinformatics provided us with a cross-reactive H. pylori epitope able to induce significant T cell clone proliferation. Clones R37 from patient 2 and clone A30 from patient 3, which shared recognition of the α836–850 H+,K+-ATPase epitope, also shared cross-reactivity with the 11-25 peptide of a lipopolysaccharide biosynthesis protein of H. pylori. In contrast, for clone C31 from patient 1 and clone A05 from patient 4, both reactive to α621–635 H+,K+-ATPase, the bioinformatic method had predicted two different series of 11 and 8 cross-reactive candidates, respectively. Indeed, clone 1.C31 reacted quite well only to the 264-278 peptide of histidine kinase, whereas clone 4.A05 showed cross-recognition of the 35-49 epitope of porphobilinogen deaminase of H. pylori. In summary, our search led to the identification of nine H. pylori proteins, each harboring a T cell epitope suitable for cross-reaction with T cell epitopes of gastric H+,K+-ATPase α chain.
T cell recognition of cross-reactive H. pylori epitopes resulted in both proliferation and expression of functional properties by cross-reactive T cell clones. In all clones, the Th1 cytokine profile expressed upon stimulation with cross-reactive H. pylori peptides paralleled that disclosed by stimulation with either bacterial lysate or entire H+,K+-ATPase or the appropriate H+,K+-ATPase epitopes. These data suggest that cross-reactive H. pylori peptides represent signals powerful enough to activate the functional program of gastric cross-reactive Th1 cells. Upon mitogen stimulation, all cross-reactive gastric clones expressed both perforin-mediated cytolysis and induction of Fas–Fas ligand-mediated apoptosis in target cells. Thus, it is tempting to hypothesize that in the inflammatory setting in which cross-reactive T cell clones are activated, parietal cells may express APC functions, becoming target, at the same time, of the cytotoxic and proapoptotic activity of cross-reactive gastric T cells. The end point of this process would be gastric corpus atrophy and hypochloridria.
One may ask whether the T cells which cross-react with H. pylori antigens are simply a chance finding and what is the probability of any T cell response showing some cross-reactivity with the total protein mixture from H. pylori. Based on the reported degeneracy of both TCR and MHC binding (29, 30), there is indeed a theoretical chance of such coincidental cross-reactivity. However, such a promiscuity of T cell responses to H. pylori lysate could not be found in a series of 206 CD4+ human T cell clones isolated from atherosclerotic plaques, which included 46 clones that recognized Chlamydia pneumoniae antigens (31), although one might expect more similarity between the proteomes of both bacteria than between the human proteome and that of H. pylori.
T cell recognition of a peptide depends on anchor residues involved in binding to the MHC molecule as well as on TCR contact residues. This suggests that the self peptide and the bacterial peptide recognized by a single T cell, should be considerably homologous in amino acid sequence. However, degeneracy in both TCR (32) and MHC binding–motives (33) reduces this sequence-specific requirement to only a few crucial residues, and it has been demonstrated that in the animal model of myocarditis cross-recognition of the autoantigen (myosin) and the microbial peptide (Chlamydia cysteine-rich outer membrane protein) depends on only four identical residues in the 16 amino acid epitope sequence (34). Another mechanism of TCR cross-recognition may work. Recently, MHC-based molecular mimicry has been reported to underlie TCR cross-reactivity in multiple sclerosis (35). A T cell clone from a patient recognizes both myelin basic protein amino acids 85–99 and Epstein-Barr virus (EBV) DNA polymerase peptide EBV627-641, but recognition of these peptides is restricted by two different DR2 molecules (i.e., DRB1*1501 and DRB5*0101, respectively). Crystal structure determination revealed structural similarities of both DR–peptide complexes at the surface presented for TCR recognition, thus explaining the mechanism that may underlie the mimicry between EBV and myelin basic protein.
Blocking experiments with anti-DR and anti-DQ antibodies showed that DR represents the MHC restriction element. However, because all our patients are heterozygous for DR alleles, we cannot exclude that the aforementioned mechanism of TCR cross-reactivity is responsible for the mimicry between H. pylori and H+,K+-ATPase observed here.
None of the bacterial epitopes recognized by the cross-reactive T cell clones in the present paper belong to H. pylori immunodominant proteins (i.e., CagA, VacA, and urease), which have been identified previously as targets of the majority of gastric T cells in H. pylori–infected patients with chronic antral gastritis (28) and peptic ulcer (26). It remains unknown to what extent the H. pylori epitopes recognized by the cross-reactive T cells described here are relevant to bacterial infection.
A clear example of epitope mimicry in humans is Lyme arthritis, in which Borrelia burgdorferi disseminates to multiple tissues, including joints. In the synovia of patients with specific MHC class II haplotypes, activation of Th1 cells reactive to the 165–173 peptide of the outer surface protein A of B. burgdorferi occurs (29, 30). Such an epitope is similar to the L332–340 peptide of the human leukocyte function-associated antigen 1α, whose expression is up-regulated on synoviocytes by the Th1-derived IFN-γ (36–38). However, some aspects of this model of human molecular mimicry are questioned because it is still unknown what precipitates the disease several months after the borrelial infection, whether the human leukocyte function-associated antigen 1α epitopes are actually being presented in the joints, and what kind of APCs would be involved (5).
With regard to the question of whether it was H. pylori infection or H. pylori–independent gastric autoimmunity that initiated disease, three hypotheses can be suggested. First, our patients, having inherited MHC haplotypes that predispose to organ-specific autoimmunity (39), already had undiagnosed or subclinical AIG and H. pylori infection, by providing a number of epitopes cross-reactive to H+,K+-ATPase, caused a Th1-mediated inflammation leading to the expansion of both cross-reactive and single (H+,K+-ATPase)-reactive gastric T cells. The outcome was increased parietal cell destruction and gastric atrophy. Second, H. pylori was the initiating factor, and primary activation of gastric Th1 cells reactive to H. pylori peptides that cross-react with H+,K+-ATPase resulted in an inflammatory process in which T cell–derived IFN-γ allowed H+,K+-ATPase-bearing parietal cells to act as APCs and to become targets of cross-reactive epitope recognition and killing and/or apoptotic suicide. Apoptotic parietal cells would allow cross-priming of T cells specific for private H+,K+-ATPase epitopes, ultimately leading to full blown AIG by epitope spreading. Third, H. pylori infection was an epiphenomenon, playing no role in the natural history of the disease.
Our data fulfil most of the criteria proposed for assessing a case of molecular mimicry (1, 5, 12). In our patients, there was a temporal association between clinical and serological evidence of AIG and H. pylori infection, at least at the time of culture of their in vivo–activated gastric T cells. Because H. pylori is commonly acquired in young life (19), the infection preceded symptoms of AIG that usually arise later in life. Taking into account the wide diffusion of H. pylori (19), one may suspect that some AIG patients, who were found H. pylori–negative at the time of their diagnosis of AIG, might have harbored the bacterium previously, and H. pylori was lost while mucosal atrophy was ongoing. Strikingly, all our AIG patients lost H. pylori during the 4-yr study period, possibly due to hypochloridria that made their gastric environment no more appreciated by the bacterium. The possibility that H. pylori gets lost due to increasing gastric atrophy and hypochloridria has already been reported (40).
We have identified a quite broad repertoire of culprit epitopes in both the pathogen and in the self gastric protein associated with AIG. The microbial cross-reactive epitopes were able to elicit vigorous responses in the same gastric T cells that responded at comparable levels to both the corresponding self H+,K+-ATPase epitopes and the entire self protein. Finally, cross-reactive T cell clones quantitatively represented a significant component of the T cell response at gastric level during the autoimmune disease and the concomitant H. pylori infection. This would argue against the possibility that the detection at gastric level of autoreactive, cytotoxic, and proapoptotic Th1 cells that cross-react to H. pylori epitopes is simply an epiphenomenon.
Together, our results support the idea that in genetically susceptible individuals, H. pylori infection triggers or accelerates the development of gastric autoimmunity via molecular mimicry.
Acknowledgments
This work was supported by grants from the Italian Ministry of University and Research, the Ministry of Health (Istituto Superiore di Sanità), and the Associazione Italiana per la Ricerca sul Cancro.
A. Amedei and M.P. Bergman contributed equally to this work.
Abbreviations used in this paper: AIG, autoimmune chronic gastritis; ATPase, adenosine triphosphatase; BLAST, basic local alignment search tool; MI, mitogenic index; ORF, open reading frame; PCA, parietal cell autoantibody.
References
- 1.Rose, N.R., and C. Bona. 1993. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol. Today. 14:426–430. [DOI] [PubMed] [Google Scholar]
- 2.Oldstone, M.B.A. 1998. Molecular mimicry and immune-mediated diseases. FASEB J. 12:1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Theofilopoulos, A.N., and D.H. Kono. 1998. Mechanisms and genetics of autoimmunity. Ann. NY Acad. Sci. 841:225–235. [DOI] [PubMed] [Google Scholar]
- 4.Lori, J.A., and R.D. Inman. 1999. Molecular mimicry and autoimmunity. N. Engl. J. Med. 341:2068–2074. [DOI] [PubMed] [Google Scholar]
- 5.Benoist, C., and D. Mathis. 2001. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat. Immunol. 2:797–801. [DOI] [PubMed] [Google Scholar]
- 6.Wucherpfennig, K.W. 2001. Mechanisms for the induction of autoimmunity by infectious agents. J. Clin. Invest. 108:1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schrer, M.T., L. Ignatowicz, G.M. Winslow, J.W. Kappler, and P. Marrack. 1993. Superantigens: bacterial and viral proteins that manipulate the immune system. Annu. Rev. Cell Biol. 9:101–128. [DOI] [PubMed] [Google Scholar]
- 8.Lehmann, P.V., T. Forsthuber, A. Miller, and E.E. Sercarz. 1992. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 358:155–157. [DOI] [PubMed] [Google Scholar]
- 9.Miller, S.D., C.L. Vanderlugt, W.S. Begolka, W. Pao, R.L. Yauch, K.L. Neville, Y. Katz-Levy, A. Carrizosa, and B.S. Kim. 1997. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 3:1133–1136. [DOI] [PubMed] [Google Scholar]
- 10.Murali-Krishna, K., J.D. Altman, M. Suresh, D.J. Sourdive, A.J. Zajac, J.D. Miller, J. Slansky, and R. Ahmed. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 8:177–187. [DOI] [PubMed] [Google Scholar]
- 11.Bachmaier, K., N. Neu, L.M. de la Maza, S. Pal, A. Hessel, and J.M. Penninger. 1999. Chlamydia infections and heart disease linked through antigenic mimicry. Science. 283:1335–1339. [DOI] [PubMed] [Google Scholar]
- 12.Rose, N.R., and I.R. Mackay. 2000. Molecular mimicry: a critical look at exemplary instances in human diseases. Cell. Mol. Life Sci. 57:542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemmer, B., B. Gran, Y. Zhao, A. Marques, J. Pascal, A. Tzou, T. Kondo, I. Cortese, B. Bielekova, S.E. Straus, et al. 1999. Identification of candidate T-cell epitopes and molecular mimics in chronic Lyme disease. Nat. Med. 5:1375–1382. [DOI] [PubMed] [Google Scholar]
- 14.Martin, R., B. Gran, Y. Zhao, S. Markovic-Plese, B. Bielekova, A. Marques, M.H. Sung, B. Hemmer, R. Simon, H.F. McFarland, and C. Pinilla. 2001. Molecular mimicry and antigen-specific T cell responses in multiple sclerosis and chronic CNS Lyme disease. J. Autoimmun. 16:187–192. [DOI] [PubMed] [Google Scholar]
- 15.Toh, B.H., I.R. van Driel, and P.A. Gleeson. 1997. Pernicious anemia. N. Engl. J. Med. 337:1441–1448. [DOI] [PubMed] [Google Scholar]
- 16.Karlsson, F.A., P. Burman, L. Loof, and S. Mardh. 1988. Major parietal cell antigen in autoimmune gastritis with pernicious anemia is the acid-producing H+,K+-adenosine triphosphatase of the stomach. J. Clin. Invest. 81:475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toh, B.H., I.R. van Driel, and P.A. Gleeson. 1992. Autoimmune gastritis: tolerance and autoimmunity to the gastric H+/K+ ATPase (proton pump). Autoimmunity. 13:165–172. [DOI] [PubMed] [Google Scholar]
- 18.D'Elios, M.M., M.P. Bergman, A. Azzurri, A. Amedei, M. Benagiano, J.J. De Pont, F. Cianchi, C.M. Vandenbroucke-Grauls, S. Romagnani, B.J. Appelmelk, and G. Del Prete. 2001. H(+),K(+)-ATPase (proton pump) is the target autoantigen of Th1-type cytotoxic T cells in autoimmune gastritis. Gastroenterology. 120:377–386. [DOI] [PubMed] [Google Scholar]
- 19.Suerbaum, S., and P. Michetti. 2002. Helicobacter pylori infection. N. Engl. J. Med. 347:1175–1186. [DOI] [PubMed] [Google Scholar]
- 20.Appelmelk, B.J., G. Faller, D. Claeys, T. Kirchner, and C.M.J.E. Vandenbroucke-Grauls. 1998. Bugs on trial: the case of Helicobacter pylori and autoimmunity. Immunol. Today. 19:296–299. [DOI] [PubMed] [Google Scholar]
- 21.Faller, G., M. Winter, H. Steininger, N. Lehn, A. Meining, E. Bayerdorffer, and T. Kirchner. 1999. Decrease of antigastric autoantibodies in Helicobacter pylori gastritis after cure of infection. Pathol. Res. Pract. 195:243–246. [DOI] [PubMed] [Google Scholar]
- 22.Swarts, H.G., T.J. Van Uem, S. Hoving, J.A. Fransen, and J.J. De Pont. 1991. Effect of free fatty acids and detergents on H,K-ATPase. The steady-state ATP phosphorylation level and the orientation of the enzyme in membrane preparations. Biochim. Biophys. Acta. 1070:283–292. [DOI] [PubMed] [Google Scholar]
- 23.Maeda, M., J. Ishizaki, and M. Futai. 1988. cDNA cloning and sequence determination of pig gastric (H+,K+)-ATPase. Biochem. Biophys. Res. Commun. 157:203–206. [DOI] [PubMed] [Google Scholar]
- 24.Van der Zee, R., S.M. Anderton, C.A.F. Buskens, E. Alonso de Velasco, and W. Van Eden. 1995. Heat shock protein T cell epitopes as immunogenic carriers in subunit vaccines. Peptides 1994, Proceedings of the Twenty-Third Peptide Symposium. H.L.S. Maya, editor. ESCOM, Leiden, Netherlands. 842.
- 25.Sing, H., and G.P.S. Raghava. 2001. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 17:1236–1237. [DOI] [PubMed] [Google Scholar]
- 26.D'Elios, M.M., M. Manghetti, M. De Carli, F. Costa, C.T. Baldari, D. Burroni, J.L. Telford, S. Romagnani, and G. Del Prete. 1997. Th1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J. Immunol. 158:962–967. [PubMed] [Google Scholar]
- 27.Vergelli, M., B. Hemmer, P.A. Muraro, L. Tranquill, W.E. Biddison, A. Sarin, H.F. McFarland, and R. Martin. 1997. Human autoreactive CD4 T cell clones use perforin or Fas/Fas ligand-mediated pathways for target cell lysis. J. Immunol. 158:2756–2761. [PubMed] [Google Scholar]
- 28.D'Elios, M.M., M. Manghetti, F. Almerigogna, A. Amedei, F. Costa, D. Burroni, C.T. Baldari, S. Romagnani, J.L. Telford, and G. Del Prete. 1997. Different cytokine profile and antigen-specificity repertoire in Helicobacter pylori-specific T cell clones from the antrum of chronic gastritis patients with or without peptic ulcer. Eur. J. Immunol. 27:1751–1755. [DOI] [PubMed] [Google Scholar]
- 29.Kersh, G.J., and P.M. Allen. 1996. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J. Exp. Med. 184:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemmer, B., M. Vergelli, C. Pinilla, R. Houghten, and R. Martin. 1998. Probing degeneracy in T-cell recognition using combinatorial peptide libraries. Immunol. Today. 19:163–168. [DOI] [PubMed] [Google Scholar]
- 31.Benagiano, M., A. Azzurri, A. Ciervo, A. Amedei, C. Tamburini, M. Ferrari, J.L. Telford, C.T. Baldari, S. Romagnani, A. Cassone, et al. 2003. T helper type 1 lymphocytes drive inflammation in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA. 100:6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wucherpfennig, K.W., D.A. Hafler, and J.L. Strominger. 1995. Structure of human T-cell receptors specific for an immunodominant myelin basic protein peptide: positioning of T-cell receptors on HLA-DR2/peptide complexes. Proc. Natl. Acad. Sci. USA. 92:8896–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wucherpfennig, K.W., A. Sette, S. Southwood, C. Oseroff, M. Matsui, J.L. Strominger, and D.A. Hafler. 1994. Structural requirements for binding of an immunodominant myelin basic protein peptide to DR2 isotypes and for its recognition by human T cell clones. J. Exp. Med. 179:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bachmaier, K., N. Neu, L.M. de la Maza, S. Pal, A. Hessel, and J.M. Penninger. 1999. Chlamydia infections and heart disease linked through antigenic mimicry. Science. 283:1335–1339. [DOI] [PubMed] [Google Scholar]
- 35.Lang, H.L., H. Jacobsen, S. Ikemizu, C. Andersson, K. Harlos, L. Madsen, P. Hjorth, L. Sondergaard, A. Svejgaard, K. Wucherpfennig, et al. 2002. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 3:940–943. [DOI] [PubMed] [Google Scholar]
- 36.Lengl-Janssen, B., A.F. Strauss, A.C. Steere, and T. Kamradt. 1994. The T helper cell response in Lyme arthritis: differential recognition of Borrelia burgdorferi outer surface protein A in patients with treatment-resistant or treatment-responsive Lyme arthritis. J. Exp. Med. 180:2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gross, D.M., T. Forsthuber, M. Tary-Lehmann, C. Etling, K. Ito, Z.A. Nagy, J.A. Field, A.C. Steere, and B.T. Huber. 1998. Identification of LFA-1 as a candidate autoantigen in treatment-resistant Lyme arthritis. Science. 281:703–706. [DOI] [PubMed] [Google Scholar]
- 38.Akin, E., J. Aversa, and A.C. Steere. 2001. Expression of adhesion molecules in synovia of patients with treatment-resistant Lyme arthritis. Infect. Immun. 69:1774–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDevitt, H.O. 2000. Discovering the role of the major histocompatibility complex in the immune response. Annu. Rev. Immunol. 18:1–17. [DOI] [PubMed] [Google Scholar]
- 40.Ma, J.-Y., K. Borch, S.E. Sjöstrand, L. Janzon, and S. Mårdh. 1994. Positive correlation between H,K-adenosine triphosphatase autoantibodies and Helicobacter pylori antibodies in patients with pernicious anemia. Scand. J. Gastroenterol. 29:961–965. [DOI] [PubMed] [Google Scholar]