

Abstract
Pax5 activity is enhanced in activated B cells and is essential for class switch recombination (CSR). We show that inhibitor of differentiation (Id)2 suppresses CSR by repressing the gene expression of activation-induced cytidine deaminase (AID), which has been shown to be indispensable for CSR. Furthermore, a putative regulatory region of AID contains E2A- and Pax5-binding sites, and the latter site is indispensable for AID gene expression. Moreover, the DNA-binding activity of Pax5 is decreased in Id2-overexpressing B cells and enhanced in Id2−/− B cells. The kinetics of Pax5, but not E2A, occupancy to AID locus is the same as AID expression in primary B cells. Finally, enforced expression of Pax5 induces AID transcription in pro–B cell lines. Our results provide evidence that the balance between Pax5 and Id2 activities has a key role in AID gene expression.
Keywords: B cell activation, class switch recombination, chromatin immunoprecipitation, histone acetylation, transcription
Introduction
Inhibitor of differentiation (Id) proteins play regulatory roles in the coordination of proliferation and differentiation (1, 2). They regulate differentiation through sequestration of basic helix-loop-helix transcription factors, so-called E proteins. On the other hand, Id proteins have also been shown to interact with other families of transcription factors, such as Pax and Ets proteins, and inhibit their DNA-binding functions (3, 4). These observations suggest that Id proteins play a more widespread regulatory role in cell differentiation and function. E2A and Pax5 are essential for the normal development of B cells (5). B cell differentiation is completely blocked in mice deficient for either of these factors (5–7). Several studies have suggested another essential role of E2A and Pax5 in mature B cell function (8, 9). In activated B cells, the levels of E2A protein and Pax5 transcript are highly augmented (9, 10). Furthermore, enforced expression of Id2, an inhibitor of E2A, or Blimp1, a repressor of Pax5, completely blocks class switch recombination (CSR) (11). Recent studies demonstrated that activation-induced cytidine deaminase (AID) is indispensable for somatic hypermutation (SHM) and CSR of immunoglobulin genes (12). These processes result in the production of high affinity antibodies against the immunizing antigen and modification of effector functions of antibodies (13). On the other hand, little is known about a possible inhibitory system for CSR that prevents an over-response of B cells. Here we show that enforced expression of Id2 reduced AID expression and inhibited CSR. By comparing human and murine sequences, we found a strikingly homologous region within 400 bp upstream of the AID gene in which there is a Pax5-binding site that is indispensable for AID gene expression. Moreover, a Pax5-containing DNA-binding complex at the AID promoter was reduced by Id2 overexpression and enhanced in Id2−/− B cells. Furthermore, we found that Pax5 was recruited to the AID locus after 2 d of stimulation of B cells in vitro, at which time AID transcription was initiated. Finally, we demonstrated that enforced expression of Pax5 but not E2A induced endogenous AID gene expression in pro–B-like cell lines. This effect was cancelled by additional introduction of Id2. Our results provide evidence that Id2 plays an important role in modulating Pax5 activity in regulation of AID expression that is functionally antagonized by Id2. This result further suggests a fundamental role in differentiation by antagonizing several sets of master gene activities.
Materials and Methods
Mice.
Id2−/− mice on the 129/Sv genetic background were maintained in specific pathogen-free conditions and used at 7–10 wk of age.
Cell Lines.
The pro–B cell line BaF/3 and the murine B cell line M12 were cultured in RPMI1640 medium containing 10% FBS and 50 μM 2-mercaptoethanol with mouse IL-3 (BaF/3) or without IL-3 (M12). IL-3 was prepared as described previously (14). Phoenix Eco cells (15) were cultured according to the protocol at http://www.uib.no/mbi/nolan/NL-phnxr.html.
Plasmids.
pcDNA-Pax5 and pcDNA-E47 were constructed replacing the fragment from pCXN2-Pax5, pCMVE47 (16, 17) into the pcDNA3 vector. To generate pMX-IRES-hCD4, the IRES-GFP fragment of pMX-IRES-GFP was replaced by the IRES-hCD4 fragment (Miltenyi Biotech). Id2, Pax5, and E47 cDNAs were cloned into pMX-IRES-hCD4. A BssHII site was introduced into the multiple cloning site of pGL3 basic vector (Promega).
Retrovirus Preparation and Infection.
Retroviruses were produced by transfecting pMX plasmids into the ecotropic Phoenix packaging cell line (15). After overnight stimulation with LPS, purified splenic B cells (18) were spin infected with the virus-containing supernatant in the presence of 10 μg/ml DOTAP and cultured with fresh complete RPMI medium plus cytokines (9).
Primers Used for RT-PCR.
The following primers were used for RT-PCR: AID, sense, 5′-ACATCTCAGACTGGGACCTG-3′ and antisense, 5′-TCAAAATCCCAACATACGAAATG-3′; GAPDH, sense, 5′-CCATCACCATCTTCCAGGAG-3′ and antisense: 5′-CCTGCTTCACCACCTTCTTG-3′; and Pax5, sense, 5′-TATTGTCACAGGCCGAGACTTGGCGAGC-3′ and antisense, 5′-CCTCGGGCTGCAGGGCTGTAATAGT-3′. Quantification of band intensity was performed using the NIH image program.
Cloning and Sequencing of Genomic Fragments of the AID Gene.
Genomic fragments of the AID gene were obtained from a mouse genomic library, TT2, using an AID cDNA probe. The probe used for screening was generated by RT-PCR using the following primers: sense, 5′-GGCACGAGCAGCACTGAAGCAGCC-3′ and antisense, 5′-GGGCGGCCGCTCAAAATCCCAACATACGAAATG-3′. The 5′-flanking region, exon 1, intron 1, and exon 2 of the AID gene were sequenced using a BigDye-terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems).
Luciferase Reporter Gene Transactivation Assay.
A series of reporter constructs was generated by PCR using an AID genomic fragment as a template and cloned into a modified pGL3 basic vector. Various mutations were introduced using a site-directed mutagenesis kit. 3 × 106 BaF/3 cells were washed twice with PBS, mixed with plasmid DNA in 400 μl of PBS, and electroporated at 960 μF, 600 V/cm. Transfectants were harvested after 18 h of incubation. Luciferase assays were performed as described previously (19). Firefly luciferase activity was normalized by Renilla luciferase activity. AID promoter constructs were amplified using the 3′ antisense primer 5′-CTCGCGCGCTGTCCATATCGGTCTC-3′ with the following 5′ primers: 0.9P, sense, 5′-CGGGGTACCGATCAGGGTTCAGATCCC-3′; 0.3P sense, 5′-CGGGGTACCCCCGGGACCACACACACACA-3′; 0.2P, sense, 5′-CGGGGTACCCGAAGGAAGTTGGACA-3′; 0.1P, sense, 5′-CGGGGTACCGACCCAACCCAGGAGG-3′; and 0.02P, sense, 5′-CGGGGTACCGTGCTCTGTCACACAA-3′. The primers used for site-directed mutagenesis were as follows: mP1, sense, 5′-CTATCGATAGGTACCGAAAAAAAAAAGGAAAAAGATGTTGGATACCTGG-3′ and antisense 5′-CCAGGTATCCAACATCTTTTTCCTTTTTTTTTTCGGTACCTATC-GATAG-3′; mP2, sense, 5′-CCAGGAGGCAGATGTTAAATAAATAATAATAGTGATGCTGTCGTG-3′ and antisense, 5′-CACGACAGCATCACTATTATTATTTATTTAACATCTGCCTCCTGG-3′; mP3, sense, 5′- GTAGTGATGCTGTCGTAAAAAAGGAAAAAACAAGAGCAAGCTCAG-3′ and antisense, 5′-CTGAGCTTGCTCTTGTTTTTTCCTTTTTTAC-GACAGCATCACTAC-3′; mP4, sense, 5′-GCAAGCTCAG-ATTTGAATAAAAAAAAAAAGTGCTCTGTCACACAAC-AGC-3′ and antisense, 5′-GCTGTTGTGTGACAGAGCA-CTTTTTTTTTTTATTCAAATCTGAGCTTGC-3′. Three independent clones of each construct were sequenced and used for experiments. The primers used for CR1 and CR2 fragments were as follows: Bam-CR1F, 5′- CGGGATCCCTGTGTTGCTAGAGATAGGAAG-3′; Bam-CR1R, 5′-CGGGATCCCCAAACGGAAGTGAGCTCAAGT-3′; Bam-CR2F, 5′-CGGGATCCGGGTCCGAGAGAAATGCATGAG-3′; Bam-CR2R, 5′-CGGGATCCATTATGAAGGAAAGGTATCCTT-3′.
Immunoblot Analysis.
Cell extracts were prepared from primary B cells or 293T cells transfected with indicated expression vectors for 48 h, and protein amounts were quantified with Micro BCR Protein Assay Reagent (Pierce Chemical Co.). For immunoblotting, we used anti-E47 antibody (G127–32; BD Biosciences), anti-Pax-5 (N19; Santa Cruz Biotechnology, Inc.), and anti–β-actin (AC74; Sigma-Aldrich).
Electrophoretic Mobility Shift Assay.
Nuclear extracts of splenic B cells were prepared after incubation with LPS (Sigma-Aldrich) plus IL4 (Peprotech) (18) and protein amounts quantified with Micro BCA Protein Assay Reagent (Pierce Chemical Co.). Double stranded oligoprobes were end labeled using T4 polynucleotide kinase and purified over a Sepharose G50 column. Nuclear extracts and labeled oligoprobes were used in a gel shift assay as described (17) with slight modifications. For the super shift experiment, nuclear extract was incubated with 5 μg of a goat anti–mouse Pax5 antibody (N-19; Santa Cruz Biotechnology, Inc.), designated antibody 1, or a mouse anti-Pax5 antibody (BD610862; BD Biosciences), designated antibody 2, or a mouse anti-E2A antibody (Yae; Santa Cruz Biotechnology, Inc.) for 20 min at room temperature before the labeled probe was added. The sequences of the oligoprobes were as follows: CD19 (competitor), 5′-CCCCCGCAGACACCCATGGTTGAGTGCCCTCCAGGCCCCTGCC-3′ and 5′-CAGGCAGGGGCCTGGAGGGCACTCAACCATGGGTGTCTGCGG-3′; Pa3, 5′-GATGCTGTCGTGGGGGAGGAGCCCACAAGAGCAAGCTCAG-3′ and 5′-CTGAGCTTGCTCTTGTGGGCTCCTCCCCCACGACAGCATC-3′; Pa3-m1, 5′-GATGCTGTCGTAAAAAAGGAGCCCACAAGAGCAAGCTCAG-3′ and 5′-CTGAGCTTGCTCTTGTGGGCTCCTTTTTTACGACAGCATC-3′; Pa3-m2, 5′-GATGCTGTCGTGGGGGAGGAAAAAACAAGAGCAAGCTCAG-3′ and 5′-CTGAGCTTGCTCTTGTTTTTTCCTCCCCCACGACAGCATC-3′; Pa3-m3, 5′-GATGCTGTCGTAAAAAAGGAAAAAACAAGAGCAAGCTCAG-3′ and 5′-CTGAGCTTGCTCTTGTTTTTTCCTTTTTTACGACAGCATC-3′; Pa3-m4, 5′-GATGCTGTCGTTTTTTAGGAGCCCACAAGAGCAAGCTCAG-3′ and 5′-CTGAGCTTGCTCTTGTGGGCTCCTAAAAAACGACAGCATC-3′; Pa3-m5, 5′-GATGCTGTCGTGGGGGAGGATTTTACAAGAGCAAGC-TCAG-3′ and 5′-CTGAGCTTGCTCTTGTAAAATCCTCCCCCACGACAGCATC-3′; Pa3-m6, 5′-GATGCTGTCGT-TTTTTAGGATTTTACAAGAGCAAGCTCAG-3′; and 5′-CTGAGCTTGCTCTTGTAAAATCCTAAAAAACGACAGCATC-3′; oct, 5′-TGTCGAATGCAAATCACTAG-3′ and 5′- TTCTAGTGATTTGCATTCGA-3′; and μE5 (competitor), 5′-TCGAAGAACACCTGCAGCAGCT-3′ and 5′-TAGAGCTG-CTGCAGGTGTTCTT-3′. Quantification of intensity was performed using a Bio Imaging Analyzer MacBAS (FUJIFILM).
Chromatin Immunoprecipitation.
ChIP assays were performed according to the protocol for the ChIP assay kit (Upstate Biotechnology; http://www.upstate.com/misc/protocols.asp?prot=chips) and as reported (20) with slight modifications. Splenic B cells (106) were subjected to chromatin cross-linking by addition of formaldehyde to the medium at a final concentration of 1% and incubation for 2 min at 37°C. The cells were then washed with ice cold PBS, resuspended in 0.2 ml of SDS-lysis buffer (50 mM Tris, pH 8.0, 1% SDS, 10 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptine, 1 mM PMSF), and incubated on ice for 10 min. Cell lysates were sonicated to reduce the DNA length to between 200 and 1,000 bp, and debris was removed by centrifugation. Sonicated lysates were then diluted by adding 1.8 ml of ChIP dilution buffer (50 mM Tris, pH 8.0, 167 mM NaCl, 1.1% Triton X-100, 0.11% sodium deoxycholate, 10 μg/ml aprotinin, 10 μg/ml leupeptine, 1 mM PMSF) and precleared according to the protocol of Upstate Biotechnology. Precleared lysates were incubated overnight at 4°C with 6 μg of the following antibodies. For E2A, mixed anti-E2A antibodies (BD554077 [BD Biosciences] and V18X [Santa Cruz Biotech]) were used. For Pax5, anti-Pax5 antibody (C-20; Santa Cruz Biotech) was used. For acetylated histone H3, α-acetyl H3 antibody (Lys9, Lys14; Upstate Biotechnology) was used. As a control, normal goat IgG (for Pax5) or mixed normal mouse and goat IgG (for E2A) or normal rabbit IgG (for acetylate H3) was used (Santa Cruz Biotechnology). Each immunocomplex was recovered with protein G–Sepharose beads (Amersham Biosciences). Each precipitate was washed serially with 1-ml aliquots of the following wash buffers. Low salt buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 2% Triton X-100, 0.2% SDS, 0.2% sodium deoxycholate), high salt buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM EDTA, 2% Triton X-100, 0.2% SDS, 0.2% sodium deoxycholate), LiCl wash buffer (250 mM LiCl, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0), and TE buffer, pH 8.0. Immunocomplexes were next eluted from beads using elution buffer (10 mM Tris-HCl, pH 8.0, 5 mM EDTA, 300 mM NaCl, 0.5% SDS), and the cross-linking was reversed by incubation in this buffer at 65°C for 16 h. The samples were then treated with RNase A (20 μg/ml, 30 min, 37°C) followed by proteinase K (50 μg/ml, 1 h, 55°C), extracted with phenol-chloroform, and precipitated with ethanol in the presence of carrier glycogen. Pellets were resuspended in 50 μl of TE buffer for input DNA and 20 μl of TE buffer for ChIP DNA. Serial dilutions of DNAs were prepared in 25 μg/ml sonicated pBSKS. Specific sequences in the immunoprecipitates were detected by PCR in which product yield was dependent on input DNA dose. Primer pairs with the following sequences were used: AID-CR2 region, sense, 5′-CGGGATCCGGGTCCGAGAGAAATGCATGAG-3′, antisense, 5′-CGGGATCCATTATGAAGGAAAGGTATCCTT-3′; AID promoter, sense, 5′-CGGGGTACCCGAAGGAAGTTGGACA-3′ and antisense, 5′-CTCGCGCGCTGTCCATATCGGTCTC-3′; mb-1, sense, 5′-CACGCACTAGAGAGAGACTCAA-3′ and antisense, 5′-CCGCCTCACTTCCTGTTCAGCCG-3′; CD3ɛ, sense, 5′-GGCCCGGATTTCCTCAGTTA-3′ and antisense, 5′-GCCATAGTAGGATGAATGAG-3′; Iɛ, sense, 5′-CAAGGAGACCTGGGATCAGACGATGGAG-3′ and antisense, 5′-TCTGCCTCAGTGCCCTCAGCTAAAAG-3′. Intensity was quantified using the NIH image program.
Results
Reduced AID Gene Expression in Id2-overexpressing B Cells.
It has been shown that enforced expression of Id2 blocks CSR completely (9). To gain insight into the molecular mechanisms underlying CSR regulation, we analyzed the levels of expression of AID, which is essential for CSR (12), in Id2-overexpressing B cells. Id2 cDNA was cloned into the pMXIRES-hCD4 retroviral vector (21), which enabled us to trace Id2 expression by monitoring the surface expression of human CD4 (Fig. 1 a). Splenic B cells were infected with the virus by spin infection (14). After 3 d of stimulation with LPS plus IL4, Id2-expressing B cells were purified using MACS by virtue of CD4 expression. RNA was extracted from them and subjected to RT-PCR analysis. As shown in Fig. 1 b, a fivefold reduction of AID expression was observed in Id2-transduced cells. This observation suggests that the inhibitory effect of Id2 on CSR is in part the result of repression of AID expression.
Figure 1.

Effect of enforced expression of Id2 on AID transcription. (a) A diagram of the Id2 retrovirus construct. Surface expression of truncated human CD4 was used as the marker of infected cells. (b) The expression levels of AID in splenic B cells stimulated in vitro. RNAs from Id2-transduced and control populations were subjected to RT-PCR analysis. (c) The expression levels of AID in splenic B cells stimulated in vitro. RNAs from Id2−/− and Id2+/+ B cells were subjected to RT-PCR analysis. One of three independent experiments is shown. Fivefold serial dilutions of cDNAs were amplified for the indicated transcripts. GAPDH was used as an internal control. (d) Time course analysis of the Pax5 and E2A proteins in in vitro–activated splenic B cells. Each lane contains the extract from the equal cell numbers (2 × 106 cells). (e) Time course analysis of the AID expression in in vitro–activated splenic B cells. One of three independent experiments is shown. Fivefold serial dilutions of cDNAs were amplified for the indicated transcripts. GAPDH was used as an internal control.
To determine whether this inhibitory effect of Id2 on AID expression indeed works in activated B cells in vivo, we examined the AID expression in Id2−/− spleen cells. Id2−/− spleen cells expressed fivefold more AID transcripts than Id2+/+ spleen cells (Fig. 1 c). These data further support the possibility that Id2 functions as a negative regulator of AID expression in vivo. E2A and Pax5 are strong candidates as inducers of AID expression because increased amounts of these factors are observed upon B cell activation. To monitor the expression of AID mRNA during switch induction of primary B cells, RT-PCR analysis was performed after cytokine treatment. AID mRNA was undetectable initially and was induced after 2 d of in vitro culturing (Fig. 1 e). To compare the kinetics of AID expression with the levels of E2A and Pax5 proteins, Western blotting was performed. About fivefold induction of E47 and sevenfold induction of Pax5 were observed at 2 d postactivation (Fig. 1 d). In addition to this observation, E2A and Pax5 are known to bind and be antagonized by Id2. These data support the idea that Id2 represses AID expression through antagonizing E2A and Pax5 activities.
Potentially Important Sequences in 5′-flanking Region and First Intron of the AID Gene.
To elucidate the mechanism of the transcriptional regulation of AID, we isolated genomic DNA fragments of the AID gene containing the 5′-flanking region and first and second exons. Because cross species conservation of regulatory region sequences is common, we first performed homology plot analysis of sequences in the 5′-flanking region and the first intron for mice and humans. As shown in Fig. 2 a, a strikingly homologous region spanning ∼400 bp was found in the region upstream of the first ATG codon, and it included a putative transcription initiation site (shown in Fig. 2, a and b) from which the most abundant transcripts were produced (not depicted). There were two additional homologous regions, which we designated CR1 and CR2, in the first intron (Fig. 2 a). An alignment of the murine and human conserved sequences is shown in Fig. 2, b, c, and d. Two E-boxes, the consensus sequence for E2A binding, were present in CR2 region in both humans and mice (Fig. 2 d). This observation further supports the involvement of E2A in AID expression.
Figure 2.
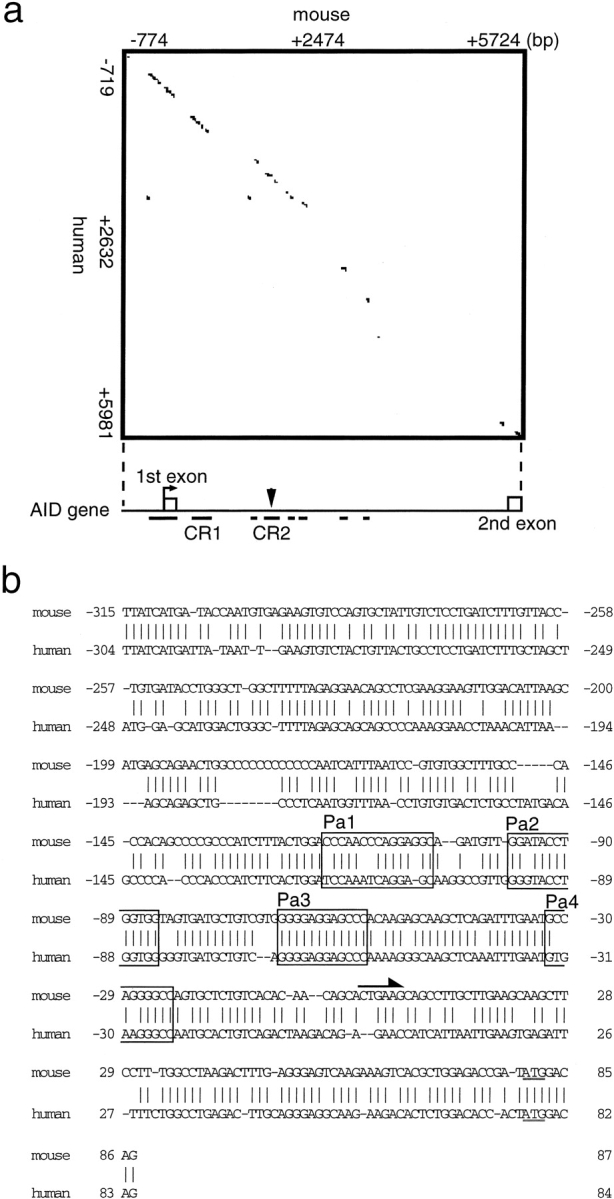

Conservation of AID promoter sequence in mice and humans. (a) Dot matrix sequence comparisons of the sequences of the mouse and human AID 5′ upstream region, exon 1, intron 1, and exon 2 using a window with 33 of 50 matches. A schematic diagram of the putative regulatory regions of the AID genes in mice and humans is shown below the matrix. The open boxes indicate exons. Underlines indicate conserved sequences. (b) Alignment of 5′-flanking conserved sequences of the AID genes of mice and humans. Vertical lines indicate conserved nucleotides in the two species. The underlined sequence is the first ATG. The arrow indicates the transcription initiation site. (c) Alignment of CR1 conserved sequences of the AID genes of mice and humans. Vertical lines indicate nucleotides conserved between the two species. (d) Alignment of CR2 conserved sequences of the AID genes of mice and humans. Vertical lines indicate nucleotides conserved between the two species. The open boxes indicate the E-box sequence.
Transactivation of AID Promoter by Pax5.
To confirm that the 900-bp 5′ fragment containing the conserved CR1 and CR2 regions was sufficient to direct developmental stage-specific AID expression, reporter assays were performed using the M12 B cell line, in which the endogenous AID gene is expressed, and the BaF/3 pro–B cell line, in which it is not (not depicted). We subcloned a 0.9-kb AID promoter region fragment including the CR1 and CR2 fragments into the pGL3 plasmid, resulting in construct 0.9P-CR1+CR2, and analyzed the promoter activity of this construct in both cell lines. The results shown in Fig. 3 a demonstrate that 0.9P-CR1+CR2 was sufficient to direct expression specifically in the M12 cell line; 0.9P-CR1+CR2 was at least fivefold better expressed in M12 cells than BaF/3 cells. This observation is consistent with the homology plot analysis. Since Id2 is a negative regulator of E protein (22) and Pax5 (4) function, reduced activity of E2A or Pax5 products may directly contribute to the loss of AID expression in cells with enforced Id2 expression. To test this possibility, the effects of these factors on transactivation of the 0.9P-CR1+CR2 construct were determined using BaF/3 cells. Pax5, but not E47, an E2A gene product, indeed induced transactivation by a factor of eleven from the WT promoter (Fig. 3 b, top column). This effect was consistent with the results shown in Fig. 3 a in which mutation of the E-boxes had no effect on transcription from the 0.9P-CR1+CR2 construct in M12 cells, which have high E2A activity (not depicted). To determine the expression of each factors, we performed Western blot analysis using transiently transfected 293T cells. As shown in Fig. 3 b, bottom column, these plasmids worked equally. In addition, E47 activity in BaF/3 cells was confirmed using E47 expression vector and ɛI promoter reporter construct, which is responsive to E47 and Pax5 (Fig. 3 b, middle column and previously reported) (23). Again, Pax5 but not E47 only has a potential to induce AID expression in BaF/3 lines. To further identify the Pax5-responsive element, 0.9P (promoter only), 0.9P-CR1 (promoter plus CR1 fragment), and 0.9P-CR2 (promoter plus CR2 fragment) were examined. As shown in Fig. 3 c, all the reporters were responsive to Pax5, indicating that a Pax5-binding sequence may exist in the 5′-flanking region of the AID gene. In addition to this observation, CR1 and CR2 seems to work as a tissue-specific repressive element of the AID gene because these regions have a repressive effect in BaF/3 cells (Fig. 3 c) but not in M12 cells (not depicted). We sometimes observed that constructs without CR1 or CR2 have higher promoter activities (Fig. 3 d) than those with CR1 or CR2, which have consistently almost no activities, in BaF/3 cells without Pax5. To further delineate the Pax5-responsive element within this region, various deletion constructs were examined. As shown in Fig. 3 d, the 0.02P promoter lost the responsiveness to Pax5, indicating that a Pax5-binding sequence may exist between −122 and −22 bp from the initiation site. We found four putative Pax5-binding sites (Pa1-Pa4) within this region (24) and analyzed the promoter activity of constructs mutated in each of these sites (Fig. 3 e). As shown in Fig. 3 e, the third site (Pa3) was indispensable for transactivation by Pax5, since Pax5 was unable to induce transactivation of the 0.1P-mPa3 promoter. To confirm that mature B cells actually use this element for AID expression, the promoter activity of each mutant construct was analyzed using the mature B cell line M12, which expresses both Pax5 and AID. Again, the third site (Pa3) was indispensable for proper expression of AID, since transactivation was reduced using the 0.1P-mPa3 promoter in M12 cells in which Pax5 activity is high (unpublished data; Fig. 3 e).
Figure 3.
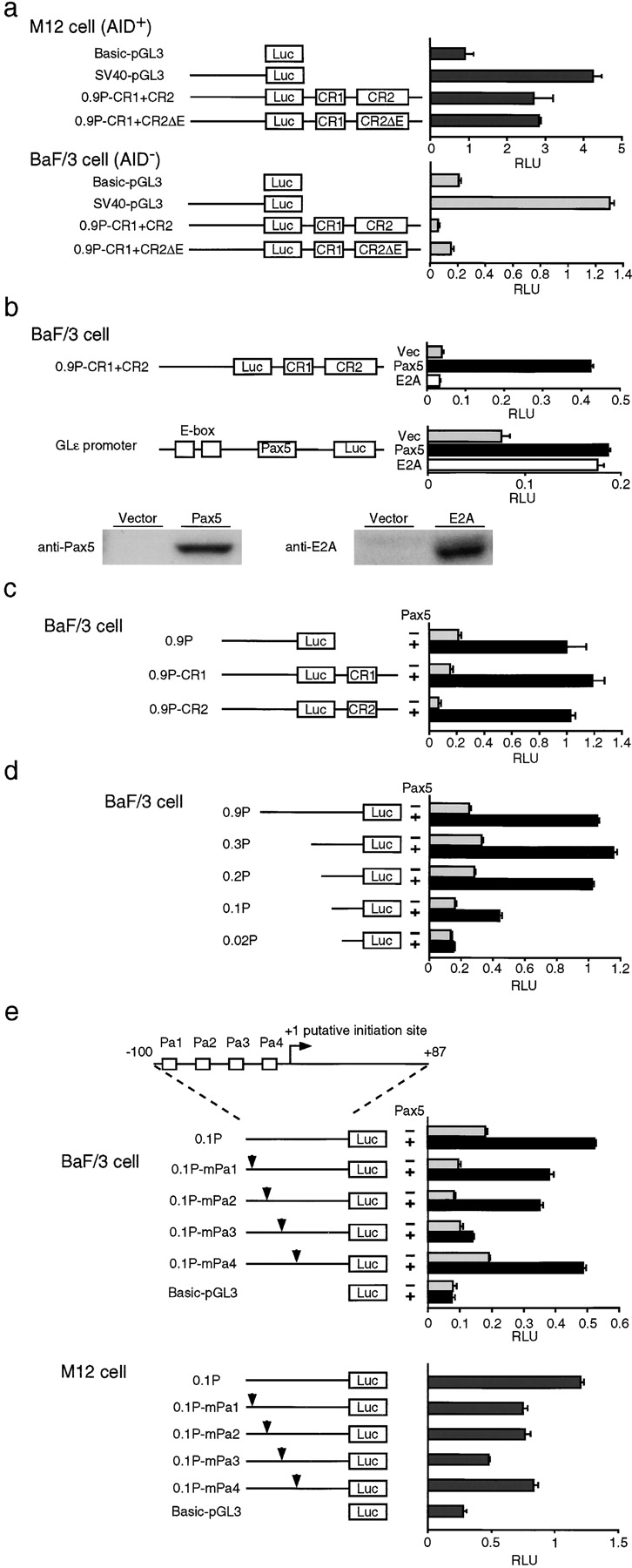
Transcriptional activation of the putative AID promoter by Pax5. (Left) Schematic diagrams of reporter constructs containing no enhancer or promoter (Basic-pGL3), only SV40 promoter (SV40-pGL3), all the conserved sequences (0.9P-CR1+CR2), all the conserved sequences with deleted E-boxes (0.9P-CR1+CR2ΔE), promoter only (0.9P), promoter plus CR1 (0.9P-CR1), promoter plus CR2 (0.9P+CR2), various promoter deletions (0.3P, 0.2P, 0.1P, and 0.02P), various potential Pax-binding site mutations (0.1P-mPa1, 0.1P-mPa2, 0.1P-mPa3, and 0.1P-mPa4). (a, right) BaF/3 cells or M12 cells were transfected with AID–luciferase constructs (20 μg). All assays were independently performed five times in duplicate. Luciferase activity is the relative luminescence units normalized by Renilla luciferase activity. (b, right) BaF/3 cells were transfected with AID–luciferase constructs (20 μg) or Glɛ promoter constructs (20 μg) in the absence (black bars) or presence of pcDNA-Pax5 (15 μg) (black bars) or pcDNA-E47 (15 μg) (white bars). Immunoblot analysis of Pax5 and E2A proteins in 293T cells transfected with indicated expression vectors. (c) BaF/3 cells were transfected with AID–luciferase constructs (20 μg) in the absence (black bars) or presence (black bars) of pcDNA-Pax5 (15 μg). (d) BaF/3 cells were transfected with various deletion AID–luciferase constructs (20 μg) in the absence (black bars) or presence of pcDNA-Pax5 (15 μg) (black bars). (e, top column) Schematic representation of AID promoter. Boxes indicate putative Pax5-binding sequences (Pa1–Pa4). (e, left) Schematic diagrams of the mutated promoter constructs. Arrowheads indicate the mutations introduced into putative Pax5-binding sites Pa1–Pa4. (e, middle right) BaF/3 cells were transfected with AID–luciferase constructs (20 μg) in the absence (black bars) or presence (black bars) of pcDNA-Pax5 (15 μg). (e, lower right) M12 cells were transfected with the indicated AID–luciferase constructs (20 μg). Luciferase activity is the relative luminescence units normalized by Renilla luciferase activity. All assays were independently performed six times in duplicate.
Pax5 Binds the AID Promoter.
To determine whether Pax5 binds to the P3 site in the AID promoter, we performed electrophoretic mobility shift assays (EMSAs) using AID promoter oligonucleotides bearing the P3 site. WT and mutant oligonucleotides used in this assay are shown in Fig. 4 a, top column. Nuclear extracts prepared from WT B cells after stimulation with LPS and IL4 were tested for binding to these oligonucleotides. One complex was detected, and inclusion of Pax5 in this complex was verified by using a specific antibody against Pax5 (Fig. 4 b, lanes 11 and 12) and competitive oligonucleotides to which Pax5 binds (Fig. 4 b, lane 10). The binding activity was reduced when mutant oligonucleotides, Pa3-m1 through Pa3-m6, were used (Fig. 4 b). On the other hand, the binding activity of Pax5 was more prominent when extract from Id2−/− B cells was analyzed. The extract from Id2−/− B cells formed about twice as much binding complex with this probe as the extract from control cells (Fig. 4 b, lanes 2 and 3). The most slowly migrated band may not contain Pax5 because this band always disappears using normal IgG (Fig. 4 b, lanes 13–15). RT-PCR demonstrated no significant difference between the expression of Pax5 in splenic B cells with the two different genotypes, suggesting that Id2 does not affect the expression of Pax5 (Fig. 4 d). As expected, Western blotting demonstrated no significant difference in the levels of Pax5 proteins in activated B cells with the two genotypes (Fig. 4 d). To further determine the actual contribution of E2A in AID expression in Id2−/− B cells, we performed EMSAs using AID enhancer oligonucleotides bearing two E-boxes (Fig. 4 c). WT and mutant oligonucleotides used in this assay are shown in Fig. 4 a, bottom column. The extract from Id2−/− B cells formed ∼1.2-fold more binding complex with this probe as the extract from control cells. Such a slight difference is unexpected, since we previously detected more prominent difference using an ɛ promoter as a probe (23). Such discrepancy of E2A-containing bands will be produced by the context-dependent differences in complex formation. Western blotting demonstrated no significant difference in the levels of E2A proteins in activated B cells with the two genotypes (Fig. 4 d). Collectively, these data further support the idea that Id2 acts as an antagonist against Pax5 and E2A within AID locus. To further determine whether Id2 indeed functions as a dominant-negative regulator of Pax5 products, nuclear extracts were prepared from Id2-overexpressing B cells and analyzed by EMSA. As expected, the level of Pax5–DNA-binding complex was reduced in the extract from Id2-transduced cells by a factor of three (Fig. 4 e). This Pax5-containing complex may contain other factors, since it migrates slowly when compared to the band using the CD19 site as a probe. Unfortunately, we couldn't identify any other components of this complex. Further studies are required to identify the other factors involved in this complex. In addition to these observations, the binding activity of Pax5 was more prominent when the CD19 site was used as a probe. Compared with the Pa3 site, ∼98-fold enhancement was observed (Fig. 4 b, lanes 1 and 3). This data indicated that the Pa3 site is the weak binding site of Pax5.
Figure 4.

Binding of Pax5 to the AID promoter. (a, top column, top panel) Pax5-binding sequence in the mb-1 promoter. The DNA sequences of the probes used in this assay (b) are shown. Pa3 represents the WT sequence. Underlined sequences of the WT P3 probe were changed as noted (Pa3-m1 to P3-m6). (a, bottom column) The DNA sequences of the probes used in this assay (c) are shown. CR2E represents the WT sequence. Underlined sequences of the WT CR2E probe were changed as noted (CR2E-m1 to P3-m3). (b) EMSA of nuclear extracts from splenic B cells of Id2−/− and control mice treated with 20 μg/ml LPS plus 10 ng/ml IL-4 for 3 d using the probes shown in panel a. The asterisk indicates the Pax5-containing complex. The arrowhead indicates a nonspecific band. An Oct probe was used as an internal control. One of three independent experiments is shown. (c) EMSA of nuclear extracts from splenic B cells of Id2−/− and control mice treated as described in panel b using the probes shown in panel a. The arrows indicate the E2A-containing complex. One of six independent experiments is shown. (d) The levels of Pax5 or E2A proteins and Pax5 transcripts. In vitro–stimulated B cells from Id2−/− and Id2+/+ mice were subjected to Western blotting or RT-PCR. One of three independent experiments is shown. Fivefold serial dilutions of cDNAs were amplified for the indicated transcripts. GAPDH or β-actin were used as an internal control. (d) Nuclear extracts prepared from hCD4-positive cells obtained as described in Fig. 1 were analyzed by EMSA using the P3 probe.
Recruitment of Pax5 to the AID Promoter in Activated B Cells.
Next, we examined whether Pax5 indeed binds to the AID promoter in activated B cells by a chromatin immunoprecipitation (ChIP) assay using α-Pax5 antiserum. Splenic B cells were isolated and stimulated in vitro with LPS plus IL-4. Serially diluted DNA samples isolated from the input, α-Pax5–bound, and control antibody-bound fractions were tested by PCR to analyze the AID promoter. AID promoter DNA was recovered from α-Pax5–bound but not from α-E2A–bound chromatin after 2 d of culturing, providing direct evidence that Pax5 but not E2A occupies the AID promoter in activated B cells (Fig. 5 a). On the other hand, the CR2 fragment, which contains two E-boxes, was recovered from α-E2A–bound chromatin at the same level at all the time points examined (Fig. 5 a). In contrast, the CR2 fragment was also recovered from α-Pax5–bound chromatin after 2 d of culturing (Fig. 5 a). For the positive and negative control loci to Pax5, mb-1 and CD3ɛ locus were selected. As shown in Fig. 5 a, mb-1 promoter but not CD3ɛ was recovered from α-Pax5–bound chromatin at the all time points examined. For the positive control for E2A, the ɛIp locus was used. As expected, ɛIp promoter was recovered from α-E2A–bound chromatin after 2 d of culturing (Fig. 5 a). Collectively, these data further support the important role of Pax5 in AID expression, considering the fact that AID expression was induced after 2 d of in vitro culturing as shown in Fig. 1 d.
Figure 5.

Recruitment of Pax5 to the AID promoter in activated B cells. (a) ChIP analyses at the indicated sites. Fivefold serial dilutions of input DNA and DNAs immunoprecipitated with anti-E2A, anti-Pax5, anti–acetylated histone H3, and control (antibodies) Abs were analyzed by PCR. One of three independent experiments is shown. (b) Pax5-induced endogenous AID expression in BaF/3 cells. Pax5 or E47 or empty vectors (pMX-IREShCD4) were introduced into BaF/3 cells via retroviral transfer. RNAs prepared from hCD4-positive cells were analyzed by RT-PCR. One of five independent experiments is shown.
Ectopic Expression of Pax5 Activates Endogenous AID Transcription in BaF/3.
Pax5 clearly transactivated the AID reporter constructs in transient transfection assays in BaF/3 cells. To test whether expression of Pax5 in non-AID–expressing cells can activate the transcription of the endogenous AID gene, BaF/3 cells were retrovirally transduced with Pax5 or E2A. RT-PCR showed that BaF/3 cells transduced with Pax5 indeed produced the AID transcript (Fig. 5 b). In contrast, BaF/3 cells transduced with E2A did not (Fig. 5 b). We further confirmed that Id2 has an antagonizing effect on AID expression induced by Pax5 in BaF/3 cells (not depicted). These observations are consistent with the results of the reporter assay, which showed that Pax5 is required for the activation of the AID promoter, and with the loss of AID expression in Id2-transduced B cells. In summary, these results demonstrate the essential and selective role of Pax5 in directing tissue- and stage-specific endogenous AID expression.
Discussion
Increasing evidence has suggested that AID expression alone is sufficient to induce SHM and CSR in B cell hybridomas, T cells, and fibroblasts (25–27). These observations also indicate that AID could act as a mutator if the negative regulation systems controlling AID expression were disrupted. On the other hand, AID is the only known factor indispensable for CSR and SHM (12), both of which are required for the proper functioning of B cells to produce high affinity antibodies with different effector functions (13). In normal B cells, AID expression is tightly regulated in an activation-dependent manner in conditions under which SHM and CSR take place (28). In this report, we clearly demonstrated that the AID promoter is a direct target of Pax5 in activated B cells. Lineage-specific transcription factors that act as positive regulators of AID expression are very likely to carry out one of the mechanisms regulating the lineage-specific expression of AID. Such a scenario would be a simple and reasonable strategy for obtaining lineage-specific expression. Within the lymphoid lineage, Pax5 is clearly restricted to B cells. On the other hand, what is the molecular switch sensing the activation stimuli and inducing the expression of the AID gene? As shown in this report, the level of Pax5 protein was increased 2 d after stimulation (10). This time course was in accord with that of AID expression. Furthermore, we have identified several other Pax5-binding sites within the regulatory regions of the AID gene (unpublished data). How, then, does the AID locus sense the augmentation of Pax5 in activated B cells? This might be achieved by low affinity binding sites within the AID regulatory regions. As shown in this paper, Pax5 occupancy at the AID locus could be detected only from the 2-d time point onward. In contrast, Pax5 occupancy at the mb-1 locus can be detected in both resting and activated B cells (Fig. 5 b). These observations are consistent with a recent study using floxed Pax5 mice in which the serum levels of isotype-switched antibodies were shown to be decreased in Pax5F/− CD19-cre mice (8). On the other hand, E47 occupancy at the AID locus was detected in both resting and activated B cells. This observation suggests that E47 may play a more general rather than B cell activation–dependent role in regulating the AID locus.
We also demonstrated that Id2 acts as a negative regulator of AID expression in activated B cells. The Id2 protein acts as a key regulator of cell fate determination. In Id2−/− mice, the number of NK cells is greatly reduced, and LNs, Peyer's patches, and NALT are absent due to the loss of CD3−CD4+CD45+ cells (29, 30). Previous reports have suggested that Id proteins play roles in B cell differentiation and maturation (31). However, Id2−/− mice show normal distributions of B cell populations in the BM and spleen (unpublished data). However, upon B cell activation Id2 plays a key role in antagonizing the Pax5 and E2A activities, both of which are required for proper B cell differentiation (5–7). In activated B cells, the levels of E2A proteins and Pax5 transcripts are highly augmented, and antagonists of these factors inhibit CSR (9–11, 32). The results suggest the involvement of E2A and Pax5 in CSR. Among them, Pax5 directly induces AID expression through a Pax5-binding sequence that exists in the putative enhancer/promoter of the AID gene (Fig. 6) . The importance of Pax5 and Id2 in regulating AID expression was further supported by our observation that enforced expression of Pax5- but not E47-induced endogenous AID expression in BaF/3 cells, and this effect was cancelled by additional induction of Id2. In contrast, whether E2A proteins are involved in the regulation of AID expression is still unclear. A recent report from Murre's group demonstrated that E2A is a positive regulator of AID gene expression (33). We were not able to reproduce such effects in our system probably because of the difference in cell lines and experimental conditions used. In considering the characters of the two cell lines used, E2A might require other factors, such as Pax5, to induce AID expression. In this context, Pax5 and E2A may act in synergy in regulating AID expression. Further experiments will be required to examine this possibility. In any case, Id2 plays a key role in the negative regulation of AID expression, as shown by the AID overexpression in Id2−/− B cells. There was no difference between Id2−/− and control B cell populations in the levels of Pax5 proteins or transcripts. However, the Pax5–AID promoter–binding complex was augmented in Id2−/− mice. Conversely, the Pax5-binding complex was reduced in Id2-transduced cells. Together, these findings suggest that the repressive effect of Id2 on AID expression was the result of antagonizing the effect of Pax5 in activated B cells. Interaction between Id2 and Pax5 as biochemical nature in vitro is actually weak and may not be direct in vivo. Binding to Pax5 may not be the only way to antagonize Pax5 activity by Id2. However, Pax5 antagonizing activity of Id2 was further supported by the observation that the repressive effect of TGF-β1 on CD19 expression was lost in Id2−/− mice (unpublished data). Further studies are required to reveal the actual mechanism involved in this antagonism. However, such regulation seems quite possible considering the mechanism of repression of the AID gene after activation. Recent studies have indicated that Blimp-1 is required for the differentiation of B cells into plasmacytes. Transcription of Blimp-1 is induced at the later stages of in vitro culturing of primary B cells (10, 32), and one of its most important functions is to repress Pax5 expression (10). Interestingly, Id2 is thought to be the target whose expression is induced by Blimp-1 (32). Collectively, these published observations and our findings in the present study suggest that after B cell activation Blimp-1 works as a molecular switch to repress AID expression through repressing and antagonizing Pax5 activity.
Figure 6.

Model for the regulation of AID and IgE gene expression by E2A, Pax5, and Id2.
We could not detect augmentation of AID expression by forced expression of either Pax5 or E2A in activated primary B cells using a retroviral transfection system (unpublished data). There are possible but not mutually exclusive reasons for this. First, there might be a safeguard or feedback system to ensure the activities of Pax5 and/or E2A do not elevate more than normal. Id2 could be involved in this system since Id2−/− B cells have elevated activities of E2A and Pax5. Second, other than a simple amount, there might be a crucial timing and/or order of activation, which we could not control by a retroviral expression system, of these factors to activate AID gene expression. Forced expression of Id2 using the same system could repress AID expression, as shown here, since unregulated overexpression could antagonize E2A and Pax5 throughout the activation process. A recent report from Murre's group showed a somewhat different result in which enforced expression of E2A enhances the AID expression by a factor of two in activated primary B cells (33). This result was obtained by an experimental system that induced very rapid, vast enhancement of E2A activity in the nuclei. They used E2A fused to estrogen receptor accumulating in cytoplasm and then forced the accumulated E2A fusion protein to move into nuclei almost at once by 4-hydroxytamoxifen treatment. Such a procedure might enhance the effective E2A activity by breaking the feedback negative regulation system. Further investigations are necessary to elucidate fine molecular mechanisms to induce AID gene expression during B cell activation.
Our results indicate that modulating the relative abundance of Pax5, E2A, and Id proteins provides a mechanism for subtle regulation of the effects of Pax5 and E2A in B cell activation (Fig. 6). The differential activities of these factors determine whether the AID locus is activated and its level of transcription. Such decision-making processes may occur at many steps of cell differentiation and function. Actually, the IgE locus is also regulated under the control of these factors in activated B cells (23, 34, 35). E2A and Pax5 indeed exist at the ɛ germline promoter in IL-4–stimulated B cells (Fig. 5 a and unpublished data). IL-4 stimulates transcription of the unrearranged γ1 and ɛ heavy chain loci (germline transcription, GLT) and induces CSR to both isotypes, but the frequency of IgE CSR is strictly regulated at a lower level. In contrast, in Id2−/− B cells, CSR to IgE is enhanced without affecting IgG1. This enhanced CSR to IgE solely depends on the augmentation of E2A and Pax5 activities, which induce ɛ GLT and CSR via E-boxes and a Pax5-binding site in the ɛ germline promoter. In WT B cells, Id2 is induced by TGF-β1 and negatively regulates CSR to IgE. This suppression does not occur in Id2-deficient B cells (23). Thus, the Pax5-E2A-Id system seems to work as a molecular switch in the B cell activation program to regulate specific transcription of various loci. Upon activation, a breakdown of repression is achieved by augmentation of Pax5 and E2A activities. Thus, Pax5 and E2A work as a transcriptional switch in B cell activation as observed at the AID and IgE loci. Moreover, Id proteins may play widespread regulatory roles in regulating gene expression and differentiation, including in activated B cells.
Although the augmentation of AID expression was observed in Id2−/− mice, the rate of SHM is not enhanced (unpublished data) and the enhancement of CSR is only observed for IgE (23). These data indicate that other regulatory mechanism(s) to prevent harmful effects of AID must exist in activated B cells. Further studies will be required to elucidate the molecular mechanisms involved in this regulatory program.
Acknowledgments
We are grateful to H. Karasuyama (Tokyo Medical and Dental University, Tokyo, Japan) for X63mIL3, A. Kudo (Tokyo Institute of Technology, Yokohama, Japan) for mPax5 expression plasmid, T. Kitamura (University of Tokyo, Tokyo, Japan) for pMX-IRESGFP plasmid, Y. Shinkai (Kyoto University, Kyoto, Japan) for the TT2 genomic library, P.B. Rothman (Columbia University, New York, NY) for M12 cells, and G.P. Nolan (Stanford University, Stanford, CA) for Phoenix Eco cells. We thank T. Ofuji for technical assistance.
This work was supported in part by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Y. Agata's present address is Horizontal Medical Research Organization, Kyoto University Faculty of Medicine, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan.
Abbreviations used in this paper: AID, activation-induced cytidine deaminase; CSR, class switch recombination; EMSA, electrophoretic mobility shift assay; Id, inhibitor of differentiation; SHM, somatic hypermutation.
References
- 1.Norton, J.D., R.W. Deed, G. Craggs, and F. Sablitzky. 1998. Id helix-loop-helix proteins in cell growth and differentiation. Trends Cell Biol. 8:58–65. [PubMed] [Google Scholar]
- 2.Yokota, Y., and S. Mori. 2002. Role of Id family proteins in growth control. J. Cell. Physiol. 190:21–28. [DOI] [PubMed] [Google Scholar]
- 3.Yates, P.R., G.T. Atherton, R.W. Deed, J.D. Norton, and A.D. Sharrocks. 1999. Id helix-loop-helix proteins inhibit nucleoprotein complex formation by the TCF ETS-domain transcription factors. EMBO J. 18:968–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts, E.C., R.W. Deed, T. Inoue, J.D. Norton, and A.D. Sharrocks. 2001. Id helix-loop-helix proteins antagonize pax transcription factor activity by inhibiting DNA binding. Mol. Cell. Biol. 21:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhuang, Y., P. Soriano, and H. Weintraub. 1994. The helix-loop-helix gene E2A is required for B cell formation. Cell. 79:875–884. [DOI] [PubMed] [Google Scholar]
- 6.Urbanek, P., Z.Q. Wang, I. Fetka, E.F. Wagner, and M. Busslinger. 1994. Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell. 79:901–912. [DOI] [PubMed] [Google Scholar]
- 7.Bain, G., E.C. Maandag, D.J. Izon, D. Amsen, A.M. Kruisbeek, B.C. Weintraub, I. Krop, M.S. Schlissel, A.J. Feeney, M. van Roon, et al. 1994. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 79:885–892. [DOI] [PubMed] [Google Scholar]
- 8.Horcher, M., A. Souabni, and M. Busslinger. 2001. Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity. 14:779–790. [DOI] [PubMed] [Google Scholar]
- 9.Quong, M.W., D.P. Harris, S.L. Swain, and C. Murre. 1999. E2A activity is induced during B-cell activation to promote immunoglobulin class switch recombination. EMBO J. 18:6307–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin, K.I., C. Angelin-Duclos, T.C. Kuo, and K. Calame. 2002. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol. Cell. Biol. 22:4771–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knodel, M., A.W. Kuss, I. Berberich, and A. Schimpl. 2001. Blimp-1 over-expression abrogates IL-4- and CD40-mediated suppression of terminal B cell differentiation but arrests isotype switching. Eur. J. Immunol. 31:1972–1980. [DOI] [PubMed] [Google Scholar]
- 12.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 13.Honjo, T., K. Kinoshita, and M. Muramatsu. 2002. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu. Rev. Immunol. 20:165–196. [DOI] [PubMed] [Google Scholar]
- 14.Ye, S.K., K. Maki, T. Kitamura, S. Sunaga, K. Akashi, J. Domen, I.L. Weissman, T. Honjo, and K. Ikuta. 1999. Induction of germline transcription in the TCRgamma locus by Stat5: implications for accessibility control by the IL-7 receptor. Immunity. 11:213–223. [DOI] [PubMed] [Google Scholar]
- 15.Kinsella, T.M., and G.P. Nolan. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405–1413. [DOI] [PubMed] [Google Scholar]
- 16.Narumi, O., S. Mori, S. Boku, Y. Tsuji, N. Hashimoto, S. Nishikawa, and Y. Yokota. 2000. OUT, a novel basic helix-loop-helix transcription factor with an Id-like inhibitory activity. J. Biol. Chem. 275:3510–3521. [DOI] [PubMed] [Google Scholar]
- 17.Sato, H., D. Wang, and A. Kudo. 2001. Dissociation of Pax-5 from KI and KII sites during kappa-chain gene rearrangement correlates with its association with the underphosphorylated form of retinoblastoma. J. Immunol. 166:6704–6710. [DOI] [PubMed] [Google Scholar]
- 18.Sugai, M., S. Kondo, A. Shimizu, and T. Honjo. 1998. Isolation of differentially expressed genes upon immunoglobulin class switching by a subtractive hybridization method using uracil DNA glycosylase. Nucleic Acids Res. 26:911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuda, E., Y. Agata, M. Sugai, T. Katakai, H. Gonda, and A. Shimizu. 2001. Targeting of Kruppel-associated box-containing zinc finger proteins to centromeric heterochromatin. Implication for the gene silencing mechanisms. J. Biol. Chem. 276:14222–14229. [DOI] [PubMed] [Google Scholar]
- 20.Orlando, V., H. Strutt, and R. Paro. 1997. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 11:205–214. [DOI] [PubMed] [Google Scholar]
- 21.Nosaka, T., T. Kawashima, K. Misawa, K. Ikuta, A.L. Mui, and T. Kitamura. 1999. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 18:4754–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massari, M.E., and C. Murre. 2000. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 20:429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugai, M., H. Gonda, T. Kusunoki, T. Katakai, Y. Yokota, and A. Shimizu. 2003. Essential role of Id2 in negative regulation of IgE class switching. Nat. Immunol. 4:25–30. [DOI] [PubMed] [Google Scholar]
- 24.Wheat, W., D. Fitzsimmons, H. Lennox, S.R. Krautkramer, L.N. Gentile, L.P. McIntosh, and J. Hagman. 1999. The highly conserved beta-hairpin of the paired DNA-binding domain is required for assembly of Pax-Ets ternary complexes. Mol. Cell. Biol. 19:2231–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okazaki, I.M., K. Kinoshita, M. Muramatsu, K. Yoshikawa, and T. Honjo. 2002. The AID enzyme induces class switch recombination in fibroblasts. Nature. 416:340–345. [DOI] [PubMed] [Google Scholar]
- 26.Yoshikawa, K., I.M. Okazaki, T. Eto, K. Kinoshita, M. Muramatsu, H. Nagaoka, and T. Honjo. 2002. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 296:2033–2036. [DOI] [PubMed] [Google Scholar]
- 27.Martin, A., P.D. Bardwell, C.J. Woo, M. Fan, M.J. Shulman, and M.D. Scharff. 2002. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature. 415:802–806. [DOI] [PubMed] [Google Scholar]
- 28.Muramatsu, M., V.S. Sankaranand, S. Anant, M. Sugai, K. Kinoshita, N.O. Davidson, and T. Honjo. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476. [DOI] [PubMed] [Google Scholar]
- 29.Yokota, Y., A. Mansouri, S. Mori, S. Sugawara, S. Adachi, S. Nishikawa, and P. Gruss. 1999. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature. 397:702–706. [DOI] [PubMed] [Google Scholar]
- 30.Fukuyama, S., T. Hiroi, Y. Yokota, P.D. Rennert, M. Yanagita, N. Kinoshita, S. Terawaki, T. Shikina, M. Yamamoto, Y. Kurono, and H. Kiyono. 2002. Initiation of NALT organogenesis is independent of the IL-7R, LTbetaR, and NIK signaling pathways but requires the Id2 gene and CD3(−)CD4(+)CD45(+) cells. Immunity. 17:31–40. [DOI] [PubMed] [Google Scholar]
- 31.Sun, X.H. 1994. Constitutive expression of the Id1 gene impairs mouse B cell development. Cell. 79:893–900. [DOI] [PubMed] [Google Scholar]
- 32.Shaffer, A.L., K.I. Lin, T.C. Kuo, X. Yu, E.M. Hurt, A. Rosenwald, J.M. Giltnane, L. Yang, H. Zhao, K. Calame, and L.M. Staudt. 2002. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 17:51–62. [DOI] [PubMed] [Google Scholar]
- 33.Sayegh, C.E., M.W. Quong, Y. Agata, and C. Murre. 2003. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat. Immunol. 4:586–593. [DOI] [PubMed] [Google Scholar]
- 34.Kusunoki, T., M. Sugai, T. Katakai, Y. Omatsu, T. Iyoda, K. Inaba, T. Nakahata, A. Shimizu, and Y. Yokota. 2003. TH2 dominance and defective development of a CD8+ dendritic cell subset in Id2-deficient mice. J. Allergy Clin. Immunol. 111:136–142. [DOI] [PubMed] [Google Scholar]
- 35.Qiu, G., and J. Stavnezer. 1998. Overexpression of BSAP/Pax-5 inhibits switching to IgA and enhances switching to IgE in the I.29 mu B cell line. J. Immunol. 161:2906–2918. [PubMed] [Google Scholar]