

Abstract
A characteristic feature of rheumatoid arthritis is the abundance of inflammatory cells in the diseased joint. Two major components of this infiltrate are neutrophils in the synovial fluid and macrophages in the synovial tissue. These cells produce cytokines including tumor necrosis factor α and other proinflammatory mediators that likely drive the disease through its effector phases. To investigate what mechanisms underlie the recruitment of these cells into the synovial fluid and tissue, we performed expression analyses of chemoattractant receptors in a related family that includes the anaphylatoxin receptors and the formyl-MetLeuPhe receptor. We then examined the effect of targeted disruption of two abundantly expressed chemoattractant receptors, the receptors for C3a and C5a, on arthritogenesis in a mouse model of disease. We report that genetic ablation of C5a receptor expression completely protects mice from arthritis.
Keywords: arthritis, C5a receptors, granulocytes, chemoattractants, monocytes
Introduction
Rheumatoid arthritis (RA)*is a chronic inflammatory disease characterized by the infiltration of T cells, B cells, macrophages, and neutrophils into the synovial lining and fluid of the periarticular spaces. The infiltrating cells produce abundant cytokines, dominated by TNFα and IL-1β, which stimulate infiltrating cells and resident fibroblast-like synoviocytes (1, 2). These interactions lead to fibroblast hyperproliferation and formation of an invasive pannus tissue that produces collagenases and stromelysins, ultimately resulting in the destruction of neighboring cartilage and bone (3). The arthritic process apparently arises through a series of discrete stages. The activation and expansion of antigen-specific T and B cells in the synovium is believed to play a role in the initiation of the disease (4). Subsequently, in the “effector” phase, macrophages, neutrophils, and lymphocytes are recruited to the joint where, in concert with resident fibroblasts, they mediate the chronic destructive events through the release of cytokines, proteases, and other mediators (1, 2). However, the mechanisms that regulate these effector responses remain to be fully elucidated and it is not clear which chemokines and chemoattractants are primarily responsible for the localization of cells into the synovium.
Inflammatory cell recruitment into the synovial fluid and the synovial tissue occurs as a result of the production of chemoattractants by activated macrophages, synovial fibroblasts, and other cells in the inflamed joint. In recent years, attention has been focused on chemokine family members such as monocyte chemoattractant protein 1, RANTES, macrophage inflammatory protein (MIP)-1α, MIP-2α, and epithelial neutrophil activator (ENA)-78 (3). Chemokines signal through a number of related G protein–coupled receptors (GPCRs) that are expressed on distinct leukocyte cell populations, including the cells recruited to the joint during arthritogenesis. Given the large number of potential recruitment pathways that could contribute to joint inflammation, it has been difficult to determine which, if any, play unique roles that cannot be compensated by other chemokines and their receptors. Studies in animal models have suggested an important role for certain chemokine interactions with their receptors. For example, experimental arthritis can be partially ameliorated with modified RANTES (met-RANTES) and monocyte chemoattractant protein 1 antagonists (5–7). As these chemokines are promiscuous in binding to several distinct chemokine receptors, the relative importance of their cognate receptors in cell recruitment remains to be fully defined.
A second family of chemoattractant molecules, which signal through a related family of GPCRs, is comprised of complement fragments and bacterial peptides such as C3a, C5a, and formyl-MetLeuPhe (8–10). Like the chemokine family, a number of these chemotactic molecules are present in the synovial tissue and/or synovial fluid of RA patients (9). The presence of elevated anaphylatoxin C3a and C5a levels is but one of several indications that the complement system is hyperactive in RA patients. For example, it has long been appreciated that antibody-driven inflammatory responses, through activation of the complement cascade or Fc-dependent cellular activation, play a crucial role in arthritis. In this context, the synovial tissue and fluid of RA patients frequently contain abundant autoantibodies such as rheumatoid factor (11). In addition, arthritis can be induced in rodents by passive transfer of anticollagen antibodies (12). The potential pathogenic role of autoantibodies has been further highlighted in recent years with the advent of the K/BxN arthritis model, in which disease can be induced by the transfer of anti–glucose-6-phosphate isomerase (GPI) autoantibodies (13).
The C5a receptor (C5aR) is recognized to be a potent chemoattractant receptor on neutrophils, monocytes, macrophages, and eosinophils (14). The liberation of C5a at sites of complement activation therefore leads to the recruitment of these cell types into sites of infection or autoantibody deposition. The C5aR is also present on a variety of other cell types, including bronchial epithelial and smooth muscle cells as well as endothelial cells and hepatocytes, where expression has been implicated in cell activation and the induction of proinflammatory mediators (15). The C3a receptor (C3aR) is also present on an array of cell types, including inflammatory cells as well as resident lung epithelium and smooth muscle cells. The C3aR and C5aR anaphylatoxin receptors are perhaps best known for their function as activators of histamine release, bronchoconstriction, and vasodilation (15). As such, the C3aR was recently implicated in the effector phase of asthma (16, 17). However, the roles of these receptors in cell recruitment pathways during the course of arthritic inflammation have not been delineated. Here, we report that genetic deletion of the C5aR completely protects mice from arthritis induced with anticollagen antibodies, indicating a central role for C5a-dependent cell recruitment and activation in the initiation of arthritis in this model. These data raise the possibility that drugs and biotherapeutics targeting the C5aR may provide a novel strategy for therapeutic intervention to disrupt the effector phase of RA.
Materials and Methods
Mice.
The generation and characterization of C3aR−/− and C5aR−/− mice has been previously described (16, 18). C3aR−/− and C5aR−/− mice backcrossed three and four generations, respectively, onto a BALB/c background and littermate controls were used for all experiments described. Animal studies were performed according to institutional and National Institutes of Health guidelines for animal use and care.
Human Cell Isolation and Culture.
Normal human peripheral blood monocytes were isolated using CD14-MicroBeads and magnetic cell separation technology according to the manufacturer's instructions (Miltenyi Biotec). Macrophages were derived by culturing purified monocytes for 10 d in the presence of 100 ng/ml GM-CSF (Pierce Chemical Co.). Granulocytes were isolated from peripheral blood via Ficoll density gradient separation. Isolated cell populations were cultured in RPMI 1640 medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen) for 4 or 24 h in the presence of one of the following stimuli: 1 μg/ml Escherichia coli 026:B6 LPS (Sigma-Aldrich), 5 μg/ml recombinant human CD40L (PeproTech), or 100 ng/ml recombinant human TNFα (PeproTech). RNA was extracted and used in TaqMan® analyses as described below.
Immunohistochemistry.
Human RA synovial tissue was obtained as discarded material from joint replacement surgery. 5-μm sections of snap frozen tissue blocks were fixed in acetone and incubated in 0.3% goat serum (Vector Laboratories)/0.3% hydrogen peroxide in PBS for 5 min. Sections were incubated in PBS/5% goat serum for 30 min followed by anti-C5aR (BD Biosciences) or control rabbit IgG (Dako) for 1 h at room temperature. The slides were developed using the VECTASTAIN® Elite ABC Rabbit IgG kit and the AEC Peroxidase Substrate kit (Vector Laboratories) and counterstained in Mayer's hematoxylin (Poly Scientific). To detect IgG deposition, mouse joints were fixed and decalcified in 4% paraformaldehyde/10% EDTA (both from Sigma-Aldrich) for 14 d and embedded in optimum cutting temperature for sectioning. 6-μm sections were incubated with FCS to block Fc receptor binding followed by biotinylated rat anti–mouse IgG (Dako). Staining was revealed using StreptABCComplex/horseradish peroxidase detection (Dako) according to the manufacturer's instructions. To detect C3 and C1q deposition, joints were fixed for 4 d in buffered 10% formalin (Fisher Scientific), decalcified for 2 wk in 5% formic acid (Sigma-Aldrich), and processed for paraffin embedding. 6-μm sections were incubated with FCS followed by anti-C3, anti-C1q (both from Connex), or control rat IgG (Dako). Sections were then incubated with biotinylated rabbit anti–rat IgG and staining was revealed using StreptABCComplex/horseradish peroxidase detection. All slides were counter stained with hematoxylin (Poly Scientific).
Antibody-induced Experimental Arthritis.
10–12-wk-old C3aR−/− or C5aR−/− mice and wild-type littermates (five mice per group) were immunized intravenously with 4 mg Arthrogen-collagen-induced arthritis (CIA) type II collagen-specific mAbs, a mixture of four mAbs that recognize individual epitopes within the CB11 fragment of type II collagen (Chemicon International, Inc.). 48–72 h later, mice were administered 25 μg LPS (Chemicon International, Inc.) intraperitoneally and monitored for clinical signs of arthritis twice weekly for 14 d. Arthritis indications were scored as follows: 0 = normal, 1 = swelling in phalangeal joints only, 2 = severe local swelling or moderate swelling over whole paw, 3 = severe swelling over whole paw, and 4 = ankylosis. Scoring was performed by a blinded observer.
Histological Analysis of Arthritis.
Knees, hind paws, and forepaws from one side of each mouse were fixed for 4 d in buffered 10% formalin (Fisher Scientific), decalcified for 2 wk in 5% formic acid (Sigma-Aldrich), and processed for paraffin embedding. 6-μm sections were stained with hematoxylin and eosin (H&E; Poly Scientific) and scored by a blinded observer for inflammation, pannus formation/cartilage loss, and bone erosion (see Table II). Severity was scored in a range from 0–4 for each parameter and the degree of involvement was determined by the percentage of articular surfaces affected by pannus formation.
Table II.
Histological Analysis of C5aR−/− and C5aR+/+ Joints
| Inflammation
|
Cartilage
|
Bone
|
||||||
|---|---|---|---|---|---|---|---|---|
| Rear | Fore | Rear | Fore | Rear | Fore | Total | Mean | |
| C5aR+/+ 1 | 4 | 3 | 3 | 4 | 3 | 4 | 21 | 21.25 |
| C5aR+/+ 2 | 4 | 4 | 3 | 4 | 3 | 4 | 22 | |
| C5aR+/+ 3 | 4 | 4 | 3 | 4 | 3 | 3 | 21 | |
| C5aR+/+ 4 | 4 | 4 | 4 | 4 | 2 | 3 | 21 | |
| C5aR−/− 1 | 1 | 1 | 0 | 0 | 0 | 0 | 2 | 2.25 |
| C5aR−/− 2 | 1 | 1 | 0 | 0 | 0 | 0 | 2 | |
| C5aR−/− 3 | 1 | 1 | 0 | 0 | 0 | 0 | 2 | |
| C5aR−/− 4 | 1 | 2 | 0 | 0 | 0 | 0 | 3 | |
RNA Extraction and PCR Analysis.
Total RNA was prepared from tissues and cells by a single step extraction method using RNA STAT-60 (Tel-Test, Inc.). After DNase I (QIAGEN) treatment, cDNA was synthesized using Multiscribe Reverse Transcription Kit (Applied Biosystems). Gene expression was measured by TaqMan® real-time PCR (Applied Biosystems) according to the manufacturer's protocols. Probes and primer sets for mouse GAPDH and mouse IL-1β were purchased from Applied Biosystems. Sequences of all other primers and probes are listed in Table I.
Table I.
Sequence of Primers and Probes
| Mouse | Forward Primer | Reverse Primer |
|---|---|---|
| C3aR | 5′-GGCCCTGGGAACCTAAGC-3′ | 5′-TGAGAGTCGCCAAATTGTCTAAAG-3′ |
| C5aR | 5′-GGGTAGCTTCCCCAAAGAGAA-3′ | 5′-AGGAGGAAGGTGTAGCAGATGTTTA-3′ |
| TNFα | 5′-ACAAGGCTGCCCCGACTAC-3′ | 5′-CGGCAGAGAGGAGGTTGACTT-3′ |
| MMP-3 | 5′-TGAGTCTTTGTGAAAGGAAGTGCTT-3′ | 5′-ATCCTTTGACAACTTGACGTTGAC-3′ |
| CD4 | 5′-TCTGCATCCTCTGCTGTGTCA-3′ | 5′-AGCCTCTTGATCTGAGACATTCG-3′ |
| CD19 | 5′-AAACCCAATTAGCCACTCAAATTCT-3′ | 5′-CCAGGATGTACTCAGACCTCAGTTC-3′ |
| CD68 | 5′-TGACCTGCTCTCTCTAAGGCTACA-3′ | 5′-AGGACCAGGCCAATGATGAG-3′ |
| OPG | 5′-ATCTCGGCCACTCGAACCT-3′ | 5′-CAATCTCTTCTGGGCTGATCTTC-3′ |
| OPGL | 5′-GCTTATGAAAAACTTACACGTGAGCTAT-3′ | 5′-TGGCATCCTCTTAATTTCAAGGTT-3′ |
| ENA-78 | 5′-TTCAGAAAATATTGGGCAGTGACA-3′ | 5′-CTATTGAACACTGGCCGTTCTTT-3′ |
| MIP-1α | 5′-TCATCGTTGACTATTTTGAAACCAG-3′ | 5′-GCCGGTTTCTCTTAGTCAGGAA-3′ |
| MIP-2α | 5′-CAAGAACATCCAGAGCTTGAGTGT-3′ | 5′-CTTGAGAGTGGCTATGACTTCTGTCT-3′ |
| ICAM-1 | 5′-CCCACAGTGGGTCGAAGGT-3′ | 5′-GATCCTCCGAGCTGGCATT-3′ |
| VCAM-1 | 5′-GTATTAAAGTCTGTGGATGGCTCGTA-3′ | 5′-TCAGTCTTAGATTCACACTCGTATATGC-3′ |
| E-selectin | 5′-GCTAGGCAGACATATTGGCTTTATC-3′ | 5′-GGACACAGAACACAATGTTCAAGAAT-3′ |
| Human | Forward Primer | Reverse Primer |
| C5aR | 5′-AGGTGGGAGAATTGCTCGAA-3′ | 5′-AGAGTGCAGTGGTGCGATCA-3′ |
| β2m | 5′-CACCCCCACTGAAAAAGATGA-3′ | 5′-CTTAACTATCTTGGGCTGTGACAAAG-3′ |
| Mouse | Probe | |
| C3aR | 5′-CACATGTCATCCATCACTGTTCACCTGAGAA-3′ | |
| C5aR | 5′-AAACCCACCATCAGCCGCAGGA-3′ | |
| TNFα | 5′-CCTCACCCACACCGTCAGCCG-3′ | |
| MMP-3 | 5′-CTCCTGTTTGGTTCTGCCATAGCACATG-3′ | |
| CD4 | 5′-CCGGCACCAACAGCGCCA-3′ | |
| CD19 | 5′-TTCCTATCCCACACAAGGGCCGG-3′ | |
| CD68 | 5′-CCCGAAGTGTCCCTTGTCAGGCA-3′ | |
| OPG | 5′-CAGGCAGGCTCTCCATCAAGGCA-3′ | |
| OPGL | 5′-TGGTCTAACCCCTGGACATGTGCCA-3′ | |
| ENA-78 | 5′-CACTGCGAGTGCATTCCGCTTAGCT-3′ | |
| MIP-1α | 5′-AGCCTTTGCTCCCAGCCAGGTGTC-3′ | |
| MIP-2α | 5′-CCCCCAGGACCCCACTGCG-3′ | |
| ICAM-1 | 5′-CCACCCACCCCGCAAGTCCAAT-3′ | |
| VCAM-1 | 5′-CCAGGCACAGCTGCAGGATGCC-3′ | |
| E-selectin | 5′-TCGGCAGCCACTGAATCCCATG-3′ | |
| Human | Probe | |
| C5aR | 5′-TGGAGGTGGAGGTTGTGGTGAGCC-3′ | |
| β2m | 5′-ATGCCTGCCGTGTGAACCACGTG-3′ |
Target gene probes were labeled with 6-carboxyfluorescein (FAM) and the internal reference probes, human β2-microglobulin and rodent GAPDH, were labeled with VIC. PCR reactions contained the forward and reverse primers (200 nM) and the probe (100 nM) for β2-microglobulin or GAPDH and the forward and reverse primers (600 nM) and probe (200 nM) for the gene of interest. The experiments were performed on an ABI PRISM 7700 Sequence Detection System (Applied Biosystems) using the following conditions: 2 min at 50°C, 10 min at 95°C, followed by two-step PCR for 40 cycles of 95°C for 15 s followed by 60°C for 1 min. The number of PCR cycles needed for FAM and VIC fluorescence to cross a threshold of a statistically significant increase in fluorescence (Ct = threshold cycle) was measured using Applied Biosystems software. Relative target gene expression was determined using this formula: Relative Expression = 2-(ΔΔCT) where ΔΔCT = (Cttarget gene – Ctreference gene in experimental cDNA sample) – (Cttarget gene – Ctreference gene in mock reverse transcribed RNA sample).
Myeloperoxidase (MPO) Assay.
Frozen joints were pulverized with a Bessman tissue pulverizer (BioSpec Products) on dry ice placed in 4 ml MPO buffer (50 mM Na2HPO4/0.5% hexadecyl trimethyl ammonium bromide/10 mM EDTA, pH 5.4) and homogenized with three 30-s bursts with a PowerGen tissue homogenizer (Fisher Scientific). The samples were subjected to three rounds of freeze/thaw at −80°C. The homogenate was sonicated briefly in a water bath sonicator at maximum intensity. Insoluble material was removed by centrifugation in a microcentrifuge at 14,000 rpm for 5 min. Total protein content of the supernatant was determined using the bicinchoninic acid protein assay (Pierce Chemical Co.) and MPO activity was measured as follows. Samples were plated in 96-well tissue culture plates (Fisher Scientific) in a volume of 25 μl/well. 25 μl 1.67 mg/ml O-dianisidine (Sigma-Aldrich) in 50 mM Na2HPO4, pH 5.4, was added to each well. 200 μl 0.009% H2O2 in 50 mM Na2HPO4, pH 5.4, was then added to each well and incubated for 10 min at room temperature. The A450 was measured and background subtracted for wells containing MPO buffer alone. The A450/mg total protein was then calculated to determine the relative MPO activity.
Results
Expression of the C3aR and C5aR in Human Cells and in RA Synovium.
To identify chemoattractant receptors expressed abundantly on key inflammatory cell types involved in arthritic pathogenesis, we performed a quantitative mRNA expression analysis using normal peripheral blood-derived monocytes, macrophages, and granulocytes. We extracted RNA from each of these populations before and after activation with relevant stimuli, synthesized cDNA, and performed TaqMan® real-time PCR analysis of the anaphylatoxin receptors C3aR and C5aR and the formyl-MetLeuPhe receptor-like GPCRs, FPRL1, and FPRL2. Together, these receptors form a chemoattractant receptor subfamily of GPCRs, although the biologic function and ligands for FPRL1 and FPRL2 have yet to be fully delineated. Of these receptors, C5aR had the highest levels of expression in granulocytes, monocytes, and macrophages (Fig. 1 A). Expression of C5aR appeared to be increased in granulocytes after a 4-h stimulation with TNFα. However, expression in monocytes and macrophages was not appreciably regulated by cell activation. C3aR expression was also notable in these three cell populations and like the C5aR, expression was augmented transiently in TNFα-stimulated granulocytes. In comparison, FPRL1 and FPRL2 were expressed at lower levels (Fig. 1 A). Given the abundant relative expression of C3aR and C5aR on these cell types, we performed additional expression analyses of these two receptors in diseased human tissue.
Figure 1.

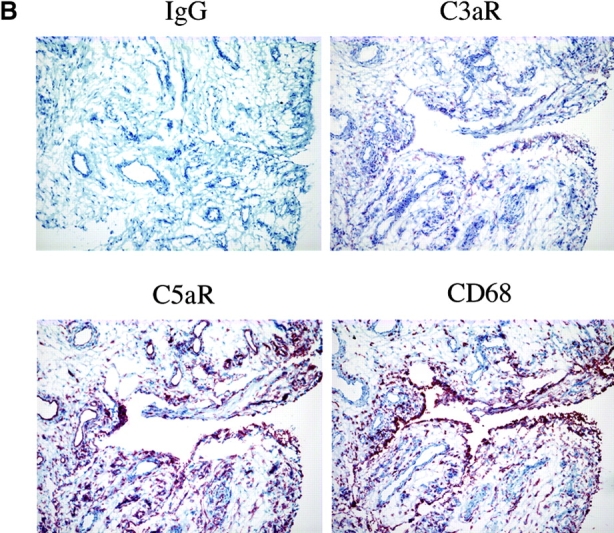

Expression of C5aR in human RA tissue and in a mouse model of arthritis. (A) Quantitative PCR analysis of C5aR, C3aR, FPRL1, and FPRL2 in untreated (UnTx) or activated (LPS, TNFα, IFNγ, or CD40L for 4 or 24 h) normal human peripheral blood monocytes (MONO), monocyte-derived macrophages (Mφ), and peripheral blood–derived granulocytes (GRAN). Data plotted are the mean ± SD of the relative expression of each receptor in RNA samples pooled from three different donors using β2-microglobulin as a reference. The results are representative of three independent experiments. (B) Immunohistochemistry analysis of human RA synovial tissue stained with anti-C5aR, anti-C3aR, anti-CD68, or control IgG. Data shown are representative of expression patterns observed of four RA synovial tissues. (C) Quantitative PCR analysis of C5aR, C3aR, and inflammatory marker expression in mouse joint RNA during the course of arthritis induction. Data are the mean ± SD of the relative expression of each gene in two joints.
To examine the distribution and expression of the C3aR and C5aR in inflamed human synovium, we performed an immunohistochemical analysis on frozen sections of human RA synovium from multiple individuals. Pronounced staining of both receptors was observed in the intimal layer, consistent with localization to macrophage-like synoviocytes, as well as scattered throughout the subintimal region (Fig. 1 B). Of the two receptors, C5aR was consistently more pronounced in expression, although this may in part reflect differences in avidities between the two antibodies. Immunohistochemistry with an anti-CD68 mAb on serial sections showed a very similar pattern of expression, with intense staining in the intimal layer and scattered positive subintimal cells. Thus, it is likely that the majority of C3aR+ and C5aR+ cells in the RA synovium are macrophages, although it is possible that C3aR+ and C5aR+ infiltrating neutrophils and other granulocytes are also present. In addition, C5aR was clearly detected on endothelial cells lining the blood vessels within synovial tissues (Fig. 1 B). The abundance of expression of these receptors in inflamed synovial tissue together with the high levels of expression on three major effector cell populations prompted us to examine the role of the C3aR and C5aR in the recruitment of inflammatory cells in a rodent model of arthritis.
Inflammatory Gene Regulation in Anticollagen Antibody-induced Arthritis.
To characterize the inflammatory changes that occur during the course of anticollagen mAb-induced arthritis, we isolated joints from mice 24, 48, and 72 h and 5, 7, 9, 11, and 13 d after intravenous injection with an arthritogenic antibody cocktail. At each time point, we assessed joint swelling and then froze isolated joints for subsequent expression analyses. We used quantitative PCR to measure the relative expression and changes in transcription of a panel of inflammatory markers, including markers for the major immune cell subsets. Most markers examined showed a consistent pattern of induction 72 h after mAb injection (24 h after an intraperitoneal LPS boost given to the animals; Fig.1 C). In particular, IL-1β and matrix metalloproteinase (MMP)-3 (stromelysin 1) showed marked induction from very low basal levels to high levels that were sustained to the last time point examined, 13 d after mAb injection. CD4 and CD68 mRNA levels increased substantially and with similar kinetics, reflecting the influx of T cells and macrophages, respectively, during the development of arthritis. Similarly, C3aR and C5aR expression dramatically increased 72 h after mAb administration, likely indicating the influx of neutrophils and monocyte/macrophage lineage cells, the principal immune cell subsets on which those receptors are found. In addition, the increase in C5aR expression may reflect augmented transcription in resident tissue macrophages and endothelial cells. Osteoprotegerin ligand (OPGL) message levels were also increased, presumably a reflection of the influx of T cells into the joint space, whereas osteoprotegerin, expressed mainly by resident chondrocytes and osteoclast precursors, remained relatively stable. In contrast, the expression of CD19, a marker for B lineage cells, did not change dramatically, indicating that B cells are not a major cell subset recruited into joints in this animal model (unpublished data).
C5aR-deficient Mice Are Resistant to Arthritis Induction.
As C3aR and C5aR expression were clearly up-regulated during the course of disease in this model of arthritis, we examined the effect of deleting expression of each of these receptors on disease induction and severity. Throughout the 14-d course of arthritogenesis, the paw swelling scores of C3aR−/− mice were nearly identical to those of C3aR+/+ littermates (Fig. 2 A). Histological analyses of joint sections taken from C3aR−/− and C3aR+/+ animals 2 wk after arthritis induction revealed similar degrees of inflammatory cell recruitment, fibroblast proliferation, and cartilage and bone destruction (Fig. 2 B). Thus, deletion of the C3aR appears to have no substantial effect on this animal model of arthritis. However, strikingly, in three independent experiments C5aR−/− mice were completely protected from disease induction as assessed by paw swelling (n = 5 per experiment). In contrast, wild-type animals consistently and synchronously developed arthritis 72 h after inoculation with the arthritogenic antibodies (Fig. 2 A). We removed joints from animals 14 d after arthritis induction and examined them for histological changes (Table II). H&E stained sections from wild-type arthritic mice had large, obvious inflammatory cell infiltrates. Pannus formation was also evident with expansion of the synovial membrane and proliferation of the synovial lining fibroblasts. Lastly, erosion of the cartilage and bone surfaces was apparent, accompanied by invasion of the pannus tissue into the bone space, reminiscent of the severe changes associated with human RA. The most severe cases showed complete destruction of the normal joint architecture. Consistent with the clinical observation that C5aR−/− animals had reduced gross inflammation, joint sections from these animals appeared normal with thin synovial lining, only rare infiltrating inflammatory cells, and normal smooth cartilage surfaces indicating a lack of cartilage- and bone-erosive processes (Fig. 2 B).
Figure 2.

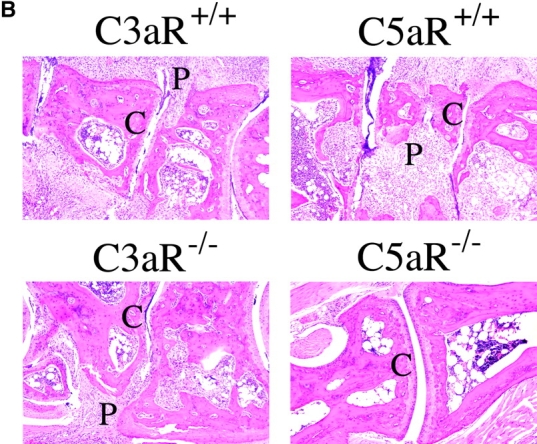

C5aR−/− mice are protected from arthritis induction. (A) Mean clinical scores at days 0, 3, 4, 7, 9, and 14 of arthritis development in C3aR−/− (bottom) or C5aR−/− (top) mice and their littermate controls. Each group of mice consisted of five animals. The results shown are representative of three separate experiments. (B) H&E stained joint sections from representative C3aR−/−, C3aR+/+, C5aR−/−, and C5aR+/+ mice 14 d after mAb transfer. Cartilage (C) surfaces are indicated in the images as are regions of pannus tissue (P) comprised of proliferating synoviocytes and infiltrating leukocytes. (C) Joints from naive C5aR+/+ mice. Ab-injected C5aR+/+ or Ab-injected C5aR−/− mice were stained with an antibody against mouse IgG to detect deposition (brown staining, top). Inflamed joints from C5aR+/+ mice 14 d after Ab transfer were stained with control IgG, anti-C3, or anti-C1q antibodies to detect the accumulation of complement components on the cartilage surfaces (bottom).
Evidence for Antibody Deposition and Complement Activation.
The failure of C5aR−/− mice to develop arthritis could be due to prevention of the localization of anticollagen antibodies to the joint in the absence of the receptor. To exclude this possibility, we analyzed joint sections from C5aR+/+ and C5aR−/− animals after anticollagen antibody transfer for deposition on the cartilage surfaces. As expected, we observed no antibody deposition in joints from naive animals. In contrast, we readily detected antibody deposition in joints from both C5aR+/+ and C5aR−/− mice (Fig. 2 C, top). To additionally examine whether anticollagen deposition on articular surfaces in this model induces complement activation, we analyzed joint sections from naive and arthritic mice for the presence of C3, a marker of activation of both alternative and classical pathways, and C1q, a marker of classical pathway activation. We detected both C3 and C1q deposition on cartilage in diseased joints (Fig. 2 C, bottom). In comparison, low background staining was observed in sections stained with a control antibody (Fig. 2 C) and in joints from naive mice (not depicted). Thus, in this acute effector model of arthritis, the deposition of autoreactive antibodies on articular cartilage leads to the activation of the classical pathway of complement activation.
Quantitative Assessment of Joint Inflammation in C5aR-deficient Mice.
Although mice lacking the C5aR were clearly much less sensitive to arthritis induction than their wild-type littermates, we felt it important to quantitatively assess the influx of inflammatory cells into the joint as well as the induction of cytokines and proteases associated with arthritis progression. Therefore, we isolated RNA from the joints of C5aR−/− and C5aR+/+ animals 14 d after arthritis induction. We then compared the relative expression of CD4, CD68, and CD19 via quantitative PCR to assess the extent of the influx of T cells, macrophages, and B cells, respectively. C5aR+/+ mice showed clear increases in mRNA levels of CD4 and CD68 relative to naive joints, consistent with the influx of inflammatory cells into the joints (Fig. 3) . In contrast, mRNA levels in the joints from C5aR−/− animals after arthritis induction were significantly lower than in wild-type animals. In fact, the levels of these markers remained within the range of naive animals, indicating a profound reduction in the number of T cells and macrophages recruited into the joint. Similarly, we analyzed the level of expression of several inflammatory markers, including IL-1β, TNFα, and MMP-3. The levels of mRNA for each of these genes were significantly increased in joints from arthritic wild-type mice in comparison to naive joints (Fig. 3). As before, mRNA levels in C5aR−/− animals remained at baseline levels 2 wk after injection of the arthritogenic mAbs. Similarly, we observed that OPGL mRNA levels increased substantially in wild-type mice during the course of disease, yet failed to change in C5aR−/− animals. This difference is likely to reflect the reduced osteoclastogenesis and bone erosion in C5aR−/− mice.
Figure 3.

Quantitative assessment of inflammatory markers in C5aR+/+ and C5aR−/− mouse joints. RNA was extracted from the joints of naive wild-type or mAb-injected C5aR+/+ (+/+ A) or mAb-injected C5aR−/− (−/− A) mice 13 d after mAb transfer and analyzed via quantitative PCR analysis for the indicated genes. The levels of IL-1β, CD68, TNFα, CD4, MMP-3, and OPGL were significantly reduced in the joints from C5aR−/− mice injected with the arthritogenic antibodies (−/− A) compared with injected C5aR+/+ mice (+/+ A; P < 0.0001, Student's t test). Data are the mean ± SD of the relative expression of each gene in ten joints each from naive mice or from mAb-injected C5aR−/− or C5aR+/+ mice. The results are representative of three independent experiments.
Reduced Influx of Neutrophils into Joints of C5aR-deficient Mice.
The C5aR is expressed on and mediates chemotaxis of neutrophils, monocytes, and macrophages. In addition to C5a, other mediators of neutrophil recruitment, including the chemokines MIP-1α, MIP-2, and ENA-78, have been found at elevated levels in RA synovial tissue, synovial fluid, and peripheral blood. To assess the effect of C5aR deletion on the recruitment of neutrophils into the joint space after arthritis induction, we measured the relative expression of several chemokines known to recruit neutrophils via receptors other than the C5aR. In wild-type animals, MIP-1α, MIP-2α, and ENA-78 levels were strongly induced during the course of arthritis development. C5aR−/− animals, however, failed to show induction of any of these chemoattractants above the background levels present in joints from naive mice (Fig. 4 A). To directly assess the influx of neutrophils into the joints of C5aR−/− and C5aR+/+ mice, we prepared protein extracts from frozen joints 2 wk after arthritis induction. We then measured the levels of MPO enzyme activity in the joint extracts as a measure of neutrophil influx. A significant decrease (fourfold, P < 0.005) in levels of MPO was observed in the C5aR−/− animals relative to wild-type littermates, indicating a reduction in the recruitment of neutrophils into the joint as a consequence of C5aR deletion (Fig. 4 B). The decreased MPO enzyme levels reflect the profound reduction in the numbers of neutrophils present in the joints from C5aR−/− animals as is readily observed in joint sections at high magnification (Fig. 4 C).
Figure 4.


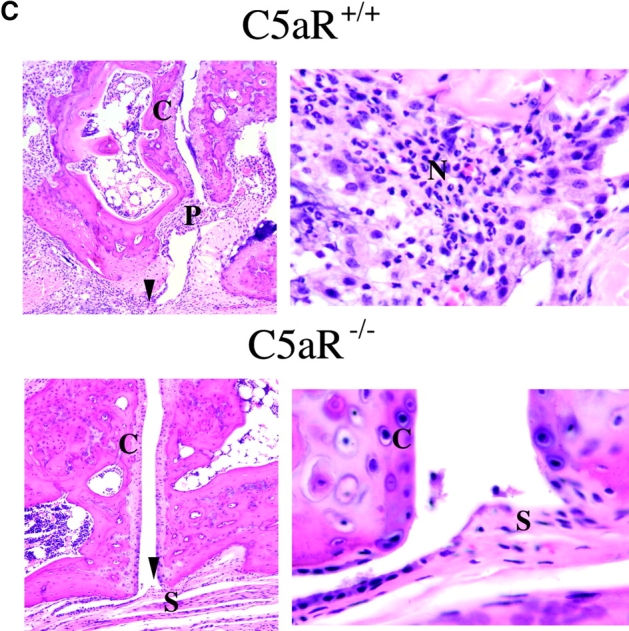
Decrease in neutrophilia, chemokines, and adhesion receptor induction in C5aR−/− mice. (A) Neutrophil chemotactic factors ENA-78 (P < 0.0001, Student's t test), MIP-1α (P < 0.0001), and MIP-2 (P < 0.0001), and adhesion molecules ICAM-1 (P < 0.002), VCAM-1 (P < 0.0001), and E-selectin (P < 0.0001) were significantly reduced in joints from C5aR−/− mice injected with the arthritogenic antibodies (−/− A) compared with injected C5aR+/+ mice (+/+ A). Quantitative PCR data are expressed as described in Fig. 3. (B) Neutrophilia is reduced in joints from C5aR−/− mice injected with the arthritogenic antibodies compared with injected C5aR+/+ mice as assessed by an assay for MPO performed with protein extracts from five joints each from C5aR+/+ and C5aR−/− mice 14 d after mAb transfer (P < 0.005). (C) Neutrophilia (N) in arthritic C5aR+/+ joints (top) is evident from examination of high powered fields of tissue sections stained with H&E. In contrast, only rare neutrophils were observed in joints from C5aR−/− animals 14 d after mAb transfer. Arrowheads indicate region of lower power fields (left) enlarged in the high power fields (right). Cartilage (C), pannus (P), and synovial lining (S) are indicated.
Reduced Induction of Adhesion Molecule Expression in the Joints of C5aR-deficient Mice.
One mechanism by which proinflammatory cytokines and chemokines recruit leukocytes into the inflamed joint is through the induction of adhesion molecules on local endothelium and synovial fibroblasts themselves. intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, and E-selectin are three of the principal adhesion receptors known to be up-regulated during the course of arthritis. Therefore, we measured the quantitative induction of mRNA for these three molecules in C5aR−/− and wild-type mice during arthritogenesis. Each of these adhesion molecules was substantially up-regulated in joints from wild-type animals. However, in joints from C5aR−/− mice, VCAM-1 and E-selectin mRNA levels remained at baseline levels (Fig. 4 A). A slight induction of ICAM-1 was observed in C5aR-deficient joints, although the levels were significantly lower than in arthritic wild-type littermates. Therefore, the induction of the major adhesion molecules involved in the extravasation and localization of inflammatory cells in the arthritic joint is inhibited in C5aR−/− mice.
Discussion
A role for the complement system in RA is suggested by a number of observations. Perhaps foremost among these is that rheumatoid factors, autoantibodies against the Fc portion of IgG, are present in a large proportion of RA patients (11). Deposition of immune complexes in these patients, as in other autoimmune diseases such as SLE, would lead to activation of the complement system. This activation would be manifested in the proinflammatory effects of anaphylatoxins C3a and C5a and complement receptor-mediated cellular activation by C3b and other proteins in the pathway. In fact, elevated levels of C3a and C5a have been noted in the synovial fluid and peripheral blood of RA patients (19–21). Additionally, other cell surface and soluble proteins relevant to complement system activation and control are induced in RA, such as the increased expression of CD55 in the intimal layer of inflamed synovia and the increased circulating C-reactive protein levels in the peripheral blood of RA patients (22). These findings point toward a hyperactivity of the complement and innate immune system pathways in human RA, perhaps indicative of a causative or perpetuating role for these systems in chronic joint inflammation.
The principal mouse models of arthritis, such as CIA and the passive antibody transfer model, also have clear antibody and complement-driven components. Although collagen is the principal self-antigen for these models, it has recently become clear that other autoantibody targets such as GPI can substitute as initiating factors in experimental arthritis (23). Therefore, as autoreactive immune complexes form and are deposited in healthy joint tissue, the complement cascade is activated leading to the liberation of C5a. Our findings indicate that subsequent C5a-dependent recruitment of neutrophils and other cells via interaction with the C5aR is a crucial step in the cascade of events leading to the arthritic environment in the joint. It has been shown that anticollagen antibodies become deposited in the joint and activate complement in the absence of C5 (24). Here, we demonstrate that anticollagen antibodies traffic to the joints of C5aR−/− mice and are deposited on articular cartilage. Furthermore, we show that in this model, antibody deposition in the joint is associated with the presence of complement components C3 and C1q, which are also detected on the cartilage surface. Therefore, the failure of C5aR−/− mice to develop arthritis after anticollagen antibody transfer does not reflect reduced antibody deposition and consequent complement activation, but rather indicates a failure of C5aR-expressing cell populations to respond to those events. Data published while this report was in preparation suggest that in a model of arthritis induced by the transfer of antibodies against the ubiquitous enzyme GPI, the classical pathway of complement activation is dispensable (25). It is possible that there are fundamental mechanistic differences between anti-CIA and anti-GPI–induced disease. Alternatively, both models may activate the classical pathway as manifest by C1q deposition on articular cartilage, but the alternative pathway can compensate in its absence.
In contrast to C5aR, targeted disruption of C3aR expression had no discernible effect on the induction of arthritis induced by anticollagen mAbs. This difference points toward neutrophil recruitment as an important component of arthritogenesis in this model because C3aR protein expression is much lower than C5aR on neutrophils and does not appear to substantially induce chemotaxis of this cell type (26–28). Here, we demonstrate that neutrophils, T cells, and macrophages are not recruited in the absence of the C5aR during arthritis induction in a model that bypasses the stage of antigen-specific T and B cell development. In addition, MIP-1α, MIP-2, and ENA-78 are not induced in the absence of the C5aR. Therefore, in this animal model, C5a-dependent stimulation of leukocytes, endothelial cells, or other cell types leads to the production of chemokines and proinflammatory mediators that contribute to the disease process. The adhesion molecules VCAM-1, ICAM-1, and E-selectin, which are induced in wild-type mice during arthritogenesis and believed to play important roles in the extravasation of inflammatory cells into the arthritic joint, are not induced in the absence of the C5aR. Because the C5aR is expressed on endothelial cells, signaling through this receptor may lead to the up-regulation of adhesion receptors important for the extravasation of inflammatory cells. Alternatively, factors produced by C5aR+ neutrophils and monocytes might be required for the induction of adhesion receptors on the endothelium. Our findings suggest that neutrophilia is a critical early event in the initiation of disease in this model and that in the absence of the C5aR, neutrophils are not recruited to sites of immune complex deposition and complement activation. Therefore, production of chemokines and other effector molecules by neutrophils might be essential for the recruitment of other inflammatory cells and the subsequent chronic stage of arthritis (Fig. 5) . Consistent with this interpretation, it was recently reported that depletion of neutrophils prevented the induction of arthritis in the K/BxN serum transfer model and depletion of neutrophils after disease initiation led to an amelioration of joint damage and inflammation (29).
Figure 5.

Model for the inflammatory cascade of arthritis. After the initiation of disease as a result of the activation of autoreactive T and B cells, rheumatoid factor and other autoantibodies are produced and deposited in the joint spaces. Immune complex formation leads to the activation of the complement cascade, ultimately yielding C5a. The C5a gradient emanating from the joint recruits and activates neutrophils and monocytes to the synovium and synovial fluid. In addition, C5a may directly activate endothelial cells to up-regulate adhesion receptor expression, promoting extravasation of inflammatory cells. Activated neutrophils liberate additional chemotactic factors that recruit additional inflammatory cell subsets, particularly monocyte/macrophage lineage cells. These latter cells, a primary source of TNFα and IL-1β, activate resident synovial fibroblasts. Activated synovial fibroblasts produce additional effector molecules, including cytokines, chemokines, and proteases. The end result is a cycle of positive feedback that amplifies the inflammation and results in cartilage and bone degradation.
Beyond the profound inhibition of inflammation, we observed no signs of cartilage and bone degradation in C5aR−/− mice. Joint histology of C5aR−/− animals after arthritogenic mAb transfer was indistinguishable from naive joints. At the molecular level, we observed a complete inhibition of induction of a key cartilage-degrading enzyme, MMP-3. In addition, OPGL expression in the joints of C5aR−/− mice remained at baseline levels, reflecting a failure to activate a key pathway believed to regulate osteoclastogenesis and bone resorption in arthritis (30, 31). Therefore, the deposition of autoantibodies and resulting activation of the complement cascade do not provide the signals necessary to progress to joint degradation in the absence of recruited inflammatory cells.
A variety of mouse strains, including NOD, are C5 deficient due to a genetic deletion encompassing a portion of the gene encoding this component of the complement cascade. These strains are resistant to arthritis induced by collagen and anti–glucose-6-phosphate autoantibodies (24, 32, 33). A role for C5 in arthritis is further supported by the observation that an anti-C5 antibody prevented CIA and ameliorated established disease (34). More recently, clinical trials with anti-C5 antibodies have been initiated to treat human RA. However, the mechanistic basis for resistance of C5-deficient mice to disease has never been delineated. The activation of the complement cascade as a result of deposition of anticollagen antibodies in the joint would lead to the liberation of C5a and the recruitment or activation of C5aR-bearing cells. In addition, the cleavage of C5 results in the formation of C5b and recruitment of additional components of the complement cascade, ultimately resulting in the formation of the membrane attack complex (MAC). The formation of the MAC in the joint could result in the necrosis of local cells and nonlytic effects of the MAC, such as induction of inflammatory mediator release and cellular proliferation, both of which could contribute to the arthritic condition. Our results indicate that the protective effect of C5 deficiency can be explained as an inability to form C5a and recruit and/or activate cells through the C5aR. Although it seems from our results that MAC-dependent processes are insufficient to cause arthritis in this model, we cannot exclude the possibility that C5b-dependent processes exacerbate disease. Given the extent to which arthritis is inhibited in an effector model of arthritis, novel inhibitors or biotherapeutics directed against the C5aR may show promise in the treatment of human RA.
Footnotes
Abbreviations used in this paper: C3aR, C3a receptor; C5aR, C5a receptor; CIA, collagen-induced arthritis; ENA, epithelial neutrophil activator; GPCR, G protein–coupled receptor; GPI, glucose-6-phosphate isomerase; H&E, hematoxylin and eosin; ICAM, intercellular adhesion molecule; MAC, membrane attack complex; MIP, macrophage inflammatory protein; MMP, matrix metalloproteinase; MPO, myeloperoxidase; OPGL, osteoprotegerin ligand; RA, rheumatoid arthritis; VCAM, vascular cell adhesion molecule.
References
- 1.Firestein, G.S. 1996. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum. 39:1781–1790. [DOI] [PubMed] [Google Scholar]
- 2.Kinne, R.W., R. Brauer, B. Stuhlmuller, E. Palombo-Kinne, and G.R. Burmester. 2000. Macrophages in rheumatoid arthritis. Arthritis Res. 2:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choy, E.H., and G.S. Panayi. 2001. Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 344:907–916. [DOI] [PubMed] [Google Scholar]
- 4.Fox, D.A. 1997. The role of T cells in the immunopathogenesis of rheumatoid arthritis: new perspectives. Arthritis Rheum. 40:598–609. [DOI] [PubMed] [Google Scholar]
- 5.Plater-Zyberk, C., A.J. Hoogewerf, A.E. Proudfoot, C.A. Power, and T.N. Wells. 1997. Effect of a CC chemokine receptor antagonist on collagen induced arthritis in DBA/1 mice. Immunol. Lett. 57:117–120. [DOI] [PubMed] [Google Scholar]
- 6.Gong, J.H., L.G. Ratkay, J.D. Waterfield, and I. Clark-Lewis. 1997. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J. Exp. Med. 186:131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogata, H., M. Takeya, T. Yoshimura, K. Takagi, and K. Takahashi. 1997. The role of monocyte chemoattractant protein-1 (MCP-1) in the pathogenesis of collagen-induced arthritis in rats. J. Pathol. 182:106–114. [DOI] [PubMed] [Google Scholar]
- 8.Gerard, C., and N.P. Gerard. 1994. The pro-inflammatory seven-transmembrane segment receptors of the leukocyte. Curr. Opin. Immunol. 6:140–145. [DOI] [PubMed] [Google Scholar]
- 9.Feldmann, M., F.M. Brennan, and R.N. Maini. 1996. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 14:397–440. [DOI] [PubMed] [Google Scholar]
- 10.Gerard, C., and B.J. Rollins. 2001. Chemokines and disease. Nat. Immunol. 2:108–115. [DOI] [PubMed] [Google Scholar]
- 11.Pope, R.M., and H. Perlman. 2000. Rheumatoid arthritis. Principles of Molecular Rheumatology. G.C. Tsokos, editor. Humana Press, Totowa, NJ. 325–361.
- 12.Stuart, J.M., and F.J. Dixon. 1983. Serum transfer of collagen-induced arthritis in mice. J. Exp. Med. 158:378–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouskoff, V., A.S. Korganow, V. Duchatelle, C. Degott, C. Benoist, and D. Mathis. 1996. Organ-specific disease provoked by systemic autoimmunity. Cell. 87:811–822. [DOI] [PubMed] [Google Scholar]
- 14.Gerard, C., and N.P. Gerard. 1994. C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu. Rev. Immunol. 12:775–808. [DOI] [PubMed] [Google Scholar]
- 15.Wetsel, R.A., J. Kildsgaard, and D.L. Haviland. 2000. Complement anaphylatoxins (C3a, C4a, C5a) and their receptors (C3aR, C5aR/CD88) as therapeutic targets in inflammation. Therapeutic Interventions in the Complement System. J.D. Lambris and V.M. Holers, editors. Humana Press, Totowa, NJ. 113–153.
- 16.Humbles, A.A., B. Lu, C.A. Nilsson, C. Lilly, E. Israel, Y. Fujiwara, N.P. Gerard, and C. Gerard. 2000. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature. 406:998–1001. [DOI] [PubMed] [Google Scholar]
- 17.Bautsch, W., H.G. Hoymann, Q. Zhang, I. Meier-Wiedenbach, U. Raschke, R.S. Ames, B. Sohns, N. Flemme, A. Meyer zu Vilsendorf, M. Grove, et al. 2000. Cutting edge: guinea pigs with a natural C3a-receptor defect exhibit decreased bronchoconstriction in allergic airway disease: evidence for an involvement of the C3a anaphylatoxin in the pathogenesis of asthma. J. Immunol. 165:5401–5405. [DOI] [PubMed] [Google Scholar]
- 18.Hopken, U.E., B. Lu, N.P. Gerard, and C. Gerard. 1996. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature. 383:86–89. [DOI] [PubMed] [Google Scholar]
- 19.Moxley, G., and S. Ruddy. 1985. Elevated C3 anaphylatoxin levels in synovial fluids from patients with rheumatoid arthritis. Arthritis Rheum. 28:1089–1095. [DOI] [PubMed] [Google Scholar]
- 20.Moxley, G., and S. Ruddy. 1987. Elevated plasma C3 anaphylatoxin levels in rheumatoid arthritis patients. Arthritis Rheum. 30:1097–1104. [DOI] [PubMed] [Google Scholar]
- 21.Jose, P.J., I.K. Moss, R.N. Maini, and T.J. Williams. 1990. Measurement of the chemotactic complement fragment C5a in rheumatoid synovial fluids by radioimmunoassay: role of C5a in the acute inflammatory phase. Ann. Rheum. Dis. 49:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamann, J., J.O. Wishaupt, R.A. van Lier, T.J. Smeets, F.C. Breedveld, and P.P. Tak. 1999. Expression of the activation antigen CD97 and its ligand CD55 in rheumatoid synovial tissue. Arthritis Rheum. 42:650–658. [DOI] [PubMed] [Google Scholar]
- 23.Korganow, A.S., H. Ji, S. Mangialaio, V. Duchatelle, R. Pelanda, T. Martin, C. Degott, H. Kikutani, K. Rajewsky, J.L. Pasquali, et al. 1999. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 10:451–461. [DOI] [PubMed] [Google Scholar]
- 24.Wang, Y., J. Kristan, L. Hao, C.S. Lenkoski, Y. Shen, and L.A. Matis. 2000. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J. Immunol. 164:4340–4347. [DOI] [PubMed] [Google Scholar]
- 25.Ji, H., K. Ohmura, U. Mahmood, D.M. Lee, F.M. Hofhuis, S.A. Boackle, K. Takahashi, V.M. Holers, M. Walport, C. Gerard, et al. 2002. Arthritis critically dependent on innate immune system players. Immunity. 16:157–168. [DOI] [PubMed] [Google Scholar]
- 26.Daffern, P.J., P.H. Pfeifer, J.A. Ember, and T.E. Hugli. 1995. C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation of neutrophils is secondary to eosinophil activation. J. Exp. Med. 181:2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin, U., D. Bock, L. Arseniev, M.A. Tornetta, R.S. Ames, W. Bautsch, J. Kohl, A. Ganser, and A. Klos. 1997. The human C3a receptor is expressed on neutrophils and monocytes, but not on B or T lymphocytes. J. Exp. Med. 186:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zwirner, J., O. Gotze, G. Begemann, A. Kapp, K. Kirchhoff, and T. Werfel. 1999. Evaluation of C3a receptor expression on human leucocytes by the use of novel monoclonal antibodies. Immunology. 97:166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wipke, B.T., and P.M. Allen. 2001. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 167:1601–1608. [DOI] [PubMed] [Google Scholar]
- 30.Kong, Y.Y., H. Yoshida, I. Sarosi, H.L. Tan, E. Timms, C. Capparelli, S. Morony, A.J. Oliveira-dos-Santos, G. Van, A. Itie, et al. 1999. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 397:315–323. [DOI] [PubMed] [Google Scholar]
- 31.Kong, Y.Y., U. Feige, I. Sarosi, B. Bolon, A. Tafuri, S. Morony, C. Capparelli, J. Li, R. Elliott, S. McCabe, et al. 1999. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 402:304–309. [DOI] [PubMed] [Google Scholar]
- 32.Johansson, A.C., M. Sundler, P. Kjellen, M. Johannesson, A. Cook, A.K. Lindqvist, B. Nakken, A.I. Bolstad, R. Jonsson, M. Alarcon-Riquelme, et al. 2001. Genetic control of collagen-induced arthritis in a cross with NOD and C57BL/10 mice is dependent on gene regions encoding complement factor 5 and FcgammaRIIb and is not associated with loci controlling diabetes. Eur. J. Immunol. 31:1847–1856. [DOI] [PubMed] [Google Scholar]
- 33.Ji, H., D. Gauguier, K. Ohmura, A. Gonzalez, V. Duchatelle, P. Danoy, H.J. Garchon, C. Degott, M. Lathrop, C. Benoist, et al. 2001. Genetic influences on the end-stage effector phase of arthritis. J. Exp. Med. 194:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, Y., S.A. Rollins, J.A. Madri, and L.A. Matis. 1995. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc. Natl. Acad. Sci. USA. 92:8955–8959. [DOI] [PMC free article] [PubMed] [Google Scholar]